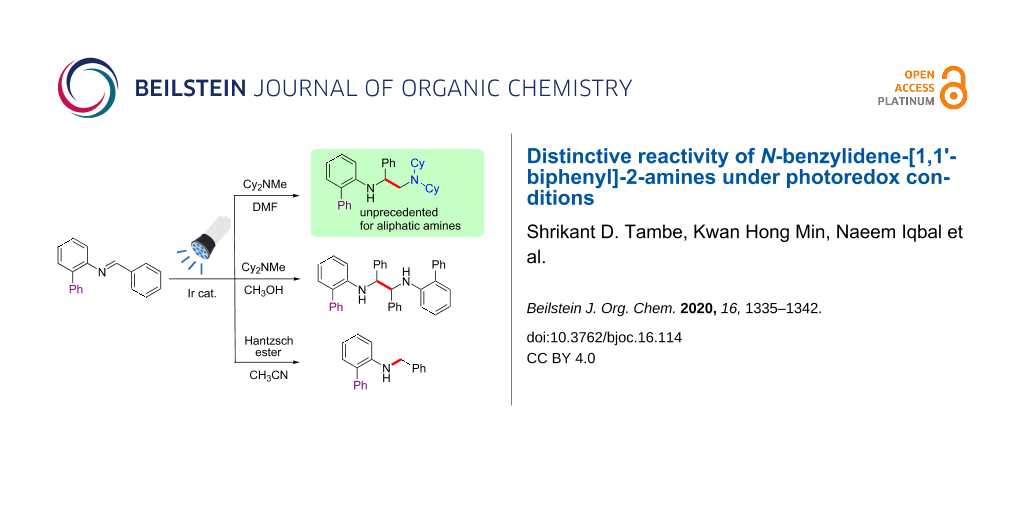
Abstract
A simple photocatalytic method was developed for the synthesis of unsymmetrical 1,2-diamines by the unprecedented reductive coupling of N-benzylidene-[1,1'-biphenyl]-2-amines with an aliphatic amine. The presence of a phenyl substituent in the aniline moiety of the substrate was critical for the reactivity. The reaction proceeded via radical–radical cross-coupling of α-amino radicals generated by proton-coupled single-electron transfer in the presence of an Ir photocatalyst. On the other hand, symmetrical 1,2-diamines were selectively produced from the same starting materials by the judicious choice of the reaction conditions, showcasing the distinct reactivity of N-benzylidene-[1,1'-biphenyl]-2-amines. The developed method can be employed for the synthesis of various bulky vicinal diamines, which are potential ligands in stereoselective synthesis.

Graphical Abstract
Introduction
The selective formation of distinct valuable compounds from the same starting material is a highly attractive divergent approach, though it represents significant synthetic challenges. Recent advances in visible-light photocatalysis, mediated by visible-light-absorbing photosensitizers, have allowed ready access to complex molecules in a controlled manner, where subtle differences in the reaction conditions opened up distinct reaction pathways [1-5].
Imines are versatile substrates that can be converted into various azo compounds, depending upon the reaction conditions [6-9]. In particular, the reactivities of N-benzylidenes have been extensively explored under visible-light photocatalysis [10-16]. N-Benzylidenes can undergo facile single-electron reduction to generate α-amino radical intermediates, which can participate in diverse processes, depending upon the nature of the substrates and the reaction conditions (Scheme 1a). Various amine systems are generated from such intermediates via a wide range of processes, including hydrogen atom abstraction [17,18], the addition to unsaturated compounds [19-21], radical–radical coupling [22-30], and cyclization reactions [31-33]. Among these diverse applications, the Rueping group has reported excellent examples of the reductive umpolung homocoupling of imines and heterocoupling with α-amino radicals for the synthesis of symmetrical and unsymmetrical vicinal diamines (Scheme 1b) [23,26]. Notably, 1,2-diamines have widespread applications as core structures in a variety of natural products, pharmaceuticals, and agrochemicals [34-38] and are valuable ligands [39,40] in stereoselective organic synthesis. Despite the availability of a plethora of synthetic methods for 1,2-diamines [41-45], the reported photocatalytic synthetic methods are mainly limited to aniline-based substrates and do not encompass aliphatic amines.
![[1860-5397-16-114-i1]](/bjoc/content/inline/1860-5397-16-114-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photocatalytic transformations of imines.
Scheme 1: Photocatalytic transformations of imines.
We planned the synthesis of 1,2-diamine compounds having an aliphatic amine moiety by the intermolecular coupling of N-benzylidines with aliphatic amines that not only act as coupling partner but also as electron donors in the photoredox cycle, and the results are reported herein. We began with the reaction of simple N-benzylideneaniline. However, this substrate did not furnish the desired products under several different photocatalysis conditions, including those reported by Rueping. We hypothesized that the structural modification of the substrate might affect its reactivity, and to our delight, the introduction of an ortho-phenyl moiety on the N-benzylideneaniline species provided the desired 1,2-diamine product, wherein the N,N-dicyclohexylmethylamine (Cy2NMe) acted as both the coupling partner and an electron donor in the photoredox cycle (Scheme 1c). It is likely that the presence of the additional phenyl group in the substrate stabilizes the α-amino radical intermediate and modulates its reactivity [46,47]. In addition to the cross-coupled 1,2-diamines, we envisioned the generation of other valuable structural motifs via the careful control of the reaction conditions for the reaction of the biphenyl imine derivative N-benzylidene-[1,1'-biphenyl]-2-amine.
Results and Discussion
With the initial results in hand, we attempted the optimization of the reaction conditions to improve the yield of the 1,2-diamine product (Table 1). Among the series of photocatalysts tested, ranging from the Ru/Ir polypyridyl complexes to organic dyes, [Ir(dtbbpy)(ppy)2]PF6 was found to be the best and afforded 2a in 60% yield, along with 11% of the homocoupled product 3a (Table 1, entries 1–9). The amount of Cy2NMe was critical for achieving selectivity, and less than two equivalents of Cy2NMe gave greater amounts of the homocoupled product 3a (Table 1, entry 10). Control experiments showed that the photocatalyst, amine base, and light source are integral aspects of the reaction (Table 1, entries 11–13). DMF was found to be the best solvent and yielded 2a selectively (Table 1, entries 15–22). On the other hand, the homocoupled 3a was selectively obtained in protic solvents (Table 1, entries 19 and 22), and in particular, CH3OH showed a good reactivity and formed 3a in 75% yield. Unexpectedly, the cyclized product with the tethered phenyl ring proposed in Scheme 1c was not generated under any of the photocatalytic conditions evaluated.
Table 1: Reaction optimization.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-114-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | photocatalyst | variation | solvent | yield (%)b | |
|---|---|---|---|---|---|
| 2a | 3a | ||||
| 1 | [Ru(bpy)3]Cl2⋅6H2O | – | CH3CN | 12 | 0 |
| 2 | [Ru(bpz)3](PF6)2 | – | CH3CN | 0 | 0 |
| 3 | Ir(dFppy)3 | – | CH3CN | 0 | 0 |
| 4 | Ir(ppy)3 | – | CH3CN | 3 | 25 |
| 5 | [Ir(dF(CF3)ppy)2(dtbpy)]PF6 | – | CH3CN | 38 | 5 |
| 6 | [Ir(dtbbpy)(ppy)2]PF6 | – | CH3CN | 60 | 11 |
| 7 | TTPP | – | CH3CN | 0 | 0 |
| 8 | crystal violet | – | CH3CN | 0 | 0 |
| 9 | eosin-Y | – | CH3CN | 38 | 0 |
| 10 | [Ir(dtbbpy)(ppy)2]PF6 | Cy2NMe (1 equiv) | CH3CN | 14 | 55 |
| 11 | [Ir(dtbbpy)(ppy)2]PF6 | no Cy2NMe | CH3CN | 0 | trace |
| 12 | no catalyst | – | CH3CN | 0 | 0 |
| 13 | [Ir(dtbbpy)(ppy)2]PF6 | no light | CH3CN | 0 | 0 |
| 14 | [Ir(dtbbpy)(ppy)2]PF6 | – | DCM | 0 | 16 |
| 15 | [Ir(dtbbpy)(ppy)2]PF6 | – | DMF | 72 (71) | 7 |
| 16 | [Ir(dtbbpy)(ppy)2]PF6 | – | DCE | 22 | 11 |
| 17 | [Ir(dtbbpy)(ppy)2]PF6 | – | DMSO | 37 | 6 |
| 18 | [Ir(dtbbpy)(ppy)2]PF6 | – | dioxane | 50 | 13 |
| 19 | [Ir(dtbbpy)(ppy)2]PF6 | – | TFE | 0 | 20 |
| 20 | [Ir(dtbbpy)(ppy)2]PF6 | – | acetone | 22 | trace |
| 21 | [Ir(dtbbpy)(ppy)2]PF6 | – | EtOAc | 36 | 8 |
| 22 | [Ir(dtbbpy)(ppy)2]PF6 | – | CH3OH | 4 | 75 (75) |
| 23 | [Ir(dtbbpy)(ppy)2]PF6 | – | DMF/CH3OH (1:1) | 35 | 29 |
| 24 | [Ir(dtbbpy)(ppy)2]PF6 | – | DMF/H2O (1:1) | 25 | 10 |
| 25 | [Ir(dtbbpy)(ppy)2]PF6 | – | CH3CN/H2O (1:1) | 15 | 14 |
aReaction conditions: 1a (0.1 mmol), under argon atmosphere. bThe yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as the internal standard, and the isolated yields are mentioned in parentheses.
With the optimized conditions in hand, the generality of the transformations was investigated using a wide variety of phenyl-substituted N-benzylideneaniline derivatives (Scheme 2). First, the C–C cross-coupling process with Cy2NMe was explored, with variations of the benzylidene moiety. The reactions with both electron-donating (2b–2e) and electron-withdrawing substituents (2i–2m) proceeded well. Several functional groups, such as benzylic ones (2b and 2c), ethers (2e), halogens (2i and 2l) [48-50], the medicinally important CF3 group (2m), and acetals (2p) were tolerated under these mild reaction conditions. The substitution pattern of the aryl groups, such as ortho (2c, 2e, and 2k), meta (2m and 2p), and para substitution (2b–2d, 2g, 2i, 2l, 2o, and 2p), did not have any significant impact on the reaction outcome. The heteroaryl ring-bearing substrates (2n and 2o) also underwent the transformation, and furnished the valuable vicinal diamine products. The modifications on the aniline moiety were also suitable, and substrates with both electron-donating (2q and 2r) and electron-withdrawing (2s) substituents underwent the cross-coupling with excellent reactivities. We also tried the transformation with aliphatic amines other than Cy2NMe, such as TMEDA, TEA, and DIPEA. However, these reactions proceeded with a poor chemoselectivity and resulted in the formation of mixtures of several types of amine compounds.
![[1860-5397-16-114-i2]](/bjoc/content/inline/1860-5397-16-114-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope for the radical cross-couplings. Reaction conditions: 1 (0.3 mmol), under argon atmosphere, isolated yields. aThe debrominated product was also detected, and the yield represents the mixture of the brominated and debrominated product.
Scheme 2: Substrate scope for the radical cross-couplings. Reaction conditions: 1 (0.3 mmol), under argon atm...
A photochemical quantum yield, determined by using the standard ferrioxalate actinometry, was 83% for the radical cross-coupling between 1a and Cy2NMe (see Supporting Information File 1 for details).
Next, the homocoupling reactions of substituted (E)-N-benzylidene-[1,1'-biphenyl]-2-amines were studied in CH3OH (Scheme 3) [51-56]. It is noteworthy that the resulting symmetrical 1,2-diamine compounds are potential ligands for a variety of organic transformations. The dimerization reactions proceeded well, regardless of the electron density or position of the substituent on the benzylidene moiety. Interestingly, the homocoupling process was highly stereoselective, resulting in the formation of only one diastereomer, probably due to the bulky substitution pattern of the substrates. The structure of the homocoupled 1,2-diamine product 3a was unambiguously confirmed using X-ray crystallography [57].
![[1860-5397-16-114-i3]](/bjoc/content/inline/1860-5397-16-114-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Substrate scope for the homocoupling. Reaction conditions: 1 (0.3 mmol), under argon atmosphere, isolated yield. aThe debrominated product was also detected, and the yield represents the mixture of the brominated and debrominated product.
Scheme 3: Substrate scope for the homocoupling. Reaction conditions: 1 (0.3 mmol), under argon atmosphere, is...
To further extend the diversity of the reactivity of the imines in this process, we also attempted the use of a reducing agent in the reaction of 1a. Pleasingly, the reaction of 1a with 2 equivalents of the Hantzsch ester in CH3CN produced the reduced amine product 4a in 67% yield (Scheme 4) [17,18,58-61].
![[1860-5397-16-114-i4]](/bjoc/content/inline/1860-5397-16-114-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reduction of the imine 1a to the amine 4a.
Scheme 4: Reduction of the imine 1a to the amine 4a.
Based on these observations, a plausible reaction mechanism was proposed for the developed transformation (Scheme 5). Upon visible-light irradiation, the excited photocatalyst [IrIII]* is formed and is reductively quenched by single-electron transfer from Cy2NMe, resulting in the generation of the highly reducing [IrII] species and the radical cation A. To validate the reductive quenching pathway, we carried out Stern−Volmer quenching experiments (Figure S1, Supporting Information File 1). The emission intensity of the excited Ir complex significantly decreased in proportion to the concentration of Cy2NMe, while it was much less affected by the concentration of 1a, confirming the proposed working mode. The formation of 2a might be attributed to the proton-coupled electron transfer [62-66] from [IrII] to the imine 1a, where the radical cation A donates a proton to 1a to form the α-amino radical intermediates B and C, which undergo cross-coupling to give the desired unsymmetrical vicinal diamine 2a. On the other hand, in CH3OH, 1a preferentially abstracts a proton from CH3OH rather than from A, which, in turn, prevents the generation of the α-amino radical C from Cy2NMe, resulting in the homocoupling of B to selectively form the symmetrical diamine product 3a.
![[1860-5397-16-114-i5]](/bjoc/content/inline/1860-5397-16-114-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
We developed a divergent synthetic approach for the valuable 1,2-diamine motif from N-benzylidene-[1,1'-biphenyl]-2-amines by slight alterations to the reaction conditions. The in situ-generated α-amino radical intermediates successfully underwent cross- and homocoupling to yield the unsymmetrical and symmetrical 1,2-diamines, respectively. The presence of the phenyl substituent at the aniline moiety was critical in the heterocoupling with aliphatic amines. Furthermore, the reduced amine product was also obtained by employing the Hantzsch ester. The developed method can be employed for the synthesis of bulky vicinal diamines with potential applications as ligands for stereoselective synthesis.
Experimental
An oven-dried resealable tube, equipped with a magnetic stir bar, was charged with the N-benzylidene-[1,1'-biphenyl]-2-amine derivative (0.3 mmol), [Ir(dtbbpy)(ppy)2]PF6 (0.003 mmol), and Cy2NMe (0.6 mmol). The reaction mixture was purged with argon for 20 min. Then, degassed DMF or CH3OH was added to the reaction mixture under inert conditions. The reaction mixture was stirred at ambient temperature for 2–15 h under visible-light irradiation with blue LEDs (18 W). The progress of the reaction was monitored by using TLC. Upon the completion of the reaction, the crude product was diluted with ethyl acetate and washed with brine. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo. The desired vicinal diamine product was purified by silica-gel column chromatography using hexane/EtOAc as the eluent.
References
-
Cheng, Q.-Q.; Yedoyan, J.; Arman, H.; Doyle, M. P. J. Am. Chem. Soc. 2016, 138, 44–47. doi:10.1021/jacs.5b10860
Return to citation in text: [1] -
Iqbal, N.; Jung, J.; Park, S.; Cho, E. J. Angew. Chem., Int. Ed. 2014, 53, 539–542. doi:10.1002/anie.201308735
Return to citation in text: [1] -
Liu, Z.; Xu, H.; Yao, T.; Zhang, J.; Liu, L. Org. Lett. 2019, 21, 7539–7543. doi:10.1021/acs.orglett.9b02810
Return to citation in text: [1] -
Mose, R.; Preegel, G.; Larsen, J.; Jakobsen, S.; Iversen, E. H.; Jørgensen, K. A. Nat. Chem. 2017, 9, 487–492. doi:10.1038/nchem.2682
Return to citation in text: [1] -
Park, D. D.; Min, K. H.; Kang, J.; Hwang, H. S.; Soni, V. K.; Cho, C.-G.; Cho, E. J. Org. Lett. 2020, 22, 1130–1134. doi:10.1021/acs.orglett.9b04646
Return to citation in text: [1] -
Miyabe, H.; Yoshioka, E.; Kohtani, S. Curr. Org. Chem. 2010, 14, 1254–1264. doi:10.2174/138527210791330477
Return to citation in text: [1] -
Belowich, M. E.; Stoddart, J. F. Chem. Soc. Rev. 2012, 41, 2003–2024. doi:10.1039/c2cs15305j
Return to citation in text: [1] -
Lei, J.; Huang, J.; Zhu, Q. Org. Biomol. Chem. 2016, 14, 2593–2602. doi:10.1039/c6ob00087h
Return to citation in text: [1] -
Otero, A.; Chapela, M.-J.; Atanassova, M.; Vieites, J. M.; Cabado, A. G. Chem. Res. Toxicol. 2011, 24, 1817–1829. doi:10.1021/tx200182m
Return to citation in text: [1] -
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1] -
Chatterjee, T.; Iqbal, N.; You, Y.; Cho, E. J. Acc. Chem. Res. 2016, 49, 2284–2294. doi:10.1021/acs.accounts.6b00248
Return to citation in text: [1] -
Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044–2056. doi:10.1039/c5cs00655d
Return to citation in text: [1] -
Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. doi:10.1002/anie.201709766
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057
Return to citation in text: [1] -
Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239176. doi:10.1126/science.1239176
Return to citation in text: [1] -
Guo, X.; Okamoto, Y.; Schreier, M. R.; Ward, T. R.; Wenger, O. S. Eur. J. Org. Chem. 2020, 1288–1293. doi:10.1002/ejoc.201900777
Return to citation in text: [1] [2] -
Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Pu, C.-D.; Shen, Y.-M.; Wu, C.-D.; Peng, X.-G. J. Org. Chem. 2018, 83, 11886–11895. doi:10.1021/acs.joc.8b01651
Return to citation in text: [1] [2] -
Lee, K. N.; Lei, Z.; Ngai, M.-Y. J. Am. Chem. Soc. 2017, 139, 5003–5006. doi:10.1021/jacs.7b01373
Return to citation in text: [1] -
Qi, L.; Chen, Y. Angew. Chem., Int. Ed. 2016, 55, 13312–13315. doi:10.1002/anie.201607813
Return to citation in text: [1] -
Wu, G.; Wang, J.; Liu, C.; Sun, M.; Zhang, L.; Ma, Y.; Cheng, R.; Ye, J. Org. Chem. Front. 2019, 6, 2245–2249. doi:10.1039/c9qo00407f
Return to citation in text: [1] -
Chen, M.; Zhao, X.; Yang, C.; Xia, W. Org. Lett. 2017, 19, 3807–3810. doi:10.1021/acs.orglett.7b01677
Return to citation in text: [1] -
Fava, E.; Millet, A.; Nakajima, M.; Loescher, S.; Rueping, M. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. doi:10.1002/anie.201511235
Return to citation in text: [1] [2] -
Hager, D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 16986–16989. doi:10.1021/ja5102695
Return to citation in text: [1] -
Jeffrey, J. L.; Petronijević, F. R.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 8404–8407. doi:10.1021/jacs.5b05376
Return to citation in text: [1] -
Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. doi:10.1002/anie.201501556
Return to citation in text: [1] [2] -
Okamoto, S.; Ariki, R.; Tsujioka, H.; Sudo, A. J. Org. Chem. 2017, 82, 9731–9736. doi:10.1021/acs.joc.7b01838
Return to citation in text: [1] -
Okamoto, S.; Kojiyama, K.; Tsujioka, H.; Sudo, A. Chem. Commun. 2016, 52, 11339–11342. doi:10.1039/c6cc05867a
Return to citation in text: [1] -
Patel, N. R.; Kelly, C. B.; Siegenfeld, A. P.; Molander, G. A. ACS Catal. 2017, 7, 1766–1770. doi:10.1021/acscatal.6b03665
Return to citation in text: [1] -
Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. J. Am. Chem. Soc. 2015, 137, 13768–13771. doi:10.1021/jacs.5b09329
Return to citation in text: [1] -
Fava, E.; Nakajima, M.; Nguyen, A. L. P.; Rueping, M. J. Org. Chem. 2016, 81, 6959–6964. doi:10.1021/acs.joc.6b01006
Return to citation in text: [1] -
Leitch, J. A.; Fuentes de Arriba, A. L.; Tan, J.; Hoff, O.; Martínez, C. M.; Dixon, D. J. Chem. Sci. 2018, 9, 6653–6658. doi:10.1039/c8sc01704b
Return to citation in text: [1] -
Rono, L. J.; Yayla, H. G.; Wang, D. Y.; Armstrong, M. F.; Knowles, R. R. J. Am. Chem. Soc. 2013, 135, 17735–17738. doi:10.1021/ja4100595
Return to citation in text: [1] -
Viso, A.; Fernández de la Pradilla, R.; García, A.; Flores, A. Chem. Rev. 2005, 105, 3167–3196. doi:10.1021/cr0406561
Return to citation in text: [1] -
Lucet, D.; Le Gall, T.; Mioskowski, C. Angew. Chem., Int. Ed. 1998, 37, 2580–2627. doi:10.1002/(sici)1521-3773(19981016)37:19<2580::aid-anie2580>3.0.co;2-l
Return to citation in text: [1] -
Bogatcheva, E.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Barbosa, F.; Einck, L.; Nacy, C. A.; Protopopova, M. J. Med. Chem. 2006, 49, 3045–3048. doi:10.1021/jm050948+
Return to citation in text: [1] -
Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 7516–7520. doi:10.1021/ja301120z
Return to citation in text: [1] -
Saibabu Kotti, S. R. S.; Timmons, C.; Li, G. Chem. Biol. Drug Des. 2006, 67, 101–114. doi:10.1111/j.1747-0285.2006.00347.x
Return to citation in text: [1] -
Kizirian, J.-C. Chem. Rev. 2008, 108, 140–205. doi:10.1021/cr040107v
Return to citation in text: [1] -
Bennani, Y. L.; Hanessian, S. Chem. Rev. 1997, 97, 3161–3196. doi:10.1021/cr9407577
Return to citation in text: [1] -
Cai, C.-Y.; Shu, X.-M.; Xu, H.-C. Nat. Commun. 2019, 10, 4953. doi:10.1038/s41467-019-13024-5
Return to citation in text: [1] -
Ichikawa, S.; Dai, X.-J.; Buchwald, S. L. Org. Lett. 2019, 21, 4370–4373. doi:10.1021/acs.orglett.9b01592
Return to citation in text: [1] -
Mwenda, E. T.; Nguyen, H. M. Org. Lett. 2017, 19, 4814–4817. doi:10.1021/acs.orglett.7b02256
Return to citation in text: [1] -
Xiao, L.; Zhao, Y.; Qiao, S.; Sun, Z.; Santoro, O.; Redshaw, C. Dalton Trans. 2020, 49, 1456–1472. doi:10.1039/c9dt04332b
Return to citation in text: [1] -
Yu, L.; Somfai, P. Angew. Chem., Int. Ed. 2019, 58, 8551–8555. doi:10.1002/anie.201902642
Return to citation in text: [1] -
Aurrecoechea, J. M.; Suero, R. ARKIVOC 2004, No. 14, 10–35. doi:10.3998/ark.5550190.0005.e02
Return to citation in text: [1] -
McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920
Return to citation in text: [1] -
Kim, H.; Lee, C. Org. Lett. 2011, 13, 2050–2053. doi:10.1021/ol200455n
Return to citation in text: [1] -
Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582
Return to citation in text: [1] -
Kim, H.; Lee, C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. doi:10.1002/anie.201203599
Return to citation in text: [1] -
Machrouhi, F.; Namy, J.-L. Tetrahedron Lett. 1999, 40, 1315–1318. doi:10.1016/s0040-4039(98)02673-2
Return to citation in text: [1] -
VellemÄe, E.; Tsubrik, O.; Mäeorg, S.; Mäeorg, U. J. Chem. Res. 2006, 149–150. doi:10.3184/030823406776330792
Return to citation in text: [1] -
Jiang, H.-F.; Huang, X.-Z. J. Supercrit. Fluids 2007, 43, 291–294. doi:10.1016/j.supflu.2007.07.005
Return to citation in text: [1] -
Kumar, A.; Samuelson, A. G. Chem. – Asian J. 2010, 5, 1830–1837. doi:10.1002/asia.200900711
Return to citation in text: [1] -
Rao, C. N.; Hoz, S. J. Am. Chem. Soc. 2011, 133, 14795–14803. doi:10.1021/ja205885q
Return to citation in text: [1] -
Zhu, Y.; Buchwald, S. L. J. Am. Chem. Soc. 2014, 136, 4500–4503. doi:10.1021/ja501560x
Return to citation in text: [1] -
CCDC 1991125 contain the supplementary crystallographic data for this article, which can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Return to citation in text: [1] -
Cheng, X.; Huang, W. Synlett 2016, 28, 148–158. doi:10.1055/s-0036-1588129
Return to citation in text: [1] -
Jung, J.; Kim, J.; Park, G.; You, Y.; Cho, E. J. Adv. Synth. Catal. 2016, 358, 74–80. doi:10.1002/adsc.201500734
Return to citation in text: [1] -
Park, G.; Yi, S. Y.; Jung, J.; Cho, E. J.; You, Y. Chem. – Eur. J. 2016, 22, 17790–17799. doi:10.1002/chem.201603517
Return to citation in text: [1] -
Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Org. Biomol. Chem. 2019, 17, 6936–6951. doi:10.1039/c9ob01289c
Return to citation in text: [1] -
Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546–1556. doi:10.1021/acs.accounts.6b00272
Return to citation in text: [1] -
Hoffmann, N. Eur. J. Org. Chem. 2017, 1982–1992. doi:10.1002/ejoc.201601445
Return to citation in text: [1] -
Jiang, H.; Studer, A. CCS Chem. 2019, 38–49. doi:10.31635/ccschem.019.20180026
Return to citation in text: [1] -
Pannwitz, A.; Wenger, O. S. Chem. Commun. 2019, 55, 4004–4014. doi:10.1039/c9cc00821g
Return to citation in text: [1] -
Weinberg, D. R.; Gagliardi, C. J.; Hull, J. F.; Murphy, C. F.; Kent, C. A.; Westlake, B. C.; Paul, A.; Ess, D. H.; McCafferty, D. G.; Meyer, T. J. Chem. Rev. 2012, 112, 4016–4093. doi:10.1021/cr200177j
Return to citation in text: [1]
| 62. | Gentry, E. C.; Knowles, R. R. Acc. Chem. Res. 2016, 49, 1546–1556. doi:10.1021/acs.accounts.6b00272 |
| 63. | Hoffmann, N. Eur. J. Org. Chem. 2017, 1982–1992. doi:10.1002/ejoc.201601445 |
| 64. | Jiang, H.; Studer, A. CCS Chem. 2019, 38–49. doi:10.31635/ccschem.019.20180026 |
| 65. | Pannwitz, A.; Wenger, O. S. Chem. Commun. 2019, 55, 4004–4014. doi:10.1039/c9cc00821g |
| 66. | Weinberg, D. R.; Gagliardi, C. J.; Hull, J. F.; Murphy, C. F.; Kent, C. A.; Westlake, B. C.; Paul, A.; Ess, D. H.; McCafferty, D. G.; Meyer, T. J. Chem. Rev. 2012, 112, 4016–4093. doi:10.1021/cr200177j |
| 1. | Cheng, Q.-Q.; Yedoyan, J.; Arman, H.; Doyle, M. P. J. Am. Chem. Soc. 2016, 138, 44–47. doi:10.1021/jacs.5b10860 |
| 2. | Iqbal, N.; Jung, J.; Park, S.; Cho, E. J. Angew. Chem., Int. Ed. 2014, 53, 539–542. doi:10.1002/anie.201308735 |
| 3. | Liu, Z.; Xu, H.; Yao, T.; Zhang, J.; Liu, L. Org. Lett. 2019, 21, 7539–7543. doi:10.1021/acs.orglett.9b02810 |
| 4. | Mose, R.; Preegel, G.; Larsen, J.; Jakobsen, S.; Iversen, E. H.; Jørgensen, K. A. Nat. Chem. 2017, 9, 487–492. doi:10.1038/nchem.2682 |
| 5. | Park, D. D.; Min, K. H.; Kang, J.; Hwang, H. S.; Soni, V. K.; Cho, C.-G.; Cho, E. J. Org. Lett. 2020, 22, 1130–1134. doi:10.1021/acs.orglett.9b04646 |
| 19. | Lee, K. N.; Lei, Z.; Ngai, M.-Y. J. Am. Chem. Soc. 2017, 139, 5003–5006. doi:10.1021/jacs.7b01373 |
| 20. | Qi, L.; Chen, Y. Angew. Chem., Int. Ed. 2016, 55, 13312–13315. doi:10.1002/anie.201607813 |
| 21. | Wu, G.; Wang, J.; Liu, C.; Sun, M.; Zhang, L.; Ma, Y.; Cheng, R.; Ye, J. Org. Chem. Front. 2019, 6, 2245–2249. doi:10.1039/c9qo00407f |
| 57. | CCDC 1991125 contain the supplementary crystallographic data for this article, which can be obtained free of charge from The Cambridge Crystallographic Data Centre. |
| 17. | Guo, X.; Okamoto, Y.; Schreier, M. R.; Ward, T. R.; Wenger, O. S. Eur. J. Org. Chem. 2020, 1288–1293. doi:10.1002/ejoc.201900777 |
| 18. | Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Pu, C.-D.; Shen, Y.-M.; Wu, C.-D.; Peng, X.-G. J. Org. Chem. 2018, 83, 11886–11895. doi:10.1021/acs.joc.8b01651 |
| 17. | Guo, X.; Okamoto, Y.; Schreier, M. R.; Ward, T. R.; Wenger, O. S. Eur. J. Org. Chem. 2020, 1288–1293. doi:10.1002/ejoc.201900777 |
| 18. | Xi, Z.-W.; Yang, L.; Wang, D.-Y.; Pu, C.-D.; Shen, Y.-M.; Wu, C.-D.; Peng, X.-G. J. Org. Chem. 2018, 83, 11886–11895. doi:10.1021/acs.joc.8b01651 |
| 58. | Cheng, X.; Huang, W. Synlett 2016, 28, 148–158. doi:10.1055/s-0036-1588129 |
| 59. | Jung, J.; Kim, J.; Park, G.; You, Y.; Cho, E. J. Adv. Synth. Catal. 2016, 358, 74–80. doi:10.1002/adsc.201500734 |
| 60. | Park, G.; Yi, S. Y.; Jung, J.; Cho, E. J.; You, Y. Chem. – Eur. J. 2016, 22, 17790–17799. doi:10.1002/chem.201603517 |
| 61. | Wang, P.-Z.; Chen, J.-R.; Xiao, W.-J. Org. Biomol. Chem. 2019, 17, 6936–6951. doi:10.1039/c9ob01289c |
| 10. | Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707 |
| 11. | Chatterjee, T.; Iqbal, N.; You, Y.; Cho, E. J. Acc. Chem. Res. 2016, 49, 2284–2294. doi:10.1021/acs.accounts.6b00248 |
| 12. | Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044–2056. doi:10.1039/c5cs00655d |
| 13. | Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. doi:10.1002/anie.201709766 |
| 14. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 15. | Romero, N. A.; Nicewicz, D. A. Chem. Rev. 2016, 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057 |
| 16. | Schultz, D. M.; Yoon, T. P. Science 2014, 343, 1239176. doi:10.1126/science.1239176 |
| 48. | Kim, H.; Lee, C. Org. Lett. 2011, 13, 2050–2053. doi:10.1021/ol200455n |
| 49. | Narayanam, J. M. R.; Tucker, J. W.; Stephenson, C. R. J. J. Am. Chem. Soc. 2009, 131, 8756–8757. doi:10.1021/ja9033582 |
| 50. | Kim, H.; Lee, C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. doi:10.1002/anie.201203599 |
| 6. | Miyabe, H.; Yoshioka, E.; Kohtani, S. Curr. Org. Chem. 2010, 14, 1254–1264. doi:10.2174/138527210791330477 |
| 7. | Belowich, M. E.; Stoddart, J. F. Chem. Soc. Rev. 2012, 41, 2003–2024. doi:10.1039/c2cs15305j |
| 8. | Lei, J.; Huang, J.; Zhu, Q. Org. Biomol. Chem. 2016, 14, 2593–2602. doi:10.1039/c6ob00087h |
| 9. | Otero, A.; Chapela, M.-J.; Atanassova, M.; Vieites, J. M.; Cabado, A. G. Chem. Res. Toxicol. 2011, 24, 1817–1829. doi:10.1021/tx200182m |
| 51. | Machrouhi, F.; Namy, J.-L. Tetrahedron Lett. 1999, 40, 1315–1318. doi:10.1016/s0040-4039(98)02673-2 |
| 52. | VellemÄe, E.; Tsubrik, O.; Mäeorg, S.; Mäeorg, U. J. Chem. Res. 2006, 149–150. doi:10.3184/030823406776330792 |
| 53. | Jiang, H.-F.; Huang, X.-Z. J. Supercrit. Fluids 2007, 43, 291–294. doi:10.1016/j.supflu.2007.07.005 |
| 54. | Kumar, A.; Samuelson, A. G. Chem. – Asian J. 2010, 5, 1830–1837. doi:10.1002/asia.200900711 |
| 55. | Rao, C. N.; Hoz, S. J. Am. Chem. Soc. 2011, 133, 14795–14803. doi:10.1021/ja205885q |
| 56. | Zhu, Y.; Buchwald, S. L. J. Am. Chem. Soc. 2014, 136, 4500–4503. doi:10.1021/ja501560x |
| 34. | Viso, A.; Fernández de la Pradilla, R.; García, A.; Flores, A. Chem. Rev. 2005, 105, 3167–3196. doi:10.1021/cr0406561 |
| 35. | Lucet, D.; Le Gall, T.; Mioskowski, C. Angew. Chem., Int. Ed. 1998, 37, 2580–2627. doi:10.1002/(sici)1521-3773(19981016)37:19<2580::aid-anie2580>3.0.co;2-l |
| 36. | Bogatcheva, E.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Barbosa, F.; Einck, L.; Nacy, C. A.; Protopopova, M. J. Med. Chem. 2006, 49, 3045–3048. doi:10.1021/jm050948+ |
| 37. | Kano, T.; Sakamoto, R.; Akakura, M.; Maruoka, K. J. Am. Chem. Soc. 2012, 134, 7516–7520. doi:10.1021/ja301120z |
| 38. | Saibabu Kotti, S. R. S.; Timmons, C.; Li, G. Chem. Biol. Drug Des. 2006, 67, 101–114. doi:10.1111/j.1747-0285.2006.00347.x |
| 41. | Cai, C.-Y.; Shu, X.-M.; Xu, H.-C. Nat. Commun. 2019, 10, 4953. doi:10.1038/s41467-019-13024-5 |
| 42. | Ichikawa, S.; Dai, X.-J.; Buchwald, S. L. Org. Lett. 2019, 21, 4370–4373. doi:10.1021/acs.orglett.9b01592 |
| 43. | Mwenda, E. T.; Nguyen, H. M. Org. Lett. 2017, 19, 4814–4817. doi:10.1021/acs.orglett.7b02256 |
| 44. | Xiao, L.; Zhao, Y.; Qiao, S.; Sun, Z.; Santoro, O.; Redshaw, C. Dalton Trans. 2020, 49, 1456–1472. doi:10.1039/c9dt04332b |
| 45. | Yu, L.; Somfai, P. Angew. Chem., Int. Ed. 2019, 58, 8551–8555. doi:10.1002/anie.201902642 |
| 23. | Fava, E.; Millet, A.; Nakajima, M.; Loescher, S.; Rueping, M. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. doi:10.1002/anie.201511235 |
| 26. | Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. doi:10.1002/anie.201501556 |
| 46. | Aurrecoechea, J. M.; Suero, R. ARKIVOC 2004, No. 14, 10–35. doi:10.3998/ark.5550190.0005.e02 |
| 47. | McNally, A.; Prier, C. K.; MacMillan, D. W. C. Science 2011, 334, 1114–1117. doi:10.1126/science.1213920 |
| 31. | Fava, E.; Nakajima, M.; Nguyen, A. L. P.; Rueping, M. J. Org. Chem. 2016, 81, 6959–6964. doi:10.1021/acs.joc.6b01006 |
| 32. | Leitch, J. A.; Fuentes de Arriba, A. L.; Tan, J.; Hoff, O.; Martínez, C. M.; Dixon, D. J. Chem. Sci. 2018, 9, 6653–6658. doi:10.1039/c8sc01704b |
| 33. | Rono, L. J.; Yayla, H. G.; Wang, D. Y.; Armstrong, M. F.; Knowles, R. R. J. Am. Chem. Soc. 2013, 135, 17735–17738. doi:10.1021/ja4100595 |
| 22. | Chen, M.; Zhao, X.; Yang, C.; Xia, W. Org. Lett. 2017, 19, 3807–3810. doi:10.1021/acs.orglett.7b01677 |
| 23. | Fava, E.; Millet, A.; Nakajima, M.; Loescher, S.; Rueping, M. Angew. Chem., Int. Ed. 2016, 55, 6776–6779. doi:10.1002/anie.201511235 |
| 24. | Hager, D.; MacMillan, D. W. C. J. Am. Chem. Soc. 2014, 136, 16986–16989. doi:10.1021/ja5102695 |
| 25. | Jeffrey, J. L.; Petronijević, F. R.; MacMillan, D. W. C. J. Am. Chem. Soc. 2015, 137, 8404–8407. doi:10.1021/jacs.5b05376 |
| 26. | Nakajima, M.; Fava, E.; Loescher, S.; Jiang, Z.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 8828–8832. doi:10.1002/anie.201501556 |
| 27. | Okamoto, S.; Ariki, R.; Tsujioka, H.; Sudo, A. J. Org. Chem. 2017, 82, 9731–9736. doi:10.1021/acs.joc.7b01838 |
| 28. | Okamoto, S.; Kojiyama, K.; Tsujioka, H.; Sudo, A. Chem. Commun. 2016, 52, 11339–11342. doi:10.1039/c6cc05867a |
| 29. | Patel, N. R.; Kelly, C. B.; Siegenfeld, A. P.; Molander, G. A. ACS Catal. 2017, 7, 1766–1770. doi:10.1021/acscatal.6b03665 |
| 30. | Uraguchi, D.; Kinoshita, N.; Kizu, T.; Ooi, T. J. Am. Chem. Soc. 2015, 137, 13768–13771. doi:10.1021/jacs.5b09329 |
| 39. | Kizirian, J.-C. Chem. Rev. 2008, 108, 140–205. doi:10.1021/cr040107v |
| 40. | Bennani, Y. L.; Hanessian, S. Chem. Rev. 1997, 97, 3161–3196. doi:10.1021/cr9407577 |
© 2020 Tambe et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)