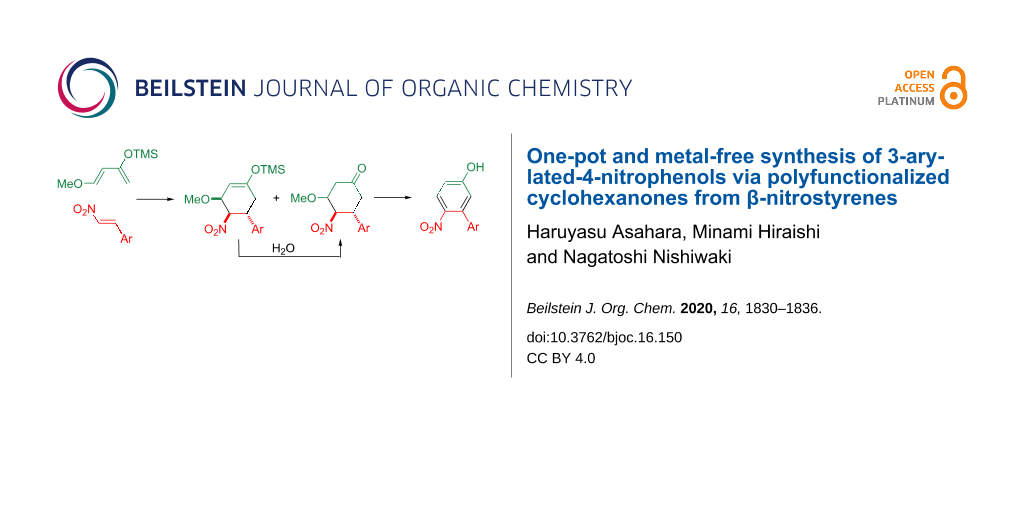
Abstract
β-Nitrostyrenes underwent a Diels–Alder reaction with Danishefsky’s diene to afford cyclohexenes together with the corresponding hydrolyzed products, 3-arylated-5-methoxy-4-nitrocyclohexanones. When the reaction was conducted in the presence of water, the cyclohexenes were efficiently hydrolyzed into cyclohexanones. Subsequent aromatization by heating the cyclohexanone with a catalytic amount of iodine in dimethyl sulfoxide gave 3-arylated-4-nitrophenols. The reaction of nitrostyrenes with Danishefsky’s diene could be conducted in one pot to directly afford the corresponding nitrophenols. Moreover, a heteroaryl group, e.g., a thienyl group could be introduced into the nitrophenol framework.

Graphical Abstract
Introduction
The 4-nitrophenol framework is characterized by a biased electron density in the ring and an acidic hydroxy group, which can be attributed to the electron-donating hydroxy and the electron-withdrawing nitro groups. The derivatives of 4-nitrophenol are widely used in various applications. In particular, 3-arylated-4-nitrophenols have attracted much attention from a biological viewpoint [1-11]; however, they cannot be synthesized by the direct modification of 4-nitrophenol because the ortho-directing hydroxy and the meta-directing nitro group hinder the electrophilic modification at the 3-position. Generally, the aryl group is introduced by a Suzuki–Miyaura cross-coupling reaction [4,5], for which 3-bromo-4-nitrophenol must be prepared by the nitration of 3-bromophenol [6,7]. An alternative approach is the nitration of 3-arylphenol [8,9]. However, these nitration methods are less effective because the yield of the desired product is reduced by the formation of regioisomers. Although the hydroxylation of 3-arylated-1-fluoro-4-nitrobenzene has also been reported as a related strategy, multistep reactions are necessary for preparing the precursor [10]. The condensation of benzyl methyl ketone with a nitrovinamidinium salt also affords 3-arylated-4-nitrophenols; however, 3-arylated-N,N-dimethyl-4-nitroanilines are competitively formed in this reaction [11]. Hence, there is an urgent demand for the development of a facile method toward the preparation of 3-arylated-4-nitrophenols.
On the other hand, nitroalkenes possess an electron-deficient double bond, and hence, they serve as an excellent Michael acceptor and dienophile [12-19]. When β-nitrostyrene (1, Ar = Ph) is subjected to the Diels–Alder reaction, a C2 unit possessing a nitro group and an aryl group at the vicinal position is incorporated into the products. This unique reactivity prompted us to probe the synthesis of the 3-arylated-4-nitrophenols 5 by the Diels–Alder reaction of nitrostyrenes 1 with Danishefsky’s diene (2, the trimethylsiloxy group of the diene can be converted to a phenolic hydroxy group by hydrolysis) [20], followed by the oxidation and aromatization of the obtained cyclohexanone 4 (Scheme 1).
![[1860-5397-16-150-i1]](/bjoc/content/inline/1860-5397-16-150-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic scheme of the 3-arylated-4-nitrophenols 5.
Scheme 1: Synthetic scheme of the 3-arylated-4-nitrophenols 5.
Results and Discussion
Heating an acetonitrile solution of nitrostyrene 1a with Danishefsky’s diene (2) at 60 °C for 18 h afforded trace amounts of 3a and 4a, both of which were obtained as a mixture of diastereomeric isomers (Table 1, entry 1). The stereochemistry of the major product 4a was determined to be trans,trans (all-equatorial) by X-ray crystallography (Figure 1). Among the solvents tested, less polar solvents, such as hexane and toluene, were found to be suitable for the reaction (Table 1, entries 1–5). Consequently, the total yield of 3a and 4a increased up to 82% when the reaction was conducted under reflux in toluene (Table 1, entries 5–7). The diastereomeric ratio increased when the reaction was conducted at temperatures higher than 90 °C, presumably due to the easier formation of the thermodynamically stable isomer. As described later, DMSO was found to be an effective solvent for the subsequent oxidation. However, toluene was determined to be the best solvent because the recovery of 1a was observed even at 120 °C (Table 1, entry 8).
Table 1: Optimization of the reaction conditions for the Diels–Alder reaction.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-150-i5.svg?max-width=637&scale=1.0)
|
||||||
| entry | solvent | T [°C] | total yield [%]b | yield [%]b | drb,c of 4a | |
| 3a | 4a | |||||
| 1 | MeCN | 60 | 6 | 3 | 3 | 67:33 |
| 2 | Et2O | 60 | 21 | 20 | 1 | 50:50 |
| 3 | CHCl3 | 60 | 41 | 31 | 10 | 50:50 |
| 4 | hexane | 60 | 39 | 28 | 11 | 55:45 |
| 5 | PhMe | 60 | 51 | 38 | 13 | 54:46 |
| 6 | PhMe | 90 | 61 | 45 | 16 | 83:17 |
| 7 | PhMe | 120 | 82 | 59 | 23 | 83:17 |
| 8 | DMSO | 120 | 30 | 30 | 0 | – |
aTol: 4-MeC6H4. bDetermined by 1H NMR spectroscopy. cDiastereomeric ratio.
![[1860-5397-16-150-1]](/bjoc/content/figures/1860-5397-16-150-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: X-ray crystallography of the major isomer of 4a. The thermal ellipsoids indicate 50% probability.
Figure 1: X-ray crystallography of the major isomer of 4a. The thermal ellipsoids indicate 50% probability.
Cyclohexene 3a was efficiently converted to the cyclohexanone 4a upon heating at 120 °C in toluene in the presence of 10 equiv water (Scheme 2). This result prompted us to synthesize 4a in one pot from 1a and 2 by Diels–Alder reaction and subsequent heating with water (Scheme 2).
![[1860-5397-16-150-i2]](/bjoc/content/inline/1860-5397-16-150-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Conversion from 3a to 4a and one-pot synthesis of 4a.
Scheme 2: Conversion from 3a to 4a and one-pot synthesis of 4a.
Cyclohexanone 4a has acidic hydrogen atoms that can facilitate the aromatization by modification, e.g., by iodination. In order to obtain further insights into this possibility, 4a was heated with deuterium oxide, but no change was observed. In contrast, the signals assigned to the protons in the 4- and 6-position disappeared in the NMR spectrum when the mixture was heated in the presence of triethylamine, indicating that the α-protons of the carbonyl and nitro groups are acidic and easily modifiable (Scheme 3, see the NMR charts in Supporting Information File 1).
![[1860-5397-16-150-i3]](/bjoc/content/inline/1860-5397-16-150-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Deuteration of cyclohexanone 4a.
Scheme 3: Deuteration of cyclohexanone 4a.
The aromatization of 4a using iodine was then attempted (Table 2). The reaction did not proceed in toluene or acetonitrile (Table 2, entries 1 and 2), but dimethyl sulfoxide (DMSO) was effective for the aromatization, and nitrophenol 5a was obtained in 26% yield (Table 2, entry 3) [21-23]. This reaction proceeded efficiently to afford 5a in 61% yield even when the amount of iodine was decreased to 10 mol %. However further decreasing the iodine amount to 5 mol % was not effective for the conversion (Table 2, entries 4 and 5). In each case, both stereoisomers of 4a were completely consumed, and several unidentified products were obtained.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-150-i6.svg?max-width=637&scale=1.0)
The aromatization is considered to proceed as shown in Scheme 4. After the iodization at the 4-position, which leads to the formation of the intermediate 6, the aromatization is achieved by the successive elimination of hydrogen iodide and methanol, with concurrent tautomerism to afford 5a. The formed hydrogen iodide is easily oxidized by DMSO to regenerate iodine, so that the reaction can be performed with a catalytic amount of iodine.
![[1860-5397-16-150-i4]](/bjoc/content/inline/1860-5397-16-150-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: A plausible mechanism for the formation of 5a.
Scheme 4: A plausible mechanism for the formation of 5a.
The optimal conditions were applied to the one-pot three-step reaction of the other nitrostyrenes 1b–e, and the corresponding 3-phenylated-4-nitrophenols 5b–e were furnished in moderate yield (Table 3, entries 1–4). Among the nitrostyrenes employed, 1b and 1d had a lower reactivity, which was presumably due to the electron-donating resonance effect of the substituents. In these cases, the resonance contributor shown in Figure 2 diminished the nitroalkene properties and consequently suppressed the Diels–Alder reaction with 2. It is noteworthy that not only the benzene ring, but also a heteroaromatic ring could be introduced into the nitrophenol framework by using this method (Table 3, entry 5).
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-150-i7.svg?max-width=637&scale=1.0)
![[1860-5397-16-150-2]](/bjoc/content/figures/1860-5397-16-150-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Resonance structure of nitroalkenes 1b and 1d.
Figure 2: Resonance structure of nitroalkenes 1b and 1d.
Conclusion
β-Nitrostyrene 1a underwent a Diels–Alder reaction with Danishefsky’s diene (2) to afford polysubstituted cyclohexene 3a and cyclohexanone 4a. The addition of water to the reaction mixture accelerated the conversion from 3a to 4a. The oxidative aromatization of 4a was achieved by the treatment with a catalytic amount of iodine in DMSO to furnish nitrophenol 5a. This protocol was also applicable to other nitroalkenes, 1b–f, to afford the corresponding 3-arylated-4-nitrophenols 5b–f. This reaction could be conducted in one pot without using any transition-metal reagent. Since the starting nitroalkenes were prepared by the condensation of a (hetero)aryl aldehyde and nitromethane, an easy modification of the aryl group at the 3-position of the 4-nitrophenol is possible. Thus, the proposed reaction would be a useful tool for the elaborate synthesis of aromatic compounds.
Experimental
General
All reagents were purchased from commercial sources and used without further purification. 1H and 13C NMR spectra were recorded on a Bruker DPX-400 spectrometer (at 400 MHz and 100 MHz, respectively) or on a JEOL JNM-LA 500 spectrometer (at 500 MHz and 125 MHz, respectively) in CDCl3 using TMS as an internal standard. The assignments of the 13C NMR signals were performed by DEPT experiments. A Shimadzu IR spectrometer equipped with an ATR detector was used to record infrared spectra. High-resolution mass spectra were obtained on an AB SCIEX Triplet TOF 4600 mass spectrometer. Melting points were recorded on an SRS-Optimelt automated melting point system and aree uncorrected.
Preparation of (E)-1-(4-methylphenyl)-2-nitroethene (1a)
To a solution of ammonium acetate (2.63 g, 34 mmol) in acetic acid (20 mL) were added nitromethane (5.25 mL, 98 mmol) and 4-methylbenzaldehyde (1.96 mL, 16 mmol), and the resulting mixture was heated at 100 °C for 6 h. After the addition of water (100 mL), the pH value was adjusted to 7 with a 2 M aqueous sodium hydroxide solution. This was extracted with ethyl acetate (50 mL × 3), and the organic layer was washed with brine (100 mL × 1), dried over magnesium sulfate, and concentrated to afford crude product (2.66 g) as yellow solid. The solid was recrystalized with a mixed solvent (hexane/dichloromethane 10:1) to afford nitrostyrene 1a [24] (1.99 g, 12.2 mmol, 76%) as yellow needles. The other nitroalkenes 1b–f [25-29] were prepared in the same way.
One-pot Diels–Alder reaction of nitrostyrene 1a and Danishefsky’s diene (2)
To a solution of Danishefsky’s diene (2, 129.2 mg, 0.75 mmol) in toluene (1 mL), nitrostyrene 1a (81.6 mg, 0.50 mmol) was added, and the resultant mixture was heated at 120 °C for 18 h in a sealed tube. Water (90 mg, 5.0 mmol) was added, and the mixture was heated at 120 °C for further 1 h in a sealed tube. After the removal of the solvent under reduced pressure, the residue was treated by column chromatography using silica gel (hexane/ethyl acetate 9:1) to afford the major isomer of cyclohexanone 4a (205 mg, 0.32 mmol, 78%) as yellow solid and the minor isomer of cyclohexanone 4a (26.3 mg, 0.10 mmol, 10%, as a mixture with the major isomer, dr = 6:1) as pale yellow solid; however, further purification of the minor isomer could not be achieved.
Major isomer: pale yellow needles; mp 122–123 °C; 1H NMR (400 MHz, CDCl3, δ) 2.32 (s, 3H), 2.56 (ddd, J = 0.6, 11.1, 14.4 Hz, 1H), 2.63 (ddd, J = 2.0, 5.1, 15.2 Hz, 1H), 2.70 (ddd, J = 0.6, 13.4, 15.2 Hz, 1H), 3.07 (ddd, J = 2.0, 5.2, 14.4 Hz, 1H), 3.37 (s, 3H), 3.41 (ddd, J = 5.1, 11.6, 13.4 Hz, 1H), 4.09 (ddd, J = 5.2, 9.2, 11.1 Hz, 1H), 4.95 (dd, J = 9.2, 11.6 Hz, 1H), 7.08 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3, δ) 21.0 (CH3), 42.8 (CH), 44.3 (CH2), 46.0 (CH2), 57.6 (CH3), 78.6 (CH), 93.6 (CH), 126.8 (CH), 130.0 (CH), 133.6 (C), 138.4 (C), 203.1 (C); IR (ATR) vmax: 536, 821, 1091, 1340, 1550, 1718 cm−1; HRESIMS-TOF (m/z): [M + Na]+ calcd for C14H17NO4Na, 286.1050; found, 286.1040.
Minor isomer: 1H NMR (400 MHz, CDCl3, δ) 2.31 (s, 3H), 2.51 (ddd, J = 0.5, 12.4, 15.2 Hz, 1H), 2.69 (ddd, J = 0.5, 3.6, 15.2 Hz, 1H), 2.71 (ddd, J = 2.4, 5.6, 15.2 Hz, 1H), 2.95 (ddd, J = 2.4, 3.6, 15.2 Hz, 1H), 3.37 (s, 3H), 4.10 (ddd, J = 5.6, 12.4, 11.4 Hz, 1H), 4.43 (ddd, J = 3.0, 3.6, 3.6 Hz, 1H), 4.66 (dd, J = 2.8, 11.4 Hz, 1H), 7.14–7.16 (m, 4H); 13C NMR (100 MHz, CDCl3, δ) 21.0 (CH3), 39.9 (CH), 42.9 (CH2), 46.5 (CH2), 57.6 (CH3), 78.8 (CH), 89.0 (CH), 126.8 (CH), 129.8 (CH), 136.4 (C), 137.6 (C), 204.2 (C).
Aromatization of cyclohexanone 4a
To a solution of the cyclohexanone 4a (52.6 mg, 0.2 mmol) in DMSO (1 mL), iodine (5.0 mg, 0.02 mmol) was added, and the resulting mixture was heated at 100 °C for 18 h. To the reaction mixture, a saturated aqueous solution of sodium thiosulfate (3 mL) was added, and the mixture was extracted with ethyl acetate (3 mL × 3). The organic layer was washed with brine (3 mL × 1), dried over magnesium sulfate, and concentrated to afford the crude product (39.4 mg) as brown oil. Further purification was performed with column chromatography on silica gel to afford 3-(4-methylphenyl)-4-nitrophenol (5a) [30], which eluted with hexane/ethyl acetate 8:2 (Rf 0.44, 28.0 mg, 0.12 mmol, 61%) as brown solid. 1H NMR (400 MHz, CDCl3, δ) 2.40 (s, 3H), 5.4–5.2 (br, 1H), 6.80 (d, J = 2.6 Hz, 1H), 6.85 (dd, J = 2.6, 8.8 Hz, 1H), 7.18 (d, J = 8.2 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H), 7.89 (d, J = 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3, δ) 21.2 (CH3), 114.4 (CH), 118.5 (CH), 127.2 (CH), 127.6 (CH), 129.3 (CH), 134.8 (C), 138.1 (C), 139.8 (C), 142.3 (C), 158.9 (C).
One-pot synthesis of 3-arylated-4-nitrophenol 5b
To a solution of Danishefsky’s diene (2, 129.2 mg, 0.75 mmol) in toluene (1 mL), nitrostyrene 1b (90.0 mg, 0.50 mmol) was added, and the resulting mixture was heated at 120 °C for 18 h in a sealed tube. Water (90 mg, 5.0 mmol) was added, and the mixture was heated at 120 °C for further 1 h in a sealed tube. After the removal of the solvent under reduced pressure, the residue was dissolved in DMSO (2.5 mL), and iodine (12.7 mg, 0.05 mmol) was added. After heating at 100 °C for 18 h, a saturated aqueous solution of sodium thiosulfate (8 mL) was added, and the mixture was extracted with ethyl acetate (8 mL × 3). The organic layer was washed with brine (8 mL × 1), dried over magnesium sulfate, and concentrated. The residue was treated by column chromatography on silica gel to afford nitrophenol 5b, which eluted with hexane/ethyl acetate 8:2 (41.7 mg, 0.17 mmol, 34%) as yellow solid. When the other nitroalkenes 1c–f were used, the reaction was conducted in the same way.
Supporting Information
| Supporting Information File 1: Spectral data for 5b–f, NMR spectra (1H, 13C, and DEPT) for 4a and 5a–f, and crystallographic data for 4a. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Trisomboon, J.; Li, C.; Suzuki, A.; Watanabe, G.; Taya, K. J. Reprod. Dev. 2015, 61, 134–137. doi:10.1262/jrd.2014-110
Return to citation in text: [1] -
Mi, Y.; Zhang, C.; Li, C.; Taneda, S.; Watanabe, G.; Suzuki, A. K.; Taya, K. Biosci., Biotechnol., Biochem. 2010, 74, 934–938. doi:10.1271/bbb.90740
Return to citation in text: [1] -
Riahi, A.; Shkoor, M.; Khera, R. A.; Reinke, H.; Langer, P. Tetrahedron Lett. 2009, 50, 3017–3019. doi:10.1016/j.tetlet.2009.03.190
Return to citation in text: [1] -
Bahta, M.; Burke, T. R., Jr. ChemMedChem 2011, 6, 1363–1370. doi:10.1002/cmdc.201100200
Return to citation in text: [1] [2] -
Zhang, Y. J.; Wei, H.; Zhang, W. Tetrahedron 2009, 65, 1281–1286. doi:10.1016/j.tet.2008.12.056
Return to citation in text: [1] [2] -
Tercel, M.; McManaway, S. P.; Liyanage, H. D. S.; Pruijn, F. B. ChemMedChem 2014, 9, 2193–2206. doi:10.1002/cmdc.201402169
Return to citation in text: [1] [2] -
Jung, K.-Y.; Vanommeslaeghe, K.; Lanning, M. E.; Yap, J. L.; Gordon, C.; Wilder, P. T.; MacKerell, A. D., Jr.; Fletcher, S. Org. Lett. 2013, 15, 3234–3237. doi:10.1021/ol401197n
Return to citation in text: [1] [2] -
Fukaya, T.; Kodo, T.; Ishiyama, T.; Kakuyama, H.; Nishikawa, H.; Baba, S.; Masumoto, S. Bioorg. Med. Chem. 2012, 20, 5568–5582. doi:10.1016/j.bmc.2012.07.023
Return to citation in text: [1] [2] -
Clapper, J. R.; Vacondio, F.; King, A. R.; Duranti, A.; Tontini, A.; Silva, C.; Sanchini, S.; Tarzia, G.; Mor, M.; Piomelli, D. ChemMedChem 2009, 4, 1505–1513. doi:10.1002/cmdc.200900210
Return to citation in text: [1] [2] -
Fier, P. S.; Maloney, K. M. Org. Lett. 2016, 18, 2244–2247. doi:10.1021/acs.orglett.6b00876
Return to citation in text: [1] [2] -
Davies, I. W.; Marcoux, J.-F.; Kuethe, J. T.; Lankshear, M. D.; Taylor, J. D. O.; Tsou, N.; Dormer, P. G.; Hughes, D. L.; Houk, K. N.; Guner, V. J. Org. Chem. 2004, 69, 1298–1308. doi:10.1021/jo035677u
Return to citation in text: [1] [2] -
Larkovich, R. V.; Ponomarev, S. A.; Aldoshin, A. S.; Tabolin, A. A.; Ioffe, S. L.; Nenajdenko, V. G. Eur. J. Org. Chem. 2020, 2479–2492. doi:10.1002/ejoc.202000054
Return to citation in text: [1] -
Alonso, D.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Molecules 2017, 22, 895. doi:10.3390/molecules22060895
Return to citation in text: [1] -
Blond, G.; Gulea, M.; Mamane, V. Curr. Org. Chem. 2016, 20, 2161–2210. doi:10.2174/1385272820666160216000401
Return to citation in text: [1] -
Halimehjani, A. Z.; Namboothiri, I. N. N.; Hooshmand, S. E. RSC Adv. 2014, 4, 31261–31299. doi:10.1039/c4ra04069d
Return to citation in text: [1] -
Halimehjani, A. Z.; Namboothiri, I. N. N.; Hooshmand, S. E. RSC Adv. 2014, 4, 48022–48084. doi:10.1039/c4ra08828j
Return to citation in text: [1] -
Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. J. Org. Chem. 2018, 83, 13691–13699. doi:10.1021/acs.joc.8b01865
Return to citation in text: [1] -
Hao, F.; Yokoyama, S.; Nishiwaki, N. Org. Biomol. Chem. 2018, 16, 2768–2775. doi:10.1039/c8ob00408k
Return to citation in text: [1] -
Hao, F.; Asahara, H.; Nishiwaki, N. Org. Lett. 2017, 19, 5442–5445. doi:10.1021/acs.orglett.7b02724
Return to citation in text: [1] -
Node, M.; Nishide, K.; Imazato, H.; Kurosaki, R.; Inoue, T.; Ikariya, T. Chem. Commun. 1996, 2559–2560. doi:10.1039/cc9960002559
Return to citation in text: [1] -
Rather, S. A.; Kumar, A.; Ahmed, Q. N. Chem. Commun. 2019, 55, 4511–4514. doi:10.1039/c9cc00346k
Return to citation in text: [1] -
Bansode, A. H.; Suryavanshi, G. ACS Omega 2019, 4, 9636–9644. doi:10.1021/acsomega.9b00833
Return to citation in text: [1] -
Miao, C.; Jiang, L.; Ren, L.; Xue, Q.; Yan, F.; Shi, W.; Li, X.; Sheng, J.; Kai, S. Tetrahedron 2019, 75, 2215–2228. doi:10.1016/j.tet.2019.02.041
Return to citation in text: [1] -
Lopchuk, J. M.; Hughes, R. P.; Gribble, G. W. Org. Lett. 2013, 15, 5218–5221. doi:10.1021/ol402385v
Return to citation in text: [1] -
Kumar, M. S.; Rajanna, K. C.; Reddy, K. R.; Venkateswarlu, M.; Venkanna, P. Synth. Commun. 2013, 43, 2672–2677. doi:10.1080/00397911.2012.735331
See for 1b.
Return to citation in text: [1] -
Saikia, A. K.; Barua, N. C.; Sharma, R. P.; Ghosh, A. C. Synthesis 1994, 685–686. doi:10.1055/s-1994-25546
See for 1c.
Return to citation in text: [1] -
Taylor, E. C.; Liu, B.; Wang, W. J. Org. Chem. 2000, 65, 4750–4752. doi:10.1021/jo000205q
See for 1d.
Return to citation in text: [1] -
Simpson, A. J.; Lam, H. W. Org. Lett. 2013, 15, 2586–2589. doi:10.1021/ol400578c
See for 1e.
Return to citation in text: [1] -
Ignatovich, L.; Muravenko, V.; Romanovs, V.; Sleiksha, I.; Shestakova, I.; Domrachova, I.; Belyakov, S.; Popelis, J.; Lukevics, E. Appl. Organomet. Chem. 2010, 24, 858–864. doi:10.1002/aoc.1712
See for 1f.
Return to citation in text: [1] -
Taneda, S.; Kamata, K.; Hayashi, H.; Toda, N.; Seki, K.; Sakushima, A.; Yoshino, S.; Yamaki, K.; Sakata, M.; Mori, Y.; Suzuki, A. K. J. Health Sci. 2004, 50, 133–141. doi:10.1248/jhs.50.133
Return to citation in text: [1]
| 1. | Trisomboon, J.; Li, C.; Suzuki, A.; Watanabe, G.; Taya, K. J. Reprod. Dev. 2015, 61, 134–137. doi:10.1262/jrd.2014-110 |
| 2. | Mi, Y.; Zhang, C.; Li, C.; Taneda, S.; Watanabe, G.; Suzuki, A. K.; Taya, K. Biosci., Biotechnol., Biochem. 2010, 74, 934–938. doi:10.1271/bbb.90740 |
| 3. | Riahi, A.; Shkoor, M.; Khera, R. A.; Reinke, H.; Langer, P. Tetrahedron Lett. 2009, 50, 3017–3019. doi:10.1016/j.tetlet.2009.03.190 |
| 4. | Bahta, M.; Burke, T. R., Jr. ChemMedChem 2011, 6, 1363–1370. doi:10.1002/cmdc.201100200 |
| 5. | Zhang, Y. J.; Wei, H.; Zhang, W. Tetrahedron 2009, 65, 1281–1286. doi:10.1016/j.tet.2008.12.056 |
| 6. | Tercel, M.; McManaway, S. P.; Liyanage, H. D. S.; Pruijn, F. B. ChemMedChem 2014, 9, 2193–2206. doi:10.1002/cmdc.201402169 |
| 7. | Jung, K.-Y.; Vanommeslaeghe, K.; Lanning, M. E.; Yap, J. L.; Gordon, C.; Wilder, P. T.; MacKerell, A. D., Jr.; Fletcher, S. Org. Lett. 2013, 15, 3234–3237. doi:10.1021/ol401197n |
| 8. | Fukaya, T.; Kodo, T.; Ishiyama, T.; Kakuyama, H.; Nishikawa, H.; Baba, S.; Masumoto, S. Bioorg. Med. Chem. 2012, 20, 5568–5582. doi:10.1016/j.bmc.2012.07.023 |
| 9. | Clapper, J. R.; Vacondio, F.; King, A. R.; Duranti, A.; Tontini, A.; Silva, C.; Sanchini, S.; Tarzia, G.; Mor, M.; Piomelli, D. ChemMedChem 2009, 4, 1505–1513. doi:10.1002/cmdc.200900210 |
| 10. | Fier, P. S.; Maloney, K. M. Org. Lett. 2016, 18, 2244–2247. doi:10.1021/acs.orglett.6b00876 |
| 11. | Davies, I. W.; Marcoux, J.-F.; Kuethe, J. T.; Lankshear, M. D.; Taylor, J. D. O.; Tsou, N.; Dormer, P. G.; Hughes, D. L.; Houk, K. N.; Guner, V. J. Org. Chem. 2004, 69, 1298–1308. doi:10.1021/jo035677u |
| 10. | Fier, P. S.; Maloney, K. M. Org. Lett. 2016, 18, 2244–2247. doi:10.1021/acs.orglett.6b00876 |
| 8. | Fukaya, T.; Kodo, T.; Ishiyama, T.; Kakuyama, H.; Nishikawa, H.; Baba, S.; Masumoto, S. Bioorg. Med. Chem. 2012, 20, 5568–5582. doi:10.1016/j.bmc.2012.07.023 |
| 9. | Clapper, J. R.; Vacondio, F.; King, A. R.; Duranti, A.; Tontini, A.; Silva, C.; Sanchini, S.; Tarzia, G.; Mor, M.; Piomelli, D. ChemMedChem 2009, 4, 1505–1513. doi:10.1002/cmdc.200900210 |
| 6. | Tercel, M.; McManaway, S. P.; Liyanage, H. D. S.; Pruijn, F. B. ChemMedChem 2014, 9, 2193–2206. doi:10.1002/cmdc.201402169 |
| 7. | Jung, K.-Y.; Vanommeslaeghe, K.; Lanning, M. E.; Yap, J. L.; Gordon, C.; Wilder, P. T.; MacKerell, A. D., Jr.; Fletcher, S. Org. Lett. 2013, 15, 3234–3237. doi:10.1021/ol401197n |
| 4. | Bahta, M.; Burke, T. R., Jr. ChemMedChem 2011, 6, 1363–1370. doi:10.1002/cmdc.201100200 |
| 5. | Zhang, Y. J.; Wei, H.; Zhang, W. Tetrahedron 2009, 65, 1281–1286. doi:10.1016/j.tet.2008.12.056 |
| 21. | Rather, S. A.; Kumar, A.; Ahmed, Q. N. Chem. Commun. 2019, 55, 4511–4514. doi:10.1039/c9cc00346k |
| 22. | Bansode, A. H.; Suryavanshi, G. ACS Omega 2019, 4, 9636–9644. doi:10.1021/acsomega.9b00833 |
| 23. | Miao, C.; Jiang, L.; Ren, L.; Xue, Q.; Yan, F.; Shi, W.; Li, X.; Sheng, J.; Kai, S. Tetrahedron 2019, 75, 2215–2228. doi:10.1016/j.tet.2019.02.041 |
| 25. |
Kumar, M. S.; Rajanna, K. C.; Reddy, K. R.; Venkateswarlu, M.; Venkanna, P. Synth. Commun. 2013, 43, 2672–2677. doi:10.1080/00397911.2012.735331
See for 1b. |
| 26. |
Saikia, A. K.; Barua, N. C.; Sharma, R. P.; Ghosh, A. C. Synthesis 1994, 685–686. doi:10.1055/s-1994-25546
See for 1c. |
| 27. |
Taylor, E. C.; Liu, B.; Wang, W. J. Org. Chem. 2000, 65, 4750–4752. doi:10.1021/jo000205q
See for 1d. |
| 28. |
Simpson, A. J.; Lam, H. W. Org. Lett. 2013, 15, 2586–2589. doi:10.1021/ol400578c
See for 1e. |
| 29. |
Ignatovich, L.; Muravenko, V.; Romanovs, V.; Sleiksha, I.; Shestakova, I.; Domrachova, I.; Belyakov, S.; Popelis, J.; Lukevics, E. Appl. Organomet. Chem. 2010, 24, 858–864. doi:10.1002/aoc.1712
See for 1f. |
| 20. | Node, M.; Nishide, K.; Imazato, H.; Kurosaki, R.; Inoue, T.; Ikariya, T. Chem. Commun. 1996, 2559–2560. doi:10.1039/cc9960002559 |
| 30. | Taneda, S.; Kamata, K.; Hayashi, H.; Toda, N.; Seki, K.; Sakushima, A.; Yoshino, S.; Yamaki, K.; Sakata, M.; Mori, Y.; Suzuki, A. K. J. Health Sci. 2004, 50, 133–141. doi:10.1248/jhs.50.133 |
| 12. | Larkovich, R. V.; Ponomarev, S. A.; Aldoshin, A. S.; Tabolin, A. A.; Ioffe, S. L.; Nenajdenko, V. G. Eur. J. Org. Chem. 2020, 2479–2492. doi:10.1002/ejoc.202000054 |
| 13. | Alonso, D.; Baeza, A.; Chinchilla, R.; Gómez, C.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Molecules 2017, 22, 895. doi:10.3390/molecules22060895 |
| 14. | Blond, G.; Gulea, M.; Mamane, V. Curr. Org. Chem. 2016, 20, 2161–2210. doi:10.2174/1385272820666160216000401 |
| 15. | Halimehjani, A. Z.; Namboothiri, I. N. N.; Hooshmand, S. E. RSC Adv. 2014, 4, 31261–31299. doi:10.1039/c4ra04069d |
| 16. | Halimehjani, A. Z.; Namboothiri, I. N. N.; Hooshmand, S. E. RSC Adv. 2014, 4, 48022–48084. doi:10.1039/c4ra08828j |
| 17. | Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. J. Org. Chem. 2018, 83, 13691–13699. doi:10.1021/acs.joc.8b01865 |
| 18. | Hao, F.; Yokoyama, S.; Nishiwaki, N. Org. Biomol. Chem. 2018, 16, 2768–2775. doi:10.1039/c8ob00408k |
| 19. | Hao, F.; Asahara, H.; Nishiwaki, N. Org. Lett. 2017, 19, 5442–5445. doi:10.1021/acs.orglett.7b02724 |
| 11. | Davies, I. W.; Marcoux, J.-F.; Kuethe, J. T.; Lankshear, M. D.; Taylor, J. D. O.; Tsou, N.; Dormer, P. G.; Hughes, D. L.; Houk, K. N.; Guner, V. J. Org. Chem. 2004, 69, 1298–1308. doi:10.1021/jo035677u |
| 24. | Lopchuk, J. M.; Hughes, R. P.; Gribble, G. W. Org. Lett. 2013, 15, 5218–5221. doi:10.1021/ol402385v |
© 2020 Asahara et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)