Abstract
A straightforward sequential synthetic strategy has been developed for the synthesis of a pentasaccharide repeating unit corresponding to the cell wall O-antigen of the Escherichia albertii O4 strain in very good yield with the desired configuration at the glycosidic linkages using thioglycosides and trichloroacetimidate derivatives as glycosyl donors and perchloric acid supported over silica (HClO4/SiO2) as a solid supported protic acid glycosyl activator. The expected configuration at the glycosidic linkages was achieved using a reasonable selection of protecting groups in the manosaccharide intermediates.

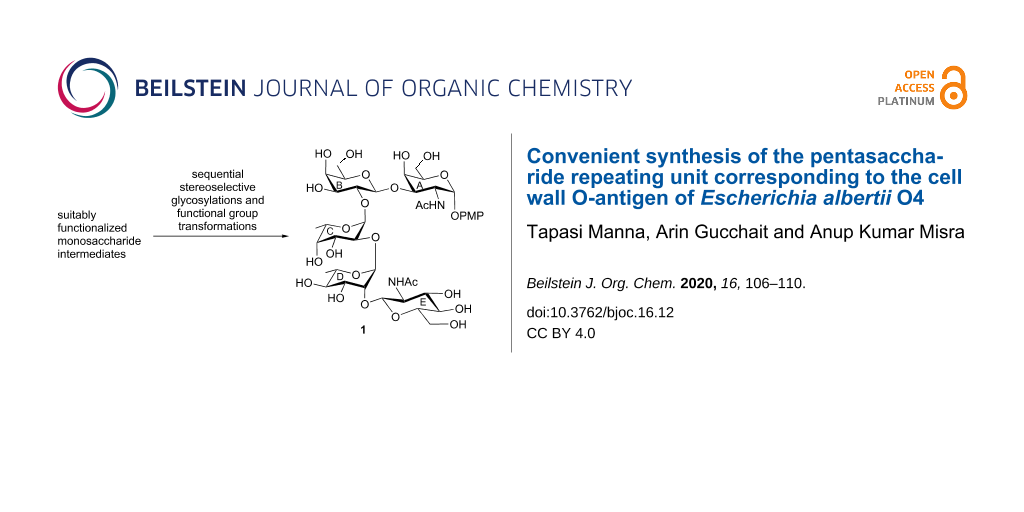
Graphical Abstract
Introduction
Diarrheal outbreaks are serious concerns all over the world particularly in the developing countries due to inadequate sanitation systems [1]. In most of the cases, the enteric infections originated due to the intake of less cooked food and contaminated water [2]. Several strains of Shigella [3], Salmonella [4] and enteropathogenic Escherichia coli [5] are commonly known for causing diarrheal infections. Besides the mainstream enteropathogenic bacterium, Escherichia albertii (E. albertii) is an emerging human pathogen causing gastroenteric infections in different countries [6]. Although, this species was identified earlier as Hafnia alvei, later it was redesignated as E. albertii [7]. E. albertii acted as a causative agent for diarrheal diseases in children with vomiting, fever and abdominal distension [8]. Several strains of E. albertii have been identified till date, which significantly contributed to the spreading of devastating diarrheal infections in different countries [9]. The role of cell wall O-polysaccharides in regulating the virulence properties of bacteria is well established [10]. Recently, Naumenko et al. [11] reported the structure of the repeating unit of the cell wall O-polysaccharide of the E. albertii O4 strain [11], which is a pentasaccharide comprising of α-linked ᴅ-galactosamine, β-linked ᴅ-glucosamine, β-linked ᴅ-galactose, α-linked ʟ-fucose and α-linked ʟ-rhamnose moieties. In the recent past, several vaccine candidates have been developed to control bacterial infections by conjugating cell wall polysaccharides with suitable proteins, which include vaccines against Haemophilia influenza type b (Hib) [12,13], meningitis [14], pneumococcal infections [15,16] and enteric diseases such as cholera [17], diarrhea [18] and urinary tract infections [19]. Despite the possibility of isolating the polysaccharides by fermentation techniques, it is difficult to get a significant quantity of polysaccharide fragments from natural sources with adequate purity. Therefore, the development of chemical synthetic strategies is quite pertinent to obtain a requisite quantity of oligosaccharide fragments with adequate purity. In this direction, the total synthesis of the pentasaccharide repeating unit corresponding to the cell wall O-antigenic polysaccharide of the E. albertii O4 strain using a sequential glycosylation strategy is presented herein (Figure 1).
![[1860-5397-16-12-1]](/bjoc/content/figures/1860-5397-16-12-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of the pentasaccharide repeating unit corresponding to the cell wall O-antigen of Escherichia albertii O4 and its synthetic intermediates.
Figure 1: Structure of the pentasaccharide repeating unit corresponding to the cell wall O-antigen of Escheri...
Results and Discussion
The synthesis of pentasaccharide 1 was achieved using a convergent as well as a block synthetic strategy. For this purpose, a series of suitably functionalized monosaccharide intermediates 2 [20], 3 [21], 4 [22], 5 [23], 6 [24] and 7 [25] were prepared from the commercially available reducing sugars utilizing the reaction conditions reported in the literature (Figure 1). Although the monosaccharide intermediates used for the construction of the pentasaccharide derivative 15 are known in the literature, preparation of these intermediates required multiple step reaction sequences. Having obtained the monosaccharide intermediates, it was decided to proceed through a step-economic block synthetic strategy to achieve the target pentasaccharide derivative. Accordingly, stereoselective glycosylation of a ᴅ-galactosamine derivative 2 with a ᴅ-galactose thioglycoside derivative 3 in the presence of a combination [26,27] of N-iodosuccinimide (NIS) and perchloric acid supported over silica (HClO4/SiO2) [28,29] furnished disaccharide derivative 8 in 79% yield, which on de-O-acetylation using sodium methoxide [30] gave the disaccharide acceptor 9 in 95% yield. NMR spectral analysis of compound 9 confirmed its formation with appropriate configuration at the glycosidic linkages [Signals at δ 5.54 (d, J = 2.5 Hz, H-1A), 5.44 (s, PhCH), 4.54 (d, J = 7.5 Hz, H-1B) in 1H NMR and at δ 105.2 (C-1B), 100.6 (PhCH), 98.2 (C-1A) in 13C NMR spectra] (Scheme 1).
![[1860-5397-16-12-i1]](/bjoc/content/inline/1860-5397-16-12-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: (a) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, −45 °C, 1 h, 79%; (b) 0.1 M CH3ONa, CH3OH, room temperature, 2 h, 95%.
Scheme 1: (a) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, −45 °C, 1 h, 79%; (b) 0.1 M CH3ONa, CH3OH, room temperature, 2...
In another experiment, ʟ-rhamnosyl trichloroacetimidate donor 5 was coupled with ʟ-fucosyl thioglycoside acceptor 4 in the presence of HClO4/SiO2 [31] as activator using an orthogonal glycosylation approach to furnish disaccharide thioglycoside derivative 10 in 76% yield, which was directly used in the next level of glycosylation. NMR spectral analysis of compound 10 unambiguously confirmed its formation [signals at δ 5.26 (d, J = 1.5 Hz, H-1D), 4.23 (d, J = 9.5 Hz, H-1C) in 1H NMR and at δ 98.4 (C-1D), 84.8 (C-1C) in 13C NMR spectra] (Scheme 2). It is worth noting that sulfide linkage at the anomeric position of compound 4 remained unaffected under the reaction conditions.
![[1860-5397-16-12-i2]](/bjoc/content/inline/1860-5397-16-12-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: (a) HClO4/SiO2, CH2Cl2, −10 °C, 1 h, 76%.
Scheme 2: (a) HClO4/SiO2, CH2Cl2, −10 °C, 1 h, 76%.
Having achieved the disaccharide acceptor 9 and the disaccharide thioglycoside donor 10, a stereoselective glycosylation between them was attempted in the presence of a combination [26,27] of NIS and HClO4/SiO2 as thiophilic activator. Unfortunately, the required tetrasaccharide derivative 11 was obtained in a poor yield (22%, Scheme 3). It was decided to follow a sequential glycosylation strategy to achieve a significant quantity of compound 11. Accordingly, a stereoselective glycosylation was carried out using compound 9 with ʟ-fucose thioglycoside derivative 6 in the presence of a combination [26,27] of NIS and HClO4/SiO2 as thiophilic activator. Gratifyingly, the trisaccharide derivative 12 was obtained in 74% yield with a newly formed 1,2-cis glycosyl linkage in it. The structural confirmation of compound 12 was established by its NMR spectral analysis [signals at δ 5.67 (d, J = 3.0 Hz, H-1A), 5.60 (d, J = 3.5 Hz, H-1C), 5.50 (s, PhCH), 4.79 (d, J = 7.5 Hz, H-1B) in 1H NMR and at δ 103.3 (C-1B), 100.8 (PhCH), 99.0 (C-1A), 97.1 (C-1C) in 13C NMR spectra]. Compound 12 was subjected to a set of reactions consisting of a one-pot [32] de-O-acetylation and benzylation using benzyl bromide and sodium hydroxide in the presence of tetrabutylammonium bromide (TBAB) followed by oxidative removal [33] of the PMB group using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give trisaccharide acceptor 13 in 72% yield. Trisaccharide acceptor 13 was then allowed to couple with ʟ-rhamnosyl trichloroacetimidate donor 5 in the presence of HClO4/SiO2 as a solid acid activator [31] to provide tetrasaccharide derivative 11 in 76% yield, which was de-O-acetylated to furnish tetrasaccharide acceptor 14 in 94% yield. The formation of compound 11 with appropriate configuration at the glycosidic linkages was supported by its NMR spectral analysis [signals at δ 5.72 (d, J = 3.5 Hz, H-1A), 5.58 (d, J = 3.5 Hz, H-1C), 5.51 (s, PhCH), 4.80 (d, J = 7.5 Hz, H-1B), 4.71 (br s, H-1D) in 1H NMR and at δ 103.3 (C-1B), 100.6 (PhCH), 99.0 (C-1C), 95.0 (C-1A), 94.1 (C-1D) in 13C NMR spectra]. Finally, NIS and HClO4/SiO2-promoted stereoselective glycosylation of compound 14 with ᴅ-glucosamine thioglycoside donor 7 furnished the desired pentasaccharide derivative 15 in 70% yield. The formation of compound 15 with appropriate configuration at the glycosidic linkages was supported by its NMR spectral analysis [signals at δ 5.64 (d, J = 3.5 Hz, H-1A), 5.54 (d, J = 3.0 Hz, H-1C), 5.50 (d, J = 8.5 Hz, H-1E), 5.48 (s, PhCH), 5.41 (s, PhCH), 4.96 (br s, H-1D), 4.69 (d, J = 8.0 Hz, H-1B) in 1H NMR and at δ 103.4 (C-1B), 101.6, 100.4 (2 C, 2 PhCH), 100.1 (C-1E), 99.0 (C-1C), 94.9 (C-1D), 94.7 (C-1A) in 13C NMR spectra]. Compound 15 was subjected to a sequence of reactions consisting of (i) reductive transformation of the azido group into an acetamido group by the treatment with thioacetic acid [34]; (ii) transformation of the N-phthalimido group into acetamido group using hydrazine hydrate followed by selective N-acetylation [35]; (iii) hydrogenolysis of benzyl ethers and benzylidene acetals over Pearlman’s catalyst [36] to furnish the desired pentasaccharide 1 in 49% overall yield (Scheme 4). The structure of compound 1 was unambiguously characterized by its NMR spectral analysis [signals at δ 5.37 (d, J = 2.0 Hz, H-1A), 5.29 (d, J = 3.5 Hz, H-1C), 5.12 (br s, H-1D), 4.73 (d, J = 7.5 Hz, H-1E), 4.61 (d, J = 8.0 Hz, H-1B) in 1H NMR and at δ 102.2 (C-1E), 102.1 (C-1B), 96.8 (C-1A), 96.5 (C-1C), 96.0 (C-1D) in 13C NMR spectra].
![[1860-5397-16-12-i3]](/bjoc/content/inline/1860-5397-16-12-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: (a) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, −40 °C, 1 h, 22%.
Scheme 3: (a) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, −40 °C, 1 h, 22%.
![[1860-5397-16-12-i4]](/bjoc/content/inline/1860-5397-16-12-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: (a) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, −45 °C, 1 h, 74%; (b) BnBr, NaOH, TBAB, THF, room temperature, 6 h; (c) DDQ, CH2Cl2/H2O (9:1), room temperature, 2 h, 72% in two steps; (d) HClO4/SiO2, CH2Cl2, −10 °C, 1 h, 76%; (e) 0.1 M CH3ONa, CH3OH, room temperature, 2 h, 94%; (f) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, –15 °C, 1 h, 70%; (g) CH3COSH, pyridine, room temperature, 16 h; (h) NH2NH2·H2O, EtOH, 80 °C, 12 h; (i) Ac2O, CH3OH, room temperature, 30 min; (j) H2, 20%-Pd(OH)2/C, CH3OH, room temperature, 24 h, 49% in four steps.
Scheme 4: (a) NIS, HClO4/SiO2, MS 4 Å, CH2Cl2, −45 °C, 1 h, 74%; (b) BnBr, NaOH, TBAB, THF, room temperature,...
Conclusion
In summary, a convenient stepwise synthetic strategy has been developed for the synthesis of the pentasaccharide repeating unit of the cell wall O-antigen of Escherichia albertii O4 in very good yield. Although the target compound can be achieved by block synthetic approach but a better yield of the product was obtained by a sequential approach. HClO4/SiO2 was used as a solid acid activator in the glycosylation reactions using trichloroacetimidate as well as thioglycoside donors. All intermediate steps were high yielding with excellent stereo outcome in the glycosidic linkages.
Supporting Information
| Supporting Information File 1: Experimental and analytical data and copies of NMR spectra. | ||
| Format: PDF | Size: 6.2 MB | Download |
References
-
Mokomane, M.; Kasvosve, I.; de Melo, E.; Pernica, J. M.; Goldfarb, D. M. Ther. Adv. Infect. Dis. 2018, 5, 29–43. doi:10.1177/2049936117744429
Return to citation in text: [1] -
Heredia, N.; García, S. Anim. Nutr. 2018, 4, 250–255. doi:10.1016/j.aninu.2018.04.006
Return to citation in text: [1] -
Baker, S.; The, H. C. Curr. Opin. Infect. Dis. 2018, 31, 449–454. doi:10.1097/qco.0000000000000475
Return to citation in text: [1] -
Kariuki, S.; Revathi, G.; Kariuki, N.; Kiiru, J.; Mwituria, J.; Hart, C. A. BMC Microbiol. 2006, 6, 101. doi:10.1186/1471-2180-6-101
Return to citation in text: [1] -
Nataro, J. P.; Kaper, J. B. Clin. Microbiol. Rev. 1998, 11, 142–201. doi:10.1128/cmr.11.1.142
Return to citation in text: [1] -
Nimri, L. F. Diagn. Microbiol. Infect. Dis. 2013, 77, 91–95. doi:10.1016/j.diagmicrobio.2013.06.028
Return to citation in text: [1] -
Huys, G.; Cnockaert, M.; Janda, J. M.; Swings, J. Int. J. Syst. Evol. Microbiol. 2003, 53, 807–810. doi:10.1099/ijs.0.02475-0
Return to citation in text: [1] -
Sharma, M.; Kniel, K. E.; Derevianko, A.; Ling, J.; Bhagwat, A. A. Appl. Environ. Microbiol. 2007, 73, 4351–4353. doi:10.1128/aem.03001-06
Return to citation in text: [1] -
Yamamoto, D.; Hernandes, R. T.; Liberatore, A. M. A.; Abe, C. M.; de Souza, R. B.; Romão, F. T.; Sperandio, V.; Koh, I. H.; Gomes, T. A. T. PLoS One 2017, 12, e0171385. doi:10.1371/journal.pone.0171385
Return to citation in text: [1] -
Silhavy, T. J.; Kahne, D.; Walker, S. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. doi:10.1101/cshperspect.a000414
Return to citation in text: [1] -
Naumenko, O. I.; Zheng, H.; Senchenkova, S. N.; Wang, H.; Li, Q.; Shashkov, A. S.; Wang, J.; Knirel, Y. A.; Xiong, Y. Carbohydr. Res. 2017, 449, 17–22. doi:10.1016/j.carres.2017.06.008
Return to citation in text: [1] [2] -
Zarei, A. E.; Almehdar, H. A.; Redwan, E. M. J. Immunol. Res. 2016, No. 7203587. doi:10.1155/2016/7203587
Return to citation in text: [1] -
Verez-Bencomo, V.; Fernández-Santana, V.; Hardy, E.; Toledo, M. E.; Rodríguez, M. C.; Heynngnezz, L.; Rodriguez, A.; Baly, A.; Herrera, L.; Izquierdo, M.; Villar, A.; Valdés, Y.; Cosme, K.; Deler, M. L.; Montane, M.; Garcia, E.; Ramos, A.; Aguilar, A.; Medina, E.; Toraño, G.; Sosa, I.; Hernandez, I.; Martínez, R.; Muzachio, A.; Carmenates, A.; Costa, L.; Cardoso, F.; Campa, C.; Diaz, M.; Roy, R. Science 2004, 305, 522–525. doi:10.1126/science.1095209
Return to citation in text: [1] -
McCarthy, P. C.; Sharyan, A.; Moghaddam, L. S. Vaccine 2018, 6, No. 12. doi:10.3390/vaccines6010012
Return to citation in text: [1] -
Reglinski, M.; Ercoli, G.; Plumptre, C.; Kay, E.; Petersen, F. C.; Paton, J. C.; Wren, B. W.; Brown, J. S. npj Vaccines 2018, 3, No. 53. doi:10.1038/s41541-018-0090-4
Return to citation in text: [1] -
Kaplonek, P.; Khan, N.; Reppe, K.; Schumann, B.; Emmadi, M.; Lisboa, M. P.; Xu, F.-F.; Calow, A. D. J.; Parameswarappa, S. G.; Witzenrath, M.; Pereira, C. L.; Seeberger, P. H. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 13353–13358. doi:10.1073/pnas.1811862115
Return to citation in text: [1] -
Kay, E.; Cuccui, J.; Wren, B. W. npj Vaccines 2019, 4, No. 16. doi:10.1038/s41541-019-0110-z
Return to citation in text: [1] -
Berti, F.; Adamo, R. Chem. Soc. Rev. 2018, 47, 9015–9025. doi:10.1039/c8cs00495a
Return to citation in text: [1] -
Bhatia, S.; Dimde, M.; Haag, R. Med. Chem. Commun. 2014, 5, 862–878. doi:10.1039/c4md00143e
Return to citation in text: [1] -
Shit, P.; Gucchait, A.; Misra, A. K. Tetrahedron 2019, 75, 130697. doi:10.1016/j.tet.2019.130697
Return to citation in text: [1] -
Chernyak, A.; Oscarson, S.; Turek, D. Carbohydr. Res. 2000, 329, 309–316. doi:10.1016/s0008-6215(00)00189-0
Return to citation in text: [1] -
Sun, J.; Han, X.; Yu, B. Synlett 2005, 437–440. doi:10.1055/s-2004-837221
Return to citation in text: [1] -
Zhang, J.; Mao, J.; Chen, H.; Cai, M. Tetrahedron: Asymmetry 1994, 5, 2283–2290. doi:10.1016/s0957-4166(00)86306-5
Return to citation in text: [1] -
Hendel, J. L.; Wang, J.-W.; Jackson, T. A.; Hardmeier, K.; De Los Santos, R.; Auzanneau, F.-I. J. Org. Chem. 2009, 74, 8321–8331. doi:10.1021/jo901616p
Return to citation in text: [1] -
Kihlberg, J. O.; Leigh, D. A.; Bundle, D. R. J. Org. Chem. 1990, 55, 2860–2863. doi:10.1021/jo00296a055
Return to citation in text: [1] -
Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119
Return to citation in text: [1] [2] [3] -
Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913
Return to citation in text: [1] [2] [3] -
Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f
Return to citation in text: [1] -
Chakraborti, A. K.; Gulhane, R. A Process for the Acylation of Various Substrates using a Solid Support Catalyst. Indian Pat. Appl. 266/DEL/2003, March 10, 2003.
Return to citation in text: [1] -
Zemplén, G. Ber. Dtsch. Chem. Ges. B 1926, 59, 1254–1266. doi:10.1002/cber.19260590626
Return to citation in text: [1] -
Mukhopadhyay, B.; Maurer, S. V.; Rudolph, N.; van Well, R. M.; Russell, D. A.; Field, R. A. J. Org. Chem. 2005, 70, 9059–9062. doi:10.1021/jo051390g
Return to citation in text: [1] [2] -
Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007
Return to citation in text: [1] -
Oikawa, Y.; Yoshioka, T.; Yonemitsu, O. Tetrahedron Lett. 1982, 23, 885–888. doi:10.1016/s0040-4039(00)86974-9
Return to citation in text: [1] -
Shangguan, N.; Katukojvala, S.; Greenberg, R.; Williams, L. J. J. Am. Chem. Soc. 2003, 125, 7754–7755. doi:10.1021/ja0294919
Return to citation in text: [1] -
Lee, H.-H.; Schwartz, D. A.; Harris, J. F.; Carver, J. P.; Krepinsky, J. J. Can. J. Chem. 1986, 64, 1912–1918. doi:10.1139/v86-315
Return to citation in text: [1] -
Pearlman, W. M. Tetrahedron Lett. 1967, 8, 1663–1664. doi:10.1016/s0040-4039(00)70335-2
Return to citation in text: [1]
| 31. | Mukhopadhyay, B.; Maurer, S. V.; Rudolph, N.; van Well, R. M.; Russell, D. A.; Field, R. A. J. Org. Chem. 2005, 70, 9059–9062. doi:10.1021/jo051390g |
| 26. | Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 27. | Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913 |
| 26. | Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 27. | Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913 |
| 1. | Mokomane, M.; Kasvosve, I.; de Melo, E.; Pernica, J. M.; Goldfarb, D. M. Ther. Adv. Infect. Dis. 2018, 5, 29–43. doi:10.1177/2049936117744429 |
| 5. | Nataro, J. P.; Kaper, J. B. Clin. Microbiol. Rev. 1998, 11, 142–201. doi:10.1128/cmr.11.1.142 |
| 15. | Reglinski, M.; Ercoli, G.; Plumptre, C.; Kay, E.; Petersen, F. C.; Paton, J. C.; Wren, B. W.; Brown, J. S. npj Vaccines 2018, 3, No. 53. doi:10.1038/s41541-018-0090-4 |
| 16. | Kaplonek, P.; Khan, N.; Reppe, K.; Schumann, B.; Emmadi, M.; Lisboa, M. P.; Xu, F.-F.; Calow, A. D. J.; Parameswarappa, S. G.; Witzenrath, M.; Pereira, C. L.; Seeberger, P. H. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 13353–13358. doi:10.1073/pnas.1811862115 |
| 4. | Kariuki, S.; Revathi, G.; Kariuki, N.; Kiiru, J.; Mwituria, J.; Hart, C. A. BMC Microbiol. 2006, 6, 101. doi:10.1186/1471-2180-6-101 |
| 17. | Kay, E.; Cuccui, J.; Wren, B. W. npj Vaccines 2019, 4, No. 16. doi:10.1038/s41541-019-0110-z |
| 3. | Baker, S.; The, H. C. Curr. Opin. Infect. Dis. 2018, 31, 449–454. doi:10.1097/qco.0000000000000475 |
| 12. | Zarei, A. E.; Almehdar, H. A.; Redwan, E. M. J. Immunol. Res. 2016, No. 7203587. doi:10.1155/2016/7203587 |
| 13. | Verez-Bencomo, V.; Fernández-Santana, V.; Hardy, E.; Toledo, M. E.; Rodríguez, M. C.; Heynngnezz, L.; Rodriguez, A.; Baly, A.; Herrera, L.; Izquierdo, M.; Villar, A.; Valdés, Y.; Cosme, K.; Deler, M. L.; Montane, M.; Garcia, E.; Ramos, A.; Aguilar, A.; Medina, E.; Toraño, G.; Sosa, I.; Hernandez, I.; Martínez, R.; Muzachio, A.; Carmenates, A.; Costa, L.; Cardoso, F.; Campa, C.; Diaz, M.; Roy, R. Science 2004, 305, 522–525. doi:10.1126/science.1095209 |
| 35. | Lee, H.-H.; Schwartz, D. A.; Harris, J. F.; Carver, J. P.; Krepinsky, J. J. Can. J. Chem. 1986, 64, 1912–1918. doi:10.1139/v86-315 |
| 2. | Heredia, N.; García, S. Anim. Nutr. 2018, 4, 250–255. doi:10.1016/j.aninu.2018.04.006 |
| 14. | McCarthy, P. C.; Sharyan, A.; Moghaddam, L. S. Vaccine 2018, 6, No. 12. doi:10.3390/vaccines6010012 |
| 36. | Pearlman, W. M. Tetrahedron Lett. 1967, 8, 1663–1664. doi:10.1016/s0040-4039(00)70335-2 |
| 9. | Yamamoto, D.; Hernandes, R. T.; Liberatore, A. M. A.; Abe, C. M.; de Souza, R. B.; Romão, F. T.; Sperandio, V.; Koh, I. H.; Gomes, T. A. T. PLoS One 2017, 12, e0171385. doi:10.1371/journal.pone.0171385 |
| 11. | Naumenko, O. I.; Zheng, H.; Senchenkova, S. N.; Wang, H.; Li, Q.; Shashkov, A. S.; Wang, J.; Knirel, Y. A.; Xiong, Y. Carbohydr. Res. 2017, 449, 17–22. doi:10.1016/j.carres.2017.06.008 |
| 31. | Mukhopadhyay, B.; Maurer, S. V.; Rudolph, N.; van Well, R. M.; Russell, D. A.; Field, R. A. J. Org. Chem. 2005, 70, 9059–9062. doi:10.1021/jo051390g |
| 8. | Sharma, M.; Kniel, K. E.; Derevianko, A.; Ling, J.; Bhagwat, A. A. Appl. Environ. Microbiol. 2007, 73, 4351–4353. doi:10.1128/aem.03001-06 |
| 11. | Naumenko, O. I.; Zheng, H.; Senchenkova, S. N.; Wang, H.; Li, Q.; Shashkov, A. S.; Wang, J.; Knirel, Y. A.; Xiong, Y. Carbohydr. Res. 2017, 449, 17–22. doi:10.1016/j.carres.2017.06.008 |
| 34. | Shangguan, N.; Katukojvala, S.; Greenberg, R.; Williams, L. J. J. Am. Chem. Soc. 2003, 125, 7754–7755. doi:10.1021/ja0294919 |
| 7. | Huys, G.; Cnockaert, M.; Janda, J. M.; Swings, J. Int. J. Syst. Evol. Microbiol. 2003, 53, 807–810. doi:10.1099/ijs.0.02475-0 |
| 32. | Madhusudan, S. K.; Agnihotri, G.; Negi, D. S.; Misra, A. K. Carbohydr. Res. 2005, 340, 1373–1377. doi:10.1016/j.carres.2005.03.007 |
| 6. | Nimri, L. F. Diagn. Microbiol. Infect. Dis. 2013, 77, 91–95. doi:10.1016/j.diagmicrobio.2013.06.028 |
| 10. | Silhavy, T. J.; Kahne, D.; Walker, S. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. doi:10.1101/cshperspect.a000414 |
| 33. | Oikawa, Y.; Yoshioka, T.; Yonemitsu, O. Tetrahedron Lett. 1982, 23, 885–888. doi:10.1016/s0040-4039(00)86974-9 |
| 20. | Shit, P.; Gucchait, A.; Misra, A. K. Tetrahedron 2019, 75, 130697. doi:10.1016/j.tet.2019.130697 |
| 18. | Berti, F.; Adamo, R. Chem. Soc. Rev. 2018, 47, 9015–9025. doi:10.1039/c8cs00495a |
| 19. | Bhatia, S.; Dimde, M.; Haag, R. Med. Chem. Commun. 2014, 5, 862–878. doi:10.1039/c4md00143e |
| 28. | Chakraborti, A. K.; Gulhane, R. Chem. Commun. 2003, 1896–1897. doi:10.1039/b304178f |
| 29. | Chakraborti, A. K.; Gulhane, R. A Process for the Acylation of Various Substrates using a Solid Support Catalyst. Indian Pat. Appl. 266/DEL/2003, March 10, 2003. |
| 30. | Zemplén, G. Ber. Dtsch. Chem. Ges. B 1926, 59, 1254–1266. doi:10.1002/cber.19260590626 |
| 25. | Kihlberg, J. O.; Leigh, D. A.; Bundle, D. R. J. Org. Chem. 1990, 55, 2860–2863. doi:10.1021/jo00296a055 |
| 26. | Mukhopadhyay, B.; Collet, B.; Field, R. A. Tetrahedron Lett. 2005, 46, 5923–5925. doi:10.1016/j.tetlet.2005.06.119 |
| 27. | Mukherjee, C.; Misra, A. K. Synthesis 2007, 683–692. doi:10.1055/s-2007-965913 |
| 23. | Zhang, J.; Mao, J.; Chen, H.; Cai, M. Tetrahedron: Asymmetry 1994, 5, 2283–2290. doi:10.1016/s0957-4166(00)86306-5 |
| 24. | Hendel, J. L.; Wang, J.-W.; Jackson, T. A.; Hardmeier, K.; De Los Santos, R.; Auzanneau, F.-I. J. Org. Chem. 2009, 74, 8321–8331. doi:10.1021/jo901616p |
| 21. | Chernyak, A.; Oscarson, S.; Turek, D. Carbohydr. Res. 2000, 329, 309–316. doi:10.1016/s0008-6215(00)00189-0 |
© 2020 Manna et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)