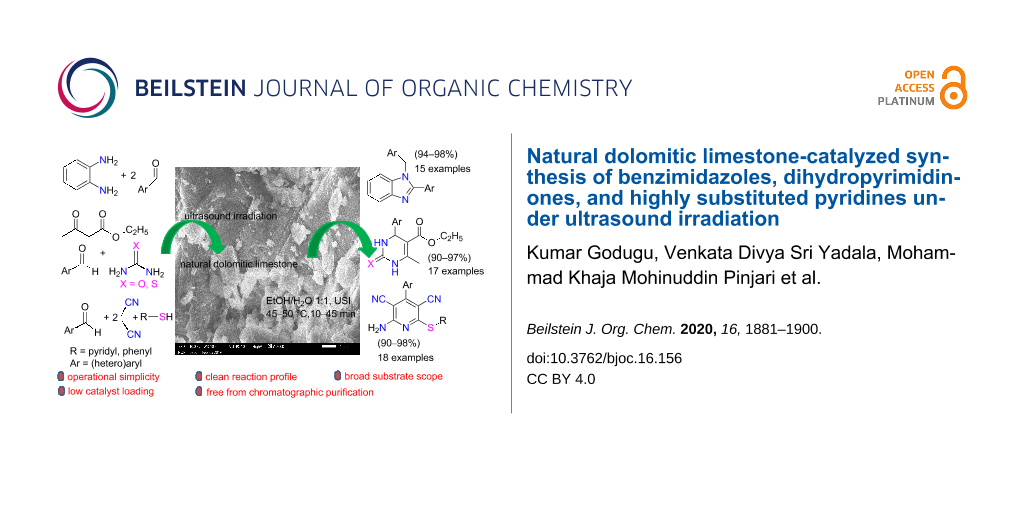
Abstract
Natural dolomitic limestone (NDL) is employed as a heterogeneous green catalyst for the synthesis of medicinally valuable benzimidazoles, dihydropyrimidinones, and highly functionalized pyridines via C–N, C–C, and C–S bond formations in a mixture of ethanol and H2O under ultrasound irradiation. The catalyst is characterized by XRD, FTIR, Raman spectroscopy, SEM, and EDAX analysis. The main advantages of this methodology include the wide substrate scope, cleaner reaction profile, short reaction times, and excellent isolated yields. The products do not require chromatographic purification, and the catalyst can be reused seven times. Therefore, the catalyst is a greener alternative for the synthesis of the above N-heterocycles compared to the existing reported catalysts.

Graphical Abstract
Introduction
Nitrogen heterocycles are recognized as “privileged medicinal scaffolds” because these compounds are found in a wide variety of bioactive natural products and pharmaceuticals [1-3]. Among them, benzimidazoles, dihydropyrimidinones, and pyridines have emerged as promising and valuable structural units in many pharmaceutical lead compounds (Figure 1) [4-9]. Hence, there is a great need for the development of a green and sustainable synthetic route to the aforesaid nitrogen-containing heterocycles.
![[1860-5397-16-156-1]](/bjoc/content/figures/1860-5397-16-156-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: The benzimidazoles I–IV, dihydropyrimidinones/-thiones V–VIII, and 2-amino-4-aryl-3,5-dicarbonitrile-6-sulfanylpyridines IX–XII as medicinally privileged structures.
Figure 1: The benzimidazoles I–IV, dihydropyrimidinones/-thiones V–VIII, and 2-amino-4-aryl-3,5-dicarbonitril...
Benzimidazoles are an important class of N-heterocycles due to their potential applications in both biology and medicinal chemistry [10-13]. These compounds are used in the treatment of diseases, such as obesity, ischemia-reperfusion injury, hypertension, etc. [14-16]. In addition, these compounds are important intermediates in a variety of organic reactions and key elements in many functional materials [17-19]. Because of their potential utility, a huge number of synthetic protocols has been developed for the preparation of benzimidazole derivatives. The most common method for the preparation of benzimidazoles is the reaction between o-phenylenediamines and carboxylic acids [20,21]. Another general synthetic route reported is the condensation reaction of o-phenylenediamine with aldehydes in the presence of various catalysts, such as Zn–proline, trimethylsilyl chloride (TMSCl), Amberlite® IR-120, indion 190, trifluoroethanol, YCl3, HClO4–SiO2, MMZY zeolite, Er(OTf)3, etc. [22-30].
Developments in already established multicomponent reactions (MCRs) are interesting topics in organic synthesis. For instance, the Biginelli reaction is a renowned and tunable MCR to synthesize the pharmacologically active 3,4-dihydropyrimidin-2-(1H)-ones (DHPMs, Biginelli products) [31]. These compounds occupy an important position in the fields of natural products and synthetic organic chemistry owing to their potential pharmacological properties [32-37]. A wide variety of Brønsted acids and Lewis acids are employed as efficient catalysts for the Biginelli reaction [38-47]. In addition, some transition metal-based catalysts and a few nonacidic inorganic salts are also utilized as catalysts for the above reaction [48-58]. Only few basic catalysts, such as t-BuOK, Ph3P, and ʟ-proline are reported for the Biginelli reaction [59-61].
2-Amino-4-aryl-3,5-dicarbonitrile-6-sulfanylpyridines have gained considerable attention due to their wide-ranging biological activities [62,63]. The most common synthetic route for the preparation of 2-amino-4-aryl-3,5-dicarbonitrile-6-thiopyridines is the condensation reaction of aldehydes, malononitrile, and thiols in the presence of a variety of catalysts [64-72]. Though the reported methods are efficient to provide the desired 1,2-disubstituted benzimidazoles, dihydropyrimidinones/-thiones and 2-amino-4-aryl-3,5-dicarbonitrile-6-sulfanylpyridines, there are still some drawbacks, which include the use of expensive catalysts, the preparation of the catalyst, long reaction times, the limited substrate scope, and complicated work-up processes; further, the products require chromatographic purification.
The mineral NDL is an irregular combination of calcium and magnesium carbonate. It is water-insoluble, environmentally benevolent, inexpensive, nontoxic, and abundant in nature. Further, dolomite is used as a heterogeneous green catalyst in very few organic transformations, such as Knoevenagel, Michael–Henry, and transesterification reactions [73,74]. To the best of our knowledge, there are no reports on the NDL-catalyzed synthesis of aforesaid N-heterocycles under ultrasonic irradiation (USI).
In this paper, we wish to report the use of NDL as a heterogeneous green catalyst for the synthesis of the 1,2-disubstituted benzimidazoles 3, the dihydropyrimidinones/-thiones 7, and the 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines 11 via C–N, C–C, and C–S bond-forming reactions, respectively, in a mixture of EtOH and H2O 1:1 under ultrasonic irradiation (Scheme 1).
![[1860-5397-16-156-i1]](/bjoc/content/inline/1860-5397-16-156-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: NDL-catalyzed synthesis of i) 1,2-disubstituted benzimidazoles 3, ii) dihydropyrimidinones/-thiones 7, and iii) 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines 11 under ultrasound irradiation.
Scheme 1: NDL-catalyzed synthesis of i) 1,2-disubstituted benzimidazoles 3, ii) dihydropyrimidinones/-thiones ...
Results and Discussion
Geological background of the NDL catalyst
The NDL catalyst was collected from V. Kothapalli village (N 14°31’54”, E 78° 02’58”), Vemula Mandal of the Cuddapah district, Rayalaseema, Andhra Pradesh, India. The rock formation in the mineralized area of this village belongs to the Vempalli Formation (VF) of the Papaghni group of the lower Cuddapah Supergroup in the Cuddapah Basin (CB). The carbonate minerals, such as limestone and dolomite, are the most abundant ones and common sedimentary rocks present in this area.
Catalyst characterization
The NDL catalyst was ground into a fine powder and then sieved in a 200-mesh sieve. The chemical composition of the catalyst was determined by standard quantitative analysis. The basic strength of the catalyst was analyzed by using Hammett indicators. The catalyst was characterized by XRD, IR, Raman, SEM, and EDAX analysis.
The chemical composition of the NDL was determined by adopting a standard quantitative analysis [75]. The obtained results are summarized in Table 1.
The basic strength of the NDL catalyst (H_) was measured using Hammett indicators, namely bromothymol blue (H_ = 7.2), phenolphthalein (H_ = 9.8), 2,4-dinitroaniline (H_ = 15.0), and nitroaniline (H_ = 18.4) as Hammett indicators. In each case, 5 mL of a methanolic solution of the Hammett indicator was added to 50 mg of the catalyst, shaken well, and then allowed to equilibrate for 2 h. No color variation of the indicators was observed. The study revealed that the basic strength of the NDL catalyst was weaker than the bromothymol blue indicator, i.e., H_ < 7.2. Hence, the NDL catalyst is a mild base, and it can activate both nucleophilic and electrophilic groups [73]. Further, the amount of basic sites on the catalyst was estimated by titration using a standard benzoic acid solution and bromothymol blue indicator. Initially, the catalyst (50 mg) was stirred with the methanolic solution of the indicator (5 mL) for 30–40 min, and then, the mixture was titrated with a 0.02 M benzoic acid solution. From the titer values of the benzoic acid solution, the amount of the basic sites was found to be 0.033 mmol/g.
The powder XRD pattern of the NDL catalyst is shown in Figure 2. The diffraction peaks at 2θ = 23.16, 29.51, 31.05, 36.02, 38.07, 39.40, 43.0, 47.2, 47.5, 48.5, 56.6, 57.6, 60.9, and 64.8° were attributed to the (012), (104), (006), (015), (110), (113), (021), (024), (018), (116), (211), (122), (214), and (030) plane, respectively, of the NDL catalyst (JCPDS card file 5–586: calcite and 11–78: dolomite) [76,77]. Small quantities of aluminium silicates (kaolinite) and iron oxides were also confirmed by the XRD pattern. The less intense diffraction peaks at 2θ = 12.3, 24.8, and 37.4 were assigned to the 001, 002, and 003 plane, respectively, of kaolinite (JCPDS card file 14-0164) [78]. The low-intense peaks at 2θ = 18.6, 26.1, 44.7, 54.6, 58.4, and 63.0 were ascribed to the 111, 211, 400, 422, 511, and 440 plane, respectively, of iron oxides (JCPDS card file 39-1346 and JCPDS card file 19-629) [79,80]. The above results were supported by FTIR and Raman characterization studies of the catalyst (vide infra).
![[1860-5397-16-156-2]](/bjoc/content/figures/1860-5397-16-156-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: XRD pattern of the NDL catalyst.
Figure 2: XRD pattern of the NDL catalyst.
The FTIR spectrum of the catalyst is shown in Figure 3. In the IR spectrum, two distinct vibrational modes of the carbonates, i.e., out-of-plane bending and in-plane bending, were observed at 875 cm−1 (ν2) and 720 cm−1 (ν4), respectively. The bands at 1086 cm−1 and 1424 cm−1 were ascribed to a symmetric stretching vibration (ν1) and an asymmetric stretching vibration (ν3) of the carbonate group, respectively. The combined bands of the carbonate group, i.e., ν1 + ν4 and ν1 + ν3 were observed at 1798 and 2524 cm−1, respectively [76,77,81]. The IR bands at 3446 cm−1 (broad) and 1674 cm−1 (sharp) indicated the presence of stretching and bending vibrations of water [82]. The impurities aluminium silicate and iron oxides in the NDL were confirmed by IR spectroscopy. The peaks located at 446, 551, 817, 952, 1247, and 1383 cm−1 were attributed to the Si–O bending, Fe–O stretching, Al–O–Si stretching, Si–OH bending, Si–O stretching, and Al–O bending, respectively [83,84]. Further, the sharp band at 3696 cm−1 indicated the presence of a well-ordered kaolinite structure [76].
![[1860-5397-16-156-3]](/bjoc/content/figures/1860-5397-16-156-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: FTIR spectrum of the NDL catalyst.
Figure 3: FTIR spectrum of the NDL catalyst.
The Raman spectrum of the NDL catalyst is shown in Figure 4. The band at 1092 cm−1 was attributed to the symmetric stretching vibration (ν1) of the carbonate group. The peaks at 714 and 1435 cm−1 were assigned to a symmetric bending (ν4) and an asymmetric stretching vibration (ν3) of carbonate. The weak peak at 1750 cm−1 was due to the combined band ν1 + ν4. The bands at 152 and 278 cm−1 were ascribed to the external vibrations of the carbonate group [76,77]. The presence of aluminium silicates and iron oxides present in the sample were confirmed by Raman spectroscopy. The bands at 418, 578, 753, and 985 cm−1 were assigned to Al–O bending, Si–O rocking, Al–O stretching, and Si–OH stretching vibrations, respectively [85]. Further, a very weak peak at 618 cm−1 was attributed to iron oxide, and a very broad peak at 1312 cm−1 (magnon) indicated the presence of magnetically ordered ferromagnetic or antiferromagnetic iron oxides [86]. The observed Raman and infrared vibrational bands of the NDL were in good agreement with the reported values. The minor shift in the band positions might be due to the presence of trace metal contents and impurities.
![[1860-5397-16-156-4]](/bjoc/content/figures/1860-5397-16-156-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Raman spectrum of the NDL catalyst.
Figure 4: Raman spectrum of the NDL catalyst.
The morphology of the NDL catalyst was analyzed by scanning electron microscopy (Figure 5). The SEM images revealed that the morphology of the NDL catalyst consists of irregular shapes and sizes with a random dispersion. Further, the elemental composition of the NDL catalyst was determined by EDAX analysis (Figure 6).
![[1860-5397-16-156-5]](/bjoc/content/figures/1860-5397-16-156-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: SEM images of the NDL catalyst.
Figure 5: SEM images of the NDL catalyst.
![[1860-5397-16-156-6]](/bjoc/content/figures/1860-5397-16-156-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: EDAX analysis of the NDL catalyst.
Figure 6: EDAX analysis of the NDL catalyst.
The catalytic activity of the NDL for the synthesis of the 1,2-disubstituted benzimidazoles 3, the dihydropyrimidinones/-thiones 7, and the 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines 11 was investigated, along with other, commercially available catalysts.
NDL-catalyzed synthesis of 1,2-disubtituted benzimidazoles 3
To check the catalytic activity of the NDL, initially, o-phenylenediamine (1) and benzaldehyde (2a) were chosen as model substrates to optimize the reaction conditions for the synthesis of 1-benzyl-2-phenyl-1H-benzo[d]imidazole (3a). At first, a control experiment was conducted by using model substrates, 1 and 2a, in H2O in the absence of catalyst under ultrasound irradiation for 60 min at 45–50 °C. It was found that the reaction did not proceed in the absence of a catalyst (Table 2, entry 1). To achieve the target compound 3a, the same reaction was repeated by employing various catalysts (2.5 wt %), such as Fe2O3, Al2O3, KF–alumina, dolomitic limestone, triethylamine, pyridine, and DABCO in different solvents, such as water, acetone, iPrOH, EtOH, and EtOH/H2O 1:1 (Table 2, entries 2–8) under ultrasound irradiation at 45–50 °C. From this study, it was observed that the NDL (2.5 wt %) was the best option, which gave the target compound 3a in a high yield (85%) in a mixture of EtOH and H2O 1:1 under ultrasound irradiation for 30 min at 45–50 °C (Table 2, entry 5). The other catalysts, Fe2O3, Al2O3, KF–alumina, triethylamine, pyridine, and DABCO, provided a moderate to low yield of the product 3a (Table 2, entries 2–4 and 6–8). The aforesaid reaction was performed under conventional stirring of the model substrates 1 and 2a in H2O in the absence of catalyst for 180 min at 45–50 °C. It was observed that the reaction did not proceed in the absence of a catalyst (Table 2, entry 1). Further, when the reaction temperature was raised from 45–50 °C to reflux, a very low yield (10%) of the product 3a was obtained after 120 min. Next, the reaction was repeated in the presence of different catalysts and solvents at reflux under conventional reaction conditions as mentioned in Table 2. The study revealed that the NDL in a mixture of EtOH and H2O 1:1 afforded a moderate yield (70%) of the product 3a (Table 2, entry 5), whereas the other catalysts, in various solvents, provided lower yields under similar reaction conditions (Table 2, entries 2–4 and 6–8). From the above observations, it was concluded that the ultrasound irradiation method is better than the conventional method in giving the maximum yield of 3a.
Table 2: Optimization of the reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-156-i3.svg?max-width=637&scale=1.0)
|
|||||||
| entry | catalyst (2.5 wt %) | solvent | product | conventional methodb | USIc | ||
| t (min) | yieldd (%) | t (min) | yieldd (%) | ||||
| 1e | no catalyst | H2O | 3a | 180 | – | 60 | – |
| 2 | Fe2O3 | H2O | 3a | 60 | 10 | 30 | 15 |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 10 | 30 | 20 | |||
| EtOH | 60 | 15 | 30 | 20 | |||
| EtOH/H2O 1:1 | 60 | 20 | 30 | 25 | |||
| 3 | Al2O3 | H2O | 3a | 60 | 20 | 30 | 20 |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 15 | 30 | 20 | |||
| EtOH | 60 | 25 | 30 | 25 | |||
| EtOH/H2O 1:1 | 60 | 30 | 30 | 40 | |||
| 4 | KF–alumina | H2O | 3a | 60 | 30 | 30 | 30 |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 25 | 30 | 30 | |||
| EtOH | 60 | 40 | 30 | 35 | |||
| EtOH/H2O 1:1 | 60 | 50 | 30 | 40 | |||
| 5 | NDL | H2O | 3a | 60 | 55 | 30 | 65 |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 35 | 30 | 45 | |||
| EtOH | 60 | 60 | 30 | 75 | |||
| EtOH/H2O 1:1 | 60 | 70 | 30 | 85 | |||
| 6 | Et3N | H2O | 3a | 60 | 10 | 30 | 10 |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 10 | 30 | 10 | |||
| EtOH | 60 | 15 | 30 | 20 | |||
| EtOH/H2O 1:1 | 60 | 10 | 30 | 10 | |||
| 7 | pyridine | H2O | 3a | 60 | – | 30 | – |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 5 | 30 | 5 | |||
| EtOH | 60 | 10 | 30 | 10 | |||
| EtOH/H2O 1:1 | 60 | 5 | 30 | 5 | |||
| 8 | DABCO | H2O | 3a | 60 | 10 | 30 | 5 |
| acetone | 60 | – | 30 | – | |||
| iPrOH | 60 | 15 | 30 | 5 | |||
| EtOH | 60 | 15 | 30 | 15 | |||
| EtOH/H2O 1:1 | 60 | 10 | 30 | 10 | |||
aReaction conditions: o-phenylenediamine (1, 1.0 mmol), benzaldehyde (2a, 2.0 mmol), catalyst (2.5 wt %), solvent (3.0 mL). bPerformed by stirring at reflux (entries 2–8). cUSI method performed at 45–50 °C. dIsolated yield. eConventional method performed by stirring at 45–50 °C.
Next, the amount of catalyst was varied (using 2.5, 5.0, 7.5, 10.0, and 12.5 wt %, respectively,) to improve the yield of 3a (Table 3). The study revealed that 5.0 wt % of the NDL was the best option to get the highest yield of the product 3a (98%) in a short reaction time (10 min, Table 3, entry 3). It was also noticed that the same yield was obtained with an increasing amount of the catalyst, i.e., 7.5, 10.0, and 12.5 wt % (Table 3, entries 4–6).
Table 3: Effect of the catalyst loading.a
| entry | NDL (wt %) | solvent | t (min) | product | yieldb (%) |
| 1 | 2.5 | EtOH/H2O 1:1 | 30 | 3a | 85 |
| 2 | 2.5 | EtOH/H2O 1:1 | 10 | 3a | 75 |
| 3 | 5.0 | EtOH/H2O 1:1 | 10 | 3a | 98 |
| 4 | 7.5 | EtOH/H2O 1:1 | 10 | 3a | 98 |
| 5 | 10.0 | EtOH/H2O 1:1 | 10 | 3a | 98 |
| 6 | 12.5 | EtOH/H2O 1:1 | 10 | 3a | 98 |
aReaction conditions: o-phenylenediamine (1, 1.0 mmol), benzaldehyde (2a, 2.0 mmol), NDL (2.5 to 12.5 wt %), EtOH/H2O 1:1 (3.0 mL), ultrasound irradiation at 45–50 °C. bIsolated yield.
In order to demonstrate the effect of the temperature on the course of the model reaction, the control experiment was performed at different temperature ranges (30–35, 35–40, 40–45, and 45–50 °C) by using the model substrates 1 and 2a in the presence of 5.0 wt % of the NDL in a mixture of ethanol and water 1:1 for 10 min under both conventional stirring and ultrasound irradiation. The obtained results are presented in Table 4. It was observed that the reaction proceeded with an improved yield of 3a (70–98%) by increasing the temperature range from 30–35 to 45–50 °C with an ultrasound irradiation method (Table 4, entries 1–4). Under conventional stirring, the yield of the product 3a increased from low to moderate when the reaction temperature was raised from 30–35 °C to reflux (Table 4, entries 1–5). From the results, it was concluded that a temperature of 45–50 °C is the optimum temperature to obtain the maximum yield of the desired product 3a within a short reaction time (10 min) under ultrasound irradiation (Table 4, entry 4).
Table 4: Effect of the temperature.a
| entry | T (°C) | product | t (min) | conventional methodb | USIc |
| yieldd (%) | yieldd (%) | ||||
| 1 | 30–35 | 3a | 10 | 10 | 70 |
| 2 | 35–40 | 3a | 10 | 14 | 79 |
| 3 | 40–45 | 3a | 10 | 20 | 87 |
| 4 | 45–50 | 3a | 10 | 26 | 98 |
| 5e | reflux | 3a | 10/60 | 35/70 | – |
aReaction conditions: o-phenylenediamine (1, 1.0 mmol), benzaldehyde (2a, 2.0 mmol), NDL (5.0 wt %), EtOH/H2O 1:1 (3.0 mL). bConventional stirring and heating with a silicone oil bath. cUltrasound irradiation in a water bath. dIsolated yield. eConventional stirring at reflux.
To demonstrate the generality and substrate scope of the present method, a variety of (hetero)aromatic aldehydes was investigated. The obtained results are presented in Table 5. o-Phenylenediamine (1) reacted well with benzaldehyde (2a) to obtain the corresponding product 3a with 98% yield (Table 5, entry 1). The reactions of o-phenylenediamine (1) with substituted benzaldehydes having activating groups (4-Me: 2b, 4-t-Bu: 2c, 2,4-dimethyl: 2d, 4-OMe: 2e, 3,4-dimethoxy: 2f, 3,4,5-trimethoxy: 2g, 4-OH-3-OMe: 2j, and 4-OH-3-OC2H5: 2k, Table 5, entries 2–7, 10 and 11), a deactivating group (4-NO2: 2l, Table 5, entry 12), or a halo group (4-F: 2m, 4-Cl: 2n, and 4-Br: 2o, Table 5, entries 13–15) in different positions provided good to excellent isolated yields of the corresponding products 3b–g and 3j–o that ranged from 94 to 98% in a stipulated period of time, as specified in Table 5. Further, heteroaromatic aldehydes, such as furan-2-aldehyde (2p) and thiophene-2-aldehyde (2q) produced the corresponding products 3p and 3q in good isolated yields within a short period of time (15 min and 13 min, respectively, Table 5, entries 16 and 17).
Table 5: NDL-catalyzed synthesis of 2-aryl-1-arylmethyl-1H-benzo[d]imidazoles 3.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-156-i4.svg?max-width=637&scale=1.0)
|
||||||
| entry | Ar | product | t (min) | yieldc (%) | mp (°C) | |
| found | reported | |||||
| 1 | phenyl: 2a | 3a | 10 | 98 | 128–131 | 133–134 [23] |
| 2 | 4-methylphenyl: 2b | 3b | 10 | 98 | 127–128 | 128–129 [23] |
| 3 | 4-tert-butylphenyl: 2c | 3c | 15 | 94 | 124–125 | 122–126 [25] |
| 4 | 2,4-dimethylphenyl: 2d | 3d | 12 | 96 | 120–122 | 119–123 [25] |
| 5 | 4-methoxyphenyl: 2e | 3e | 11 | 98 | 157–159 | 158–160 [23] |
| 6 | 3,4-dimethoxyphenyl: 2f | 3f | 12 | 95 | 167–169 | 171–173 [24] |
| 7 | 3,4,5-trimethoxyphenyl: 2g | 3g | 15 | 94 | 261–262 | 262–263 [22] |
| 8b | 2-hydroxyphenyl: 2h | 3h | 10 | 98 | 167–168 | 160–162 [23] |
| 9b | 2-hydroxy-3-ethoxyphenyl: 2i | 3i | 12 | 96 | 285–287 | – |
| 10 | 4-hydroxy-3-methoxyphenyl: 2j | 3j | 12 | 96 | 181–183 | 184–186 [24] |
| 11 | 4-hydroxy-3-ethoxyphenyl: 2k | 3k | 10 | 97 | 205–207 | 200–201 [26] |
| 12 | 4-nitrophenyl: 2l | 3l | 10 | 98 | 190–192 | 189–191 [23] |
| 13 | 4-fluorophenyl: 2m | 3m | 10 | 98 | 108–109 | 110–112 [23] |
| 14 | 4-chlorophenyl: 2n | 3n | 10 | 98 | 138–140 | 137–139 [23] |
| 15 | 4-bromophenyl: 2o | 3o | 12 | 96 | 158–160 | 160–162 [23] |
| 16 | 2-furanyl: 2p | 3p | 15 | 95 | 90–92 | 88–89 [23] |
| 17 | 2-thienyl: 2q | 3q | 13 | 96 | 149–150 | 150–152 [23] |
aReaction conditions: o-phenylenediamine (1, 1.0 mmol), aldehyde (2, 2.0 mmol), NDL (5.0 wt %), EtOH/H2O 1:1 (3.0 mL), USI, 45–50 °C. bThe reaction stopped at the bisimine I, i.e., 3h/i stage. cIsolated yield.
However, salicylaldehyde (2h) afforded the unexpected product 2,2'-((1E,1'E)-(1,2-phenylenebis(azanylylidene))bis(methanylylidene))diphenol (3h, bisimine I) within 10 min (Table 5, entry 8). The reaction was expected to proceed through the activation of the carbonyl group of 2h (of which 2.0 mmol were used) by the cations (Ca2+ and Mg2+, respectively) of the NDL. This was followed by a nucleophilic attack of the NH2 groups of o-phenylenediamine (1, of which 1.0 mmol was used), which are activated by the carbonate part of the NDL, followed by dehydration to obtain 3h (Scheme 2). Due to the mild basic nature of the NDL catalyst, it acts as a dual activator of the electrophilic carbonyl and the nucleophilic NH2 groups. The formation of the bisimine I was confirmed by 1H NMR spectral studies (Figure 7). In the 1H NMR spectrum (DMSO-d6), the two hydroxy protons of the bisimine I appeared as a broad, strongly downfield-shifted singlet at δ 13.19. The sharp singlet at δ 8.66 indicated the two imine protons (–N=CH) of the bisimine I. From this result, it was confirmed that the reaction stopped at the bisimine I stage. This was due to the intramolecular hydrogen bonding between the hydrogen atom of the ortho-hydroxy group and the nitrogen atom of the imine group in a six-membered ring transition state [87]. Similarly, the reaction between 3-ethoxysalicylaldehyde (2i) and o-phenylenediamine (1) also ended with the intermediate 6,6'-((1E,1'E)-(1,2-phenylenebis(azanylylidene))bis(methanylylidene))bis(2-ethoxyphenol) (3i) stage (Table 4, entry 9 and Supporting Information File 1, Figure S13). Most of the synthesized compounds are known and were identified easily by comparison of the melting point and spectroscopic data with those reported.
![[1860-5397-16-156-i2]](/bjoc/content/inline/1860-5397-16-156-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Unexpected formation of the bisimine I, 3h, from o-phenylenediamine (1) and salicylaldehyde (2h).
Scheme 2: Unexpected formation of the bisimine I, 3h, from o-phenylenediamine (1) and salicylaldehyde (2h).
![[1860-5397-16-156-7]](/bjoc/content/figures/1860-5397-16-156-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: 1H NMR spectrum of 2,2'-((1E,1'E)-(1,2-phenylenebis(azanylylidene))bis (methanylylidene))diphenol (bisimine I, 3h).
Figure 7: 1H NMR spectrum of 2,2'-((1E,1'E)-(1,2-phenylenebis(azanylylidene))bis (methanylylidene))diphenol (...
NDL-catalyzed synthesis of dihydropyrimidinones/-thiones 7
The results encouraged us to further investigate the catalytic activity of the NDL in the Biginelli reaction. To check the feasibility, a control experiment was performed by using the model substrates benzaldehyde (2a, 1.0 mmol), ethyl acetoacetate (4, 1.0 mmol), and urea (5, 1.0 mmol) in H2O (3.0 mL) in the absence of a catalyst under ultrasound irradiation at 45–50 °C for 60 min. It was observed that the reaction proceeded with a very low yield (20%) of product 7a. The same reaction was repeated in the presence of the NDL catalyst (5.0 wt %) in EtOH/H2O 1:1 under ultrasound irradiation at 45–50 °C for 15 min, which resulted in 97 % yield of 7a.
To exploit the substrate scope and generality of the method, various (hetero)aromatic aldehydes 2 were examined. The obtained results are summarized in Table 6. Benzaldehyde (2a) underwent the reaction with ethyl acetoacetate (4) and urea (5) to obtain the corresponding dihydropyrimidinone 7a in 97% yield (Table 6, entry 1). Benzaldehyde derivatives bearing electron-donating groups, such as 4-Me (2b), 4-OMe (2e), 3,4-dimethoxy (2f), 3-OH (2r), and 2-OH (2h), respectively, at different positions on the ring reacted well with ethyl acetoacetate (4) and urea (5) to produce the products, 7b–f in good isolated yields that ranged from 92–96% (Table 6, entries 2–6). A benzaldehyde derivative with an electron-accepting nitro group (2l) at the para position on the ring showed a good reactivity with ethyl acetoacetate (4) and urea (5) to afford the product 7g in an excellent isolated yield (94%, Table 6, entry 7). Halogen atoms at different positions on the ring of benzaldehyde derivatives (4-F: 2m, 4-Cl; 2n, and 3-Br: 2s) underwent the reaction with ethyl acetoacetate (4) and urea (5) to form the corresponding products (7h–j) in good isolated yields that ranged from 93–96% (Table 6, entries 8–10). Heteroaromatic aldehydes, such as furan-2-aldehyde (2p) and thiophene-2-aldehyde (2q) showed a good reactivity, with good yields of 7k (90%) and 7l (92%), respectively (Table 6, entries 11 and 12). From this study, it was concluded that the optimized reaction conditions are suitable for monosubstituted (both electron-rich and electron-deficient) and disubstituted benzaldehyde derivatives as well as heteroaromatic aldehydes. To expand the scope of this method, thiourea (6) was also investigated (Table 6, entries 13–17). Benzaldehyde (2a) reacted with ethyl acetoacetate (4) and thiourea (6) to give the product 7m in an excellent isolated yield (96%, Table 6, entry 13). Benzaldehyde derivatives bearing electron-donating groups, such as 4-Me (2b) and 4-OMe (2c) exhibited a good reactivity with ethyl acetoacetate (4) and thiourea (6) to produce the products 7n (95%) and 7o (95%) in excellent yields, respectively (Table 6, entries 14 and 15). Benzaldehyde with electron-withdrawing groups, such as 4-NO2 (2f) and 4-Cl (2i) at the para position reacted well with ethyl acetoacetate (4) and thiourea (6) to afford the corresponding products 7p and 7q in good isolated yields (94 and 95%) (Table 6, entries 16 and 17). Most of the synthesized compounds are known and were identified easily by comparison of the melting point and spectroscopic data with those reported.
Table 6: NDL-catalyzed synthesis of dihydropyrimidinone/-thione derivatives 7.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-156-i5.svg?max-width=637&scale=1.0)
|
|||||||
| entry | Ar | X | product | t (min) | yieldb (%) | mp (°C) | |
| found | reported | ||||||
| 1 | phenyl: 2a | O | 7a | 15 | 97 | 207–209 | 209–210 [50] |
| 2 | 4-methylphenyl: 2b | O | 7b | 15 | 96 | 213–214 | 215–216 [38] |
| 3 | 4-methoxyphenyl: 2e | O | 7c | 17 | 96 | 200–201 | 199–202 [48] |
| 4 | 3,4-dimethoxyphenyl: 2f | O | 7d | 18 | 94 | 213–215 | 212–214 [52] |
| 5 | 3-hydroxyphenyl: 2r | O | 7e | 17 | 95 | 162–164 | 163–165 [38] |
| 6 | 2-hydroxyphenyl: 2h | O | 7f | 15 | 92 | 198–200 | 199–201 [49] |
| 7 | 4-nitrophenyl: 2l | O | 7g | 12 | 94 | 210–211 | 209–212 [48] |
| 8 | 4-fluorophenyl: 2m | O | 7h | 13 | 95 | 176–179 | 175–177 [37] |
| 9 | 4-chlorophenyl: 2n | O | 7i | 12 | 96 | 208–210 | 209–211 [48] |
| 10 | 3-bromophenyl: 2s | O | 7j | 18 | 93 | 184–185 | 185–186 [47] |
| 11 | 2-furanyl: 2p | O | 7k | 20 | 90 | 204–206 | 203–205 [48] |
| 12 | 2-thienyl: 2q | O | 7l | 20 | 92 | 216–218 | 215–217 [38] |
| 13 | phenyl: 2a | S | 7m | 15 | 96 | 211–212 | 208–210 [38] |
| 14 | 4-methylphenyl: 2b | S | 7n | 15 | 95 | 189–190 | 192–194 [38] |
| 15 | 4-methoxyphenyl: 2e | S | 7o | 17 | 95 | 148–150 | 150–152 [38] |
| 16 | 4-nitrophenyl: 2l | S | 7p | 10 | 94 | 113–114 | 109–111 [38] |
| 17 | 4-chlorophenyl: 2n | S | 7q | 11 | 95 | 190–191 | 192–194 [38] |
aReaction conditions: aldehyde (2, 1.0 mmol), ethyl acetoacetate (4, 1.0 mmol), urea/thiourea (5/6, 1.0 mmol), NDL (5.0 wt %), ethanol/H2O 1:1 (3.0 mL), USI at 45–50 °C. bIsolated yield.
NDL-catalyzed synthesis of 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines 11
We further examined the catalytic efficacy of the NDL catalyst in the synthesis of the medicinally privileged highly functionalized pyridines 11. For this purpose, a control experiment in the absence of a catalyst was conducted by using the model substrates benzaldehyde (2a, 1.0 mmol), malononitrile (8, 2.0 mmol), and 2-mercaptopyridine (9, 1.0 mmol) in H2O (3.0 mL) under ultrasound irradiation at 45–50 °C for 60 min. It was observed that the reaction did not afford any product in the absence of a catalyst. The above reaction was carried out in the presence of the NDL (5.0 wt %) in EtOH/H2O 1:1 (3.0 mL) under ultrasound irradiation for 10 min, which resulted in 70% yield of 11a. To improve the yield of 11a, the same reaction was repeated at different time intervals; 15, 20, 25, 30, 35, and 40 min, respectively, at 45–50 °C, and the yields of 11a obtained were 75, 83, 89, 96%, 96, and 96%, respectively. From this study, it was found that the maximum yield of 11a (96%) was obtained in 30 min, and the yields remained the same when the reaction time was increased from 30 to 40 min.
The optimized procedure was successfully applied for the synthesis of a series of highly substituted pyridines (11b–r, Table 7) by utilizing a range of (hetero)aromatic aldehydes 2, malononitrile (8), and the thiols 9 and 10, respectively, as starting materials. Benzaldehyde (2a) underwent the reaction with malononitrile (8) and 2-mercaptopyridine (9) to form product 11a in 96% yield (Table 7, entry 1). Benzaldehyde derivatives containing a range of functional groups, such as electron-donating groups (4-OMe: 2e, 3,4,5-trimethoxy: 2g, and 3-OH: 2r), an electron-withdrawing group (4-NO2: 2l), and halogen atoms (4-F: 2m, 4-Cl: 2n, and 3,4-difluoro: 2t), respectively, at different positions on the aromatic ring showed a good reactivity with the said reactants and afforded the corresponding products 11b–h that ranged from 90 to 96% (Table 7, entries 2–8). Further, the use of pyridine-2-aldehyde (2u) resulted in a good isolated yield of 11i (93%, Table 7, entry 9). In a similar way, the reaction of benzaldehyde (2a) with malononitrile (8) and thiophenol (10) gave the product 11j in 98% yield (Table 7, entry 10). Benzaldehyde derivatives bearing various functional groups, such as electron-donating groups (4-Me: 2b, 4-OMe: 2e, and 3,4,5-trimethoxy: 2g), an electron-accepting group (4-NO2: 2l), and halogen atoms (4-F: 2m, 4-Cl: 2n, and 3-Br: 2s), respectively, at different positions on the aromatic ring displayed a good reactivity with malononitrile (8) and thiophenol (10) to give the corresponding products (11k–q) in good yields, ranging from 94 to 98% (Table 7, entries 11–17). Pyridine-2-aldehyde (2u) also provided the product 11r in a good yield (94%, Table 7, entry 18). It was observed from the above results that all reactions proceeded well irrespective of the substituents present on the (hetero)aromatic aldehyde and afforded the highly substituted pyridines 11 in good isolated yields that ranged from 90 to 98%. Most of the synthesized compounds are known and were identified easily by comparison of the melting point and spectroscopic data with those reported.
Table 7: NDL-catalyzed synthesis of 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines 11.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-156-i6.svg?max-width=637&scale=1.0)
|
|||||||
| entry | Ar | R | product | t (min) | yieldb (%) | mp (°C) | |
| found | reported | ||||||
| 1 | phenyl: 2a | pyridyl 9 | 11a | 30 | 96 | 222–223 | 224–227 [70] |
| 2 | 4-methoxyphenyl: 2e | pyridyl 9 | 11b | 35 | 96 | 248–249 | 250–253 [70] |
| 3 | 3,4,5-trimethoxyphenyl: 2g | pyridyl 9 | 11c | 40 | 92 | 267–269 | 265–268 [70] |
| 4 | 3-hydroxyphenyl: 2r | pyridyl 9 | 11d | 35 | 94 | 223–224 | 222–226 [70] |
| 5 | 4-nitrophenyl: 2l | pyridyl 9 | 11e | 32 | 96 | 241–243 | 245–248 [70] |
| 6 | 4-fluorophenyl: 2m | pyridyl: 9 | 11f | 32 | 95 | 248–250 | 246–249 [70] |
| 7 | 4-bromophenyl: 2o | pyridyl: 9 | 11g | 30 | 94 | 257–258 | 260–263 [70] |
| 8 | 3,4-difluorophenyl: 2t | pyridyl: 9 | 11h | 37 | 90 | 252–253 | 251–254 [70] |
| 9 | pyridyl: 2u | pyridyl: 9 | 11i | 45 | 93 | 230–231 | 233–235 [70] |
| 10 | phenyl: 2a | phenyl: 10 | 11j | 30 | 98 | 210–212 | 215–216 [63] |
| 11 | 4-methylphenyl: 2b | phenyl: 10 | 11k | 30 | 98 | 206–207 | 208–210 [69] |
| 12 | 4-methoxyphenyl: 2e | phenyl: 10 | 11l | 35 | 97 | 234–235 | 236–238 [64] |
| 13 | 3,4,5-trimethoxyphenyl: 2g | phenyl: 10 | 11m | 38 | 94 | 240–241 | 238–239 [63] |
| 14 | 4-nitrophenyl: 2l | phenyl: 10 | 11n | 30 | 95 | 280–282 | 286–287 [63] |
| 15 | 4-fluorophenyl: 2m | phenyl: 10 | 11o | 30 | 96 | 127–128 | 224–225 [69] |
| 16 | 4-chlorophenyl: 2n | phenyl: 10 | 11p | 30 | 96 | 220–221 | 222–223 [69] |
| 17 | 3-bromophenyl: 2s | phenyl: 10 | 11q | 34 | 94 | 250–253 | 256–258 [65] |
| 18 | pyridyl: 2u | phenyl: 10 | 11r | 42 | 94 | 300–302 | 305–306 [63] |
aReaction conditions: aldehyde (2, 1.0 mmol), malononitrile (8, 2.0 mmol), thiol 9 or 10 (1.0 mmol), NDL (5.0 wt %), EtOH/H2O 1:1 (3.0 mL), USI at 45–50 °C. bIsolated yield.
Evaluation of the green chemistry metrics for the synthesis of benzimidazoles 3, dihydropyrimidinones 7, and highly functionalized pyridines 11
In order to evaluate the “greenness” of the proposed methodologies, the green chemistry metrics, such as the atom economy (AE), E-factor, process mass intensity (PMI), Curzon’s reaction mass efficiency (RME), and generalized or global reaction mass efficiency (gRME) were evaluated by adopting established standard empirical formulae [88,89]. The obtained results are summarized in Tables 8–10. This study revealed that the reactions displayed a good to excellent AE (88–95%) and Curzon’s RME (78–93%) as well as a low to moderate E-factor (26.202–50.760) and PMI (27.202–51.760). The detailed calculations of the green chemistry metrics (AE, E-factor, PMI, Curzon’s RME, and gRME) for the synthesis of the compounds 3a, 7a, and 11a (Table 8, entry 1, Table 9, entry 1, and Table 10, entry 1) are presented in Supporting Information File 1 (see Reaction-S1–Reaction-S3).
Table 8: Green chemistry metrics for the synthesis of 2-aryl-1-arylmethyl-1H-benzo[d]imidazoles 3.
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-156-i7.svg?max-width=637&scale=1.0)
|
|||||||
| entry | Ar | product | AEa (%) | E-factorb | PMIc |
Curzon’s RMEd
(%) |
gRMEe
(%) |
| 1 | phenyl: 2a | 3a | 89 | 40.864 | 41.864 | 87 | 2.4 |
| 2 | 4-methylphenyl: 2b | 3b | 90 | 37.261 | 38.261 | 88 | 2.6 |
| 3 | 4-tert-butylphenyl: 2c | 3c | 92 | 30.614 | 31.614 | 86 | 3.2 |
| 4 | 2,4-dimethylphenyl: 2d | 3d | 90 | 35.044 | 36.044 | 86 | 2.8 |
| 5 | 4-methoxyphenyl: 2e | 3e | 91 | 33.837 | 34.837 | 89 | 2.9 |
| 6 | 3,4-dimethoxyphenyl: 2f | 3f | 92 | 29.729 | 30.729 | 87 | 3.3 |
| 7 | 3,4,5-trimethoxyphenyl: 2g | 3g | 93 | 26.202 | 27.202 | 87 | 3.7 |
| 8 | 2-hydroxyphenyl: 2h | 3h | 90 | 36.781 | 37.781 | 88 | 2.6 |
| 9 | 2-hydroxy-3-ethoxyphenyl: 2i | 3i | 92 | 29.412 | 30.412 | 88 | 3.3 |
| 10 | 4-hydroxy-3-methoxyphenyl: 2j | 3j | 91 | 31.609 | 32.609 | 88 | 3.1 |
| 11 | 4-hydroxy-3-ethoxyphenyl: 2k | 3k | 92 | 29.102 | 30.102 | 89 | 3.3 |
| 12 | 4-nitrophenyl: 2l | 3l | 91 | 31.017 | 32.017 | 89 | 3.1 |
| 13 | 4-fluorophenyl: 2m | 3m | 90 | 36.312 | 37.312 | 88 | 2.7 |
| 14 | 4-chlorophenyl: 2n | 3n | 91 | 33.052 | 34.052 | 89 | 2.9 |
| 15 | 4-bromophenyl: 2o | 3o | 93 | 26.920 | 27.920 | 89 | 3.6 |
| 16 | 2-furanyl: 2p | 3p | 88 | 45.454 | 46.454 | 84 | 2.2 |
| 17 | 2-thienyl: 2q | 3q | 89 | 40.169 | 41.169 | 85 | 2.4 |
aAE = 100⋅(GMW of the product/sum of the GMWs of the reactants); GMW = gram molecular weight. bE-factor = total input mass (minputs)f − mass of the target product (m3) − mass of the recovered materials/m3. cPMI = (minputs − mass of the recovered materials)/m3 or 1 + E-factor. dCurzon’s RME = m3/ m1 + m2 or yield × AE × 1/stoichiometric factor (SF); SF = 1. egRME = 100⋅(m3/(minputs − mass of the recovered materials)) or 100⋅(1/(1 + E-factor)). fTotal input mass, including water (minputs) = m1 + m2 + msolvent (S) + mcatalyst (C) + mwork-up materials (WPM) + mpurification materials (PM).
Table 9: Green chemistry metrics for the synthesis of dihydropyrimidinones/-thiones 7.
![[Graphic 6]](/bjoc/content/inline/1860-5397-16-156-i8.svg?max-width=637&scale=1.0)
|
||||||||
| entry | reactants | product | AE (%) | E-factora | PMIb | Curzon’s RMEc (%) |
gRMEd
(%) |
|
| Ar | 5/6 | |||||||
| 1 | phenyl: 2a | 5 | 7a | 88 | 45.254 | 46.254 | 85 | 2.2 |
| 2 | 4-methylphenyl: 2b | 5 | 7b | 88 | 43.373 | 44.373 | 84 | 2.3 |
| 3 | 4-methoxyphenyl: 2e | 5 | 7c | 89 | 41.036 | 42.036 | 85 | 2.4 |
| 4 | 3,4-dimethoxyphenyl: 2f | 5 | 7d | 90 | 37.924 | 38.924 | 85 | 2.6 |
| 5 | 3-hydroxyphenyl: 2r | 5 | 7e | 89 | 43.550 | 44.550 | 85 | 2.2 |
| 6 | 2-hydroxyphenyl: 2h | 5 | 7f | 89 | 44.953 | 45.953 | 82 | 2.2 |
| 7 | 4-nitrophenyl: 2l | 5 | 7g | 89 | 39.770 | 40.770 | 84 | 2.5 |
| 8 | 4-fluorophenyl: 2m | 5 | 7h | 89 | 43.220 | 44.220 | 85 | 2.3 |
| 9 | 4-chlorophenyl: 2n | 5 | 7i | 89 | 40.311 | 41.311 | 85 | 2.4 |
| 10 | 3-bromophenyl: 2s | 5 | 7j | 90 | 36.254 | 37.254 | 84 | 2.7 |
| 11 | 2-furanyl: 2p | 5 | 7k | 87 | 50.760 | 51.760 | 78 | 1.9 |
| 12 | 2-thienyl: 2q | 5 | 7l | 88 | 46.600 | 47.600 | 81 | 2.1 |
| 13 | phenyl: 2a | 6 | 7m | 89 | 43.045 | 44.045 | 85 | 2.3 |
| 14 | 4-methylphenyl: 2b | 6 | 7n | 89 | 41.341 | 41.341 | 85 | 2.4 |
| 15 | 4-methoxyphenyl: 2e | 6 | 7o | 90 | 39.213 | 40.213 | 86 | 2.5 |
| 16 | 4-nitrophenyl: 2l | 6 | 7p | 90 | 37.978 | 38.978 | 85 | 2.6 |
| 17 | 4-chlorophenyl: 2n | 6 | 7q | 90 | 38.685 | 39.685 | 86 | 2.5 |
aE-factor = minputse − mass of the target product (m7) − mass of the recovered materials/m7. bPMI = (minputs − mass of the recovered materials)/m7 or 1 + E-factor. cCurzon’s RME = m7/m2 + m 4 + m5/6 or yield × AE × 1/SF; SF = 1. dgRME = 100⋅(m7/(minputs − mass of the recovered materials)) or 100⋅(1/(1 + E-factor)). eminputs = m2 + m4 + m5/6 + mS + mC + mWPM + mpurification materials (PM).
Table 10: Green chemistry metrics for the synthesis of 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines 11.
![[Graphic 7]](/bjoc/content/inline/1860-5397-16-156-i9.svg?max-width=637&scale=1.0)
|
||||||||
| entry | reactants | product | AE (%) | E-factora | PMIb |
Curzon’s RMEc
(%) |
gRMEd (%) | |
| Ar | R | |||||||
| 1 | phenyl: 2a | pyridyl: 9 | 11a | 94 | 36.054 | 37.054 | 90 | 2.7 |
| 2 | 4-methoxyphenyl: 2e | pyridyl: 9 | 11b | 95 | 33.026. | 34.026 | 91 | 2.9 |
| 3 | 3,4,5-trimethoxyphenyl: 2g | pyridyl: 9 | 11c | 95 | 29.647 | 30.647 | 87 | 3.3 |
| 4 | 3-hydroxyphenyl: 2r | pyridyl: 9 | 11d | 95 | 35.188 | 36.188 | 89 | 2.8 |
| 5 | 4-nitrophenyl: 2l | pyridyl: 9 | 11e | 95 | 31.741 | 32.741 | 91 | 3.1 |
| 6 | 4-fluorophenyl: 2m | pyridyl: 9 | 11f | 95 | 34.356 | 35.356 | 90 | 2.8 |
| 7 | 4-bromophenyl: 2o | pyridyl: 9 | 11g | 95 | 29.698 | 30.698 | 89 | 3.3 |
| 8 | 3,4-difluorophenyl: 2t | pyridyl: 9 | 11h | 95 | 34.699 | 35.699 | 86 | 2.8 |
| 9 | pyridyl: 2u | pyridyl: 9 | 11i | 94 | 37.143 | 38.143 | 87 | 2.6 |
| 10 | Phenyl: 2a | phenyl: 10 | 11j | 94 | 35.474 | 36.474 | 92 | 2.7 |
| 11 | 4-methylphenyl: 2b | phenyl: 10 | 11k | 95 | 33.991 | 34.991 | 93 | 2.9 |
| 12 | 4-methoxyphenyl: 2e | phenyl: 10 | 11l | 95 | 32.827 | 33.827 | 92 | 3.0 |
| 13 | 3,4,5-trimethoxyphenyl: 2g | phenyl: 10 | 11m | 95 | 29.020 | 30.020 | 89 | 3.3 |
| 14 | 4-nitrophenyl: 2l | phenyl: 10 | 11n | 95 | 32.021 | 33.021 | 90 | 3.0 |
| 15 | 4-fluorophenyl: 2m | phenyl: 10 | 11o | 95 | 34.319 | 35.319 | 91 | 2.8 |
| 16 | 4-chlorophenyl: 2n | phenyl: 10 | 11p | 95 | 32.744 | 33.744 | 91 | 3.0 |
| 17 | 3-bromophenyl: 2s | phenyl: 10 | 11q | 95 | 29.775 | 30.775 | 89 | 3.2 |
| 18 | pyridyl: 2u | phenyl: 10 | 11r | 94 | 36.893 | 37.893 | 88 | 2.6 |
aE-factor = minputsf − mass of the target product (m11) − mass of the recovered materials/m11. bPMI = (minputs − mass of the recovered materials)/m11 or 1 + E-factor. cCurzon’s RME = m11/ m2 + m8 + m9/10 or yield × AE × 1/SF; SF = 1. dgRME = 100⋅(m11/(minputs − mass of the recovered materials)) or 100⋅(1/(1 + E-factor)). eminputs = m2 + m8 + m9/10 + mS + mC + mWPM + mPM.
Catalyst reusability experiments
Catalyst reusability tests were performed showcasing the synthesis of the compounds 3k, 7a, and 11e under the optimized reaction conditions.
Catalyst reusability experiments in the synthesis of compounds 3k, 7a, and 11e
The catalyst was tested for reusability in the preparation of 3k using o-phenylenediamine (1) and 3-ethoxy-4-hydroxybenzaldehyde (2k) under USI for 10 min. After completion of the first reaction cycle, the reaction mass was allowed to cool to rt, and ethyl acetate (4.0 mL) was added. Then, the catalyst was separated by vacuum filtration, washed with ethyl acetate (1.0 mL), dried under vacuum, and reused in the next cycles. The study revealed that the obtained yields of the product, 3k were 98, 98, 97, 97, 96, 97, and 98% for the first, second, third, fourth, fifth, sixth, and seventh cycle, respectively. Catalyst reusability tests were then conducted for the synthesis of compound 7a using benzaldehyde (2a), ethyl acetoacetate (4), and urea (5) under USI for 15 min and for 11e using 4-nitrobenzaldehyde (2l), malononitrile (8), and 2-mercaptopyridine (9) under USI for 32 min by following the same procedure as adopted for 3k. The yields obtained for the compounds were 97, 97, 97, 96, 97, 97, and 97% for 7a as well as 96, 96, 96, 97, 97, 97, and 98% for 11e for the first, second, third, fourth, fifth, sixth, and seventh cycle, respectively. From this study, it was noticed that the catalyst could successfully be reused (at least) 7 times in the synthesis of the compounds 3k, 7a, and 11e without a significant loss of the catalytic activity.
Effect of ultrasonication on the structure of the catalyst
The recovered catalyst after the 7th cycle of each synthesis was characterized by XRD to study the structural changes due to ultrasonication. As can be seen in Figure 8, the diffraction peak positions of the catalyst recovered after the synthesis of the compounds 3k, 7a, and 11e (Figure 8b–d), respectively, remained the same as compared to the fresh catalyst (Figure 8a). It was also noticed that the broadening in the XRD pattern of the recovered catalyst had increased with an increase of the ultrasonication time. This clearly indicated that the amorphization of the recovered catalyst was enhanced by increasing the ultrasonication time.
![[1860-5397-16-156-8]](/bjoc/content/figures/1860-5397-16-156-8.png?scale=1.5&max-width=1024&background=FFFFFF)
Figure 8: XRD pattern of a) the fresh NDL catalyst; b) the recovered NDL catalyst after the 7th cycle of the ultrasonic synthesis of 3k; c) the recovered NDL catalyst after the 7th cycle of the ultrasonic synthesis of 7a; and d) the recovered NDL catalyst after the 7th cycle of the ultrasonic synthesis of 11e.
Figure 8: XRD pattern of a) the fresh NDL catalyst; b) the recovered NDL catalyst after the 7th cycle of the ...
Conclusion
An environmentally benign NDL catalyst was characterized and utilized as a heterogeneous catalyst for the synthesis of 2-aryl-1-arylmethyl-1H-benzo[d]imidazoles, dihydropyrimidinones/ -thiones, and 2-amino-4-(hetero)aryl-3,5-dicarbonitrile-6-sulfanylpyridines in a mixture of ethanol and H2O 1:1 under ultrasound irradiation. Notable advantages of this methodology include the clean reaction profile, broad substrate scope, simplicity of the process and handling, low catalyst loading, and the easy and quick isolation of the products in good to excellent yield. Besides, the products obtained were in an adequate purity without the need for chromatographic separation, and the catalyst was reused 7 times without a significant loss of the catalytic activity. Hence, the catalyst is a greener alternative for the synthesis of 1,2-disubstituted benzimidazoles, dihydropyrimidinones/-thiones, and highly substituted pyridines when compared to the existing reported catalysts. Further, the expansion of the catalyst scope and the generality for the synthesis of other privileged nitrogen- and sulfur-based heterocycles is under progress in our laboratory.
Experimental
See Supporting Information File 1 for full experimental data of compounds 3, 7, and 11.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, and copies of the 1H and 13C NMR, mass, and HRMS spectra of 3, 7, and 11. | ||
| Format: PDF | Size: 3.4 MB | Download |
Acknowledgements
The authors thank Dr. R. V. J. Kashyap, Dept. of English, Yogi Vemana University, for critical reading of the manuscript to ensure linguistic correctness.
Funding
The authors gratefully acknowledge the financial support for this work from the Department of Atomic Energy Board of Research in Nuclear Sciences (Bhabha Atomic Research Centre), Mumbai, Government of India, through a major research project (No. 2011/37C/52/BRNS/2264) and the Council of Scientific and Industrial Research (CSIR), New Delhi, Government of India, under a major research project (No. 01 (2391)/10/EMR-II).
References
-
Joule, J. A.; Mills, K. Heterocyclic Chemistry, 4th ed.; Blackwell: Oxford, U.K., 2000.
Return to citation in text: [1] -
Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds. Comprehensive Heterocyclic Chemistry III; Elsevier: Oxford, U.K., 2008; Vol. 7.
Return to citation in text: [1] -
Bull, J. A.; Mousseau, J. J.; Pelletier, G.; Charette, A. B. Chem. Rev. 2012, 112, 2642–2713. doi:10.1021/cr200251d
And references therein.
Return to citation in text: [1] -
Nagarajaiah, H.; Mukhopadhyay, A.; Moorthy, J. N. Tetrahedron Lett. 2016, 57, 5135–5149. doi:10.1016/j.tetlet.2016.09.047
Return to citation in text: [1] -
Suresh; Sandhu, J. S. ARKIVOC 2012, No. i, 66–133. doi:10.3998/ark.5550190.0013.103
Return to citation in text: [1] -
Perrier, V.; Wallace, A. C.; Kaneko, K.; Safar, J.; Prusiner, S. B.; Cohen, F. E. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 6073–6078. doi:10.1073/pnas.97.11.6073
Return to citation in text: [1] -
Fredholm, B. B.; Ijzerman, A. P.; Jacobson, K. A.; Klotz, K.-N.; Linden, J. Pharmacol. Rev. 2001, 53, 527–552.
Return to citation in text: [1] -
Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. J. Am. Chem. Soc. 2013, 135, 118–121. doi:10.1021/ja311780a
Return to citation in text: [1] -
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b
Return to citation in text: [1] -
Preston, P. N. Chem. Rev. 1974, 74, 279–314. doi:10.1021/cr60289a001
Return to citation in text: [1] -
Scott, L. J.; Dunn, C. J.; Mallarkey, G.; Sharpe, M. Drugs 2002, 62, 1503–1538. doi:10.2165/00003495-200262100-00006
Return to citation in text: [1] -
Carcanague, D.; Shue, Y.-K.; Wuonola, M. A.; Uria-Nickelsen, M.; Joubran, C.; Abedi, J. K.; Jones, J.; Kühler, T. C. J. Med. Chem. 2002, 45, 4300–4309. doi:10.1021/jm020868v
Return to citation in text: [1] -
Boiani, M.; Gonzalez, M. Mini-Rev. Med. Chem. 2005, 5, 409–424. doi:10.2174/1389557053544047
And references therein.
Return to citation in text: [1] -
Shah, D. I.; Sharma, M.; Bansal, Y.; Bansal, G.; Singh, M. Eur. J. Med. Chem. 2008, 43, 1808–1812. doi:10.1016/j.ejmech.2007.11.008
Return to citation in text: [1] -
Zhu, G.-D.; Gandhi, V. B.; Gong, J.; Thomas, S.; Luo, Y.; Liu, X.; Shi, Y.; Klinghofer, V.; Johnson, E. F.; Frost, D.; Donawho, C.; Jarvis, K.; Bouska, J.; Marsh, K. C.; Rosenberg, S. H.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2008, 18, 3955–3958. doi:10.1016/j.bmcl.2008.06.023
Return to citation in text: [1] -
Ogino, Y.; Ohtake, N.; Nagae, Y.; Matsuda, K.; Moriya, M.; Suga, T.; Ishikawa, M.; Kanesaka, M.; Mitobe, Y.; Ito, J.; Kanno, T.; Ishihara, A.; Iwaasa, H.; Ohe, T.; Kanatani, A.; Fukami, T. Bioorg. Med. Chem. Lett. 2008, 18, 5010–5014. doi:10.1016/j.bmcl.2008.08.018
Return to citation in text: [1] -
Molander, G. A.; Ajayi, K. Org. Lett. 2012, 14, 4242–4245. doi:10.1021/ol301956p
Return to citation in text: [1] -
Asensio, J. A.; Gómez-Romero, P. Fuel Cells 2005, 5, 336–343. doi:10.1002/fuce.200400081
Return to citation in text: [1] -
Schwartz, G.; Fehse, K.; Pfeiffer, M.; Walzer, K.; Leo, K. Appl. Phys. Lett. 2006, 89, 083509. doi:10.1063/1.2338588
Return to citation in text: [1] -
Mariappan, G.; Hazarika, R.; Alam, F.; Karki, R.; Patangia, U.; Nath, S. Arabian J. Chem. 2015, 8, 715–719. doi:10.1016/j.arabjc.2011.11.008
And referenes therein.
Return to citation in text: [1] -
Salahuddin; Shaharyar, M.; Mazumder, A.; Ahsan, M. J. Arabian J. Chem. 2014, 7, 418–424. doi:10.1016/j.arabjc.2013.02.001
And references therein.
Return to citation in text: [1] -
Ravi, V.; Ramu, E.; Vijay, K.; Srinivas Rao, A. Chem. Pharm. Bull. 2007, 55, 1254–1257. doi:10.1248/cpb.55.1254
Return to citation in text: [1] [2] -
Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Sharma, S. D.; Konwar, D. Synth. Commun. 2009, 39, 980–991. doi:10.1080/00397910802448440
Return to citation in text: [1] [2] [3] -
Reddy, L. S.; Reddy, N. C. G.; Reddy, T. R.; Lingappa, Y.; Mohan, R. B. J. Korean Chem. Soc. 2011, 55, 304–307. doi:10.5012/jkcs.2011.55.2.304
Return to citation in text: [1] [2] [3] -
Chebolu, R.; Kommi, D. N.; Kumar, D.; Bollineni, N.; Chakraborti, A. K. J. Org. Chem. 2012, 77, 10158–10167. doi:10.1021/jo301793z
Return to citation in text: [1] [2] -
Zhang, L.-J.; Xia, J.; Zhou, Y.-Q.; Wang, H.; Wang, S.-W. Synth. Commun. 2012, 42, 328–336. doi:10.1080/00397911.2010.524337
And references therein.
Return to citation in text: [1] -
Kumar, D.; Kommi, D. N.; Chebolu, R.; Garg, S. K.; Kumar, R.; Chakraborti, A. K. RSC Adv. 2013, 3, 91–98. doi:10.1039/c2ra21994h
Return to citation in text: [1] -
Senthilkumar, S.; Kumarraja, M. Tetrahedron Lett. 2014, 55, 1971–1974. doi:10.1016/j.tetlet.2014.01.140
And references therein.
Return to citation in text: [1] -
Herrera Cano, N.; Uranga, J. G.; Nardi, M.; Procopio, A.; Wunderlin, D. A.; Santiago, A. N. Beilstein J. Org. Chem. 2016, 12, 2410–2419. doi:10.3762/bjoc.12.235
And references therein.
Return to citation in text: [1] -
Costanzo, P.; Nardi, M.; Oliverio, M. Eur. J. Org. Chem. 2020, 3954–3964. doi:10.1002/ejoc.201901923
And references therein.
Return to citation in text: [1] -
Berlinck, R. G. S.; Burtoloso, A. C. B.; Kossuga, M. H. Nat. Prod. Rep. 2008, 25, 919–954. doi:10.1039/b507874c
Return to citation in text: [1] -
Hu, E. H.; Sidler, D. R.; Dolling, U.-H. J. Org. Chem. 1998, 63, 3454–3457. doi:10.1021/jo970846u
Return to citation in text: [1] -
Sakata, K.-I.; Someya, M.; Matsumoto, Y.; Tauchi, H.; Kai, M.; Toyota, M.; Takagi, M.; Hareyama, M.; Fukushima, M. Cancer Sci. 2011, 102, 1712–1716. doi:10.1111/j.1349-7006.2011.02004.x
Return to citation in text: [1] -
Ramesh, B.; Bhalgat, C. M. Eur. J. Med. Chem. 2011, 46, 1882–1891. doi:10.1016/j.ejmech.2011.02.052
Return to citation in text: [1] -
Kaira, K.; Serizawa, M.; Koh, Y.; Miura, S.; Kaira, R.; Abe, M.; Nakagawa, K.; Ohde, Y.; Okumura, T.; Murakami, H.; Tsuya, A.; Nakamura, Y.; Naito, T.; Takahashi, T.; Kondo, H.; Nakajima, T.; Endo, M.; Yamamoto, N. Lung Cancer 2011, 74, 419–425. doi:10.1016/j.lungcan.2011.04.001
Return to citation in text: [1] -
Schroeder, P. E.; Hasinoff, B. B. Drug Metab. Dispos. 2005, 33, 1367–1372. doi:10.1124/dmd.105.005546
Return to citation in text: [1] [2] -
Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Fu, N.-Y.; Yuan, Y.-F.; Cao, Z.; Wang, S.-W.; Wang, J.-T.; Peppe, C. Tetrahedron 2002, 58, 4801–4807. doi:10.1016/s0040-4020(02)00455-6
Return to citation in text: [1] -
Rodríguez-Domínguez, J. C.; Bernardi, D.; Kirsch, G. Tetrahedron Lett. 2007, 48, 5777–5780. doi:10.1016/j.tetlet.2007.06.104
Return to citation in text: [1] -
Ahmed, N.; van Lier, J. E. Tetrahedron Lett. 2007, 48, 5407–5409. doi:10.1016/j.tetlet.2007.06.005
Return to citation in text: [1] -
Chen, X.-H.; Xu, X.-Y.; Liu, H.; Cun, L.-F.; Gong, L.-Z. J. Am. Chem. Soc. 2006, 128, 14802–14803. doi:10.1021/ja065267y
Return to citation in text: [1] -
Ryabukhin, S. V.; Plaskon, A. S.; Ostapchuk, E. N.; Volochnyuk, D. M.; Shishkin, O. V.; Shivanyuk, A. N.; Tolmachev, A. A. Org. Lett. 2007, 9, 4215–4218. doi:10.1021/ol701782v
Return to citation in text: [1] -
Li, N.; Chen, X.-H.; Song, J.; Luo, S.-W.; Fan, W.; Gong, L.-Z. J. Am. Chem. Soc. 2009, 131, 15301–15310. doi:10.1021/ja905320q
Return to citation in text: [1] -
Guggilapu, S. D.; Prajapati, S. K.; Nagarsenkar, A.; Lalita, G.; Vegi, G. M. N.; Babu, B. N. New J. Chem. 2016, 40, 838–843. doi:10.1039/c5nj02444g
And references therein.
Return to citation in text: [1] -
Barbero, M.; Cadamuro, S.; Dughera, S. Green Chem. 2017, 19, 1529–1535. doi:10.1039/c6gc03274e
And references therein.
Return to citation in text: [1] -
Oliverio, M.; Costanzo, P.; Nardi, M.; Rivalta, I.; Procopio, A. ACS Sustainable Chem. Eng. 2014, 2, 1228–1233. doi:10.1021/sc5000682
Return to citation in text: [1] [2] -
Ranu, B. C.; Hajra, A.; Jana, U. J. Org. Chem. 2000, 65, 6270–6272. doi:10.1021/jo000711f
Return to citation in text: [1] [2] [3] [4] [5] -
Lu, J.; Bai, Y. Synthesis 2002, 466–470. doi:10.1055/s-2002-20956
Return to citation in text: [1] [2] -
Gangadasu, B.; Palaniappan, S.; Rao, V. J. Synlett 2004, 1285–1287. doi:10.1055/s-2004-822925
Return to citation in text: [1] [2] -
Russowsky, D.; Lopes, F. A.; da Silva, V. S. S.; Canto, K. F. S.; Montes D'Oca, M. G.; Godoi, M. N. J. Braz. Chem. Soc. 2004, 15, 165–169. doi:10.1590/s0103-50532004000200002
Return to citation in text: [1] -
Tu, S.; Fang, F.; Zhu, S.; Li, T.; Zhang, X.; Zhuang, Q. Synlett 2004, 537–539. doi:10.1055/s-2004-815419
Return to citation in text: [1] [2] -
Fazaeli, R.; Tangestaninejad, S.; Aliyan, H.; Moghadam, M. Appl. Catal., A 2006, 309, 44–51. doi:10.1016/j.apcata.2006.04.043
Return to citation in text: [1] -
Ahmed, B.; Khan, R. A.; Habibullah; Keshari, M. Tetrahedron Lett. 2009, 50, 2889–2892. doi:10.1016/j.tetlet.2009.03.177
Return to citation in text: [1] -
Phukan, M.; Kalita, M. K.; Borah, R. Green Chem. Lett. Rev. 2010, 3, 329–334. doi:10.1080/17518253.2010.487841
Return to citation in text: [1] -
Zhang, X.; Gu, X.; Gao, Y.; Nie, S.; Lu, H. Appl. Organomet. Chem. 2017, 31, e3590. doi:10.1002/aoc.3590
And references therein.
Return to citation in text: [1] -
Chitra, S.; Pandiarajan, K. Tetrahedron Lett. 2009, 50, 2222–2224. doi:10.1016/j.tetlet.2009.02.162
Return to citation in text: [1] -
Han, B.; Han, R.-F.; Ren, Y.-W.; Duan, X.-Y.; Xu, Y.-C.; Zhang, W. Tetrahedron 2011, 67, 5615–5620. doi:10.1016/j.tet.2011.05.105
Return to citation in text: [1] -
Shen, Z.-L.; Xu, X.-P.; Ji, S.-J. J. Org. Chem. 2010, 75, 1162–1167. doi:10.1021/jo902394y
Return to citation in text: [1] -
Sheik Mansoor, S.; Syed Shafi, S.; Zaheer Ahmed, S. Arabian J. Chem. 2016, 9, S846–S851. doi:10.1016/j.arabjc.2011.09.018
Return to citation in text: [1] -
Pandey, J.; Anand, N.; Tripathi, R. P. Tetrahedron 2009, 65, 9350–9356. doi:10.1016/j.tet.2009.09.002
Return to citation in text: [1] -
Cocco, M. T.; Congiu, C.; Lilliu, V.; Onnis, V. Eur. J. Med. Chem. 2005, 40, 1365–1372. doi:10.1016/j.ejmech.2005.07.005
Return to citation in text: [1] -
May, B. C. H.; Zorn, J. A.; Witkop, J.; Sherrill, J.; Wallace, A. C.; Legname, G.; Prusiner, S. B.; Cohen, F. E. J. Med. Chem. 2007, 50, 65–73. doi:10.1021/jm061045z
Return to citation in text: [1] [2] [3] [4] [5] -
Reddy, T. R. K.; Mutter, R.; Heal, W.; Guo, K.; Gillet, V. J.; Pratt, S.; Chen, B. J. Med. Chem. 2006, 49, 607–615. doi:10.1021/jm050610f
Return to citation in text: [1] [2] -
Evdokimov, N. M.; Magedov, I. V.; Kireev, A. S.; Kornienko, A. Org. Lett. 2006, 8, 899–902. doi:10.1021/ol052994+
Return to citation in text: [1] [2] -
Evdokimov, N. M.; Kireev, A. S.; Yakovenko, A. A.; Antipin, M. Y.; Magedov, I. V.; Kornienko, A. J. Org. Chem. 2007, 72, 3443–3453. doi:10.1021/jo070114u
Return to citation in text: [1] -
Ranu, B. C.; Jana, R.; Sowmiah, S. J. Org. Chem. 2007, 72, 3152–3154. doi:10.1021/jo070015g
Return to citation in text: [1] -
Mamgain, R.; Singh, R.; Rawat, D. S. J. Heterocycl. Chem. 2009, 46, 69–73. doi:10.1002/jhet.32
Return to citation in text: [1] -
Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b
Return to citation in text: [1] [2] [3] [4] -
Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Shinde, P. V.; Sonar, S. S.; Shingate, B. B.; Shingare, M. S. Tetrahedron Lett. 2010, 51, 1309–1312. doi:10.1016/j.tetlet.2009.12.146
Return to citation in text: [1] -
Srinivasula Reddy, L.; Ram Reddy, T.; Mohan, R. B.; Mahesh, A.; Lingappa, Y.; Gangi Reddy, N. C. Chem. Pharm. Bull. 2013, 61, 1114–1120. doi:10.1248/cpb.c13-00412
Return to citation in text: [1] -
Tamaddon, F.; Tayefi, M.; Hosseini, E.; Zare, E. J. Mol. Catal. A: Chem. 2013, 366, 36–42. doi:10.1016/j.molcata.2012.08.027
Return to citation in text: [1] [2] -
Correia, L. M.; de Sousa Campelo, N.; Novaes, D. S.; Cavalcante, C. L., Jr.; Cecilia, J. A.; Rodríguez-Castellón, E.; Vieira, R. S. Chem. Eng. J. 2015, 269, 35–43. doi:10.1016/j.cej.2015.01.097
Return to citation in text: [1] -
Vogel, A. I. Quantitative inorganic analysis; Longmans: London, U.K., 1951; p 582.
Return to citation in text: [1] -
Gunasekaran, S.; Anbalagan, G.; Pandi, S. J. Raman Spectrosc. 2006, 37, 892–899. doi:10.1002/jrs.1518
Return to citation in text: [1] [2] [3] [4] -
Xu, B.; Poduska, K. M. Phys. Chem. Chem. Phys. 2014, 16, 17634–17639. doi:10.1039/c4cp01772b
Return to citation in text: [1] [2] [3] -
Shawky, A.; El-Sheikh, S. M.; Rashed, M. N.; Abdo, S. M.; El-Dosoqy, T. I. J. Environ. Chem. Eng. 2019, 7, 103174. doi:10.1016/j.jece.2019.103174
Return to citation in text: [1] -
Darezereshki, E.; Ranjbar, M.; Bakhtiari, F. J. Alloys Compd. 2010, 502, 257–260. doi:10.1016/j.jallcom.2010.04.163
Return to citation in text: [1] -
Shao, M.; Ning, F.; Zhao, J.; Wei, M.; Evans, D. G.; Duan, X. J. Am. Chem. Soc. 2012, 134, 1071–1077. doi:10.1021/ja2086323
Return to citation in text: [1] -
White, W. B.; Karr, C., Jr., Eds. Infrared and Raman Spectroscopy of Lunar and Terrestrial Minerals; Academic Press: New York, NY, USA, 1975.
Return to citation in text: [1] -
Ramasesha, K.; De Marco, L.; Mandal, A.; Tokmakoff, A. Nat. Chem. 2013, 5, 935–940. doi:10.1038/nchem.1757
Return to citation in text: [1] -
Shalaby, N. H.; Elsalamony, R. A.; El Naggar, A. M. A. New J. Chem. 2018, 42, 9177–9186. doi:10.1039/c8nj01479e
Return to citation in text: [1] -
Lin, Y.-F.; Chen, H.-W.; Chang, C.-C.; Hung, W.-C.; Chiou, C.-S. J. Chem. Technol. Biotechnol. 2011, 86, 1449–1456. doi:10.1002/jctb.2665
Return to citation in text: [1] -
Yadav, A. K.; Singh, P. RSC Adv. 2015, 5, 67583–67609. doi:10.1039/c5ra13043c
And references therein.
Return to citation in text: [1] -
Debure, M.; Andreazza, P.; Canizarès, A.; Grangeon, S.; Lerouge, C.; Mack, P.; Madé, B.; Simon, P.; Veron, E.; Warmont, F.; Vayer, M. ACS Earth Space Chem. 2017, 1, 442–454. doi:10.1021/acsearthspacechem.7b00073
Return to citation in text: [1] -
Makal, A.; Schilf, W.; Kamieński, B.; Szady-Chelmieniecka, A.; Grech, E.; Woźniak, K. Dalton Trans. 2011, 40, 421–430. doi:10.1039/c0dt00298d
Return to citation in text: [1] -
Andraos, J.; Hent, A. J. Chem. Educ. 2015, 92, 1820–1830. doi:10.1021/acs.jchemed.5b00058
Return to citation in text: [1] -
Dicks, A. P.; Hent, A. Green Chemistry Metrics; Springer International Publishing: Cham, Switzerland, 2015. doi:10.1007/978-3-319-10500-0
Return to citation in text: [1]
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 64. | Reddy, T. R. K.; Mutter, R.; Heal, W.; Guo, K.; Gillet, V. J.; Pratt, S.; Chen, B. J. Med. Chem. 2006, 49, 607–615. doi:10.1021/jm050610f |
| 25. | Reddy, L. S.; Reddy, N. C. G.; Reddy, T. R.; Lingappa, Y.; Mohan, R. B. J. Korean Chem. Soc. 2011, 55, 304–307. doi:10.5012/jkcs.2011.55.2.304 |
| 63. | May, B. C. H.; Zorn, J. A.; Witkop, J.; Sherrill, J.; Wallace, A. C.; Legname, G.; Prusiner, S. B.; Cohen, F. E. J. Med. Chem. 2007, 50, 65–73. doi:10.1021/jm061045z |
| 63. | May, B. C. H.; Zorn, J. A.; Witkop, J.; Sherrill, J.; Wallace, A. C.; Legname, G.; Prusiner, S. B.; Cohen, F. E. J. Med. Chem. 2007, 50, 65–73. doi:10.1021/jm061045z |
| 69. | Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 26. | Chebolu, R.; Kommi, D. N.; Kumar, D.; Bollineni, N.; Chakraborti, A. K. J. Org. Chem. 2012, 77, 10158–10167. doi:10.1021/jo301793z |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 63. | May, B. C. H.; Zorn, J. A.; Witkop, J.; Sherrill, J.; Wallace, A. C.; Legname, G.; Prusiner, S. B.; Cohen, F. E. J. Med. Chem. 2007, 50, 65–73. doi:10.1021/jm061045z |
| 24. | Sharma, S. D.; Konwar, D. Synth. Commun. 2009, 39, 980–991. doi:10.1080/00397910802448440 |
| 24. | Sharma, S. D.; Konwar, D. Synth. Commun. 2009, 39, 980–991. doi:10.1080/00397910802448440 |
| 69. | Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b |
| 22. | Ravi, V.; Ramu, E.; Vijay, K.; Srinivas Rao, A. Chem. Pharm. Bull. 2007, 55, 1254–1257. doi:10.1248/cpb.55.1254 |
| 65. | Evdokimov, N. M.; Magedov, I. V.; Kireev, A. S.; Kornienko, A. Org. Lett. 2006, 8, 899–902. doi:10.1021/ol052994+ |
| 25. | Reddy, L. S.; Reddy, N. C. G.; Reddy, T. R.; Lingappa, Y.; Mohan, R. B. J. Korean Chem. Soc. 2011, 55, 304–307. doi:10.5012/jkcs.2011.55.2.304 |
| 63. | May, B. C. H.; Zorn, J. A.; Witkop, J.; Sherrill, J.; Wallace, A. C.; Legname, G.; Prusiner, S. B.; Cohen, F. E. J. Med. Chem. 2007, 50, 65–73. doi:10.1021/jm061045z |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 69. | Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 88. | Andraos, J.; Hent, A. J. Chem. Educ. 2015, 92, 1820–1830. doi:10.1021/acs.jchemed.5b00058 |
| 89. | Dicks, A. P.; Hent, A. Green Chemistry Metrics; Springer International Publishing: Cham, Switzerland, 2015. doi:10.1007/978-3-319-10500-0 |
| 52. | Tu, S.; Fang, F.; Zhu, S.; Li, T.; Zhang, X.; Zhuang, Q. Synlett 2004, 537–539. doi:10.1055/s-2004-815419 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 48. | Ranu, B. C.; Hajra, A.; Jana, U. J. Org. Chem. 2000, 65, 6270–6272. doi:10.1021/jo000711f |
| 87. | Makal, A.; Schilf, W.; Kamieński, B.; Szady-Chelmieniecka, A.; Grech, E.; Woźniak, K. Dalton Trans. 2011, 40, 421–430. doi:10.1039/c0dt00298d |
| 50. | Gangadasu, B.; Palaniappan, S.; Rao, V. J. Synlett 2004, 1285–1287. doi:10.1055/s-2004-822925 |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 48. | Ranu, B. C.; Hajra, A.; Jana, U. J. Org. Chem. 2000, 65, 6270–6272. doi:10.1021/jo000711f |
| 37. | Schroeder, P. E.; Hasinoff, B. B. Drug Metab. Dispos. 2005, 33, 1367–1372. doi:10.1124/dmd.105.005546 |
| 1. | Joule, J. A.; Mills, K. Heterocyclic Chemistry, 4th ed.; Blackwell: Oxford, U.K., 2000. |
| 2. | Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds. Comprehensive Heterocyclic Chemistry III; Elsevier: Oxford, U.K., 2008; Vol. 7. |
| 3. |
Bull, J. A.; Mousseau, J. J.; Pelletier, G.; Charette, A. B. Chem. Rev. 2012, 112, 2642–2713. doi:10.1021/cr200251d
And references therein. |
| 17. | Molander, G. A.; Ajayi, K. Org. Lett. 2012, 14, 4242–4245. doi:10.1021/ol301956p |
| 18. | Asensio, J. A.; Gómez-Romero, P. Fuel Cells 2005, 5, 336–343. doi:10.1002/fuce.200400081 |
| 19. | Schwartz, G.; Fehse, K.; Pfeiffer, M.; Walzer, K.; Leo, K. Appl. Phys. Lett. 2006, 89, 083509. doi:10.1063/1.2338588 |
| 73. | Tamaddon, F.; Tayefi, M.; Hosseini, E.; Zare, E. J. Mol. Catal. A: Chem. 2013, 366, 36–42. doi:10.1016/j.molcata.2012.08.027 |
| 74. | Correia, L. M.; de Sousa Campelo, N.; Novaes, D. S.; Cavalcante, C. L., Jr.; Cecilia, J. A.; Rodríguez-Castellón, E.; Vieira, R. S. Chem. Eng. J. 2015, 269, 35–43. doi:10.1016/j.cej.2015.01.097 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 14. | Shah, D. I.; Sharma, M.; Bansal, Y.; Bansal, G.; Singh, M. Eur. J. Med. Chem. 2008, 43, 1808–1812. doi:10.1016/j.ejmech.2007.11.008 |
| 15. | Zhu, G.-D.; Gandhi, V. B.; Gong, J.; Thomas, S.; Luo, Y.; Liu, X.; Shi, Y.; Klinghofer, V.; Johnson, E. F.; Frost, D.; Donawho, C.; Jarvis, K.; Bouska, J.; Marsh, K. C.; Rosenberg, S. H.; Giranda, V. L.; Penning, T. D. Bioorg. Med. Chem. Lett. 2008, 18, 3955–3958. doi:10.1016/j.bmcl.2008.06.023 |
| 16. | Ogino, Y.; Ohtake, N.; Nagae, Y.; Matsuda, K.; Moriya, M.; Suga, T.; Ishikawa, M.; Kanesaka, M.; Mitobe, Y.; Ito, J.; Kanno, T.; Ishihara, A.; Iwaasa, H.; Ohe, T.; Kanatani, A.; Fukami, T. Bioorg. Med. Chem. Lett. 2008, 18, 5010–5014. doi:10.1016/j.bmcl.2008.08.018 |
| 75. | Vogel, A. I. Quantitative inorganic analysis; Longmans: London, U.K., 1951; p 582. |
| 10. | Preston, P. N. Chem. Rev. 1974, 74, 279–314. doi:10.1021/cr60289a001 |
| 11. | Scott, L. J.; Dunn, C. J.; Mallarkey, G.; Sharpe, M. Drugs 2002, 62, 1503–1538. doi:10.2165/00003495-200262100-00006 |
| 12. | Carcanague, D.; Shue, Y.-K.; Wuonola, M. A.; Uria-Nickelsen, M.; Joubran, C.; Abedi, J. K.; Jones, J.; Kühler, T. C. J. Med. Chem. 2002, 45, 4300–4309. doi:10.1021/jm020868v |
| 13. |
Boiani, M.; Gonzalez, M. Mini-Rev. Med. Chem. 2005, 5, 409–424. doi:10.2174/1389557053544047
And references therein. |
| 62. | Cocco, M. T.; Congiu, C.; Lilliu, V.; Onnis, V. Eur. J. Med. Chem. 2005, 40, 1365–1372. doi:10.1016/j.ejmech.2005.07.005 |
| 63. | May, B. C. H.; Zorn, J. A.; Witkop, J.; Sherrill, J.; Wallace, A. C.; Legname, G.; Prusiner, S. B.; Cohen, F. E. J. Med. Chem. 2007, 50, 65–73. doi:10.1021/jm061045z |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 4. | Nagarajaiah, H.; Mukhopadhyay, A.; Moorthy, J. N. Tetrahedron Lett. 2016, 57, 5135–5149. doi:10.1016/j.tetlet.2016.09.047 |
| 5. | Suresh; Sandhu, J. S. ARKIVOC 2012, No. i, 66–133. doi:10.3998/ark.5550190.0013.103 |
| 6. | Perrier, V.; Wallace, A. C.; Kaneko, K.; Safar, J.; Prusiner, S. B.; Cohen, F. E. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 6073–6078. doi:10.1073/pnas.97.11.6073 |
| 7. | Fredholm, B. B.; Ijzerman, A. P.; Jacobson, K. A.; Klotz, K.-N.; Linden, J. Pharmacol. Rev. 2001, 53, 527–552. |
| 8. | Nguyen, T. B.; Ermolenko, L.; Al-Mourabit, A. J. Am. Chem. Soc. 2013, 135, 118–121. doi:10.1021/ja311780a |
| 9. | Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b |
| 64. | Reddy, T. R. K.; Mutter, R.; Heal, W.; Guo, K.; Gillet, V. J.; Pratt, S.; Chen, B. J. Med. Chem. 2006, 49, 607–615. doi:10.1021/jm050610f |
| 65. | Evdokimov, N. M.; Magedov, I. V.; Kireev, A. S.; Kornienko, A. Org. Lett. 2006, 8, 899–902. doi:10.1021/ol052994+ |
| 66. | Evdokimov, N. M.; Kireev, A. S.; Yakovenko, A. A.; Antipin, M. Y.; Magedov, I. V.; Kornienko, A. J. Org. Chem. 2007, 72, 3443–3453. doi:10.1021/jo070114u |
| 67. | Ranu, B. C.; Jana, R.; Sowmiah, S. J. Org. Chem. 2007, 72, 3152–3154. doi:10.1021/jo070015g |
| 68. | Mamgain, R.; Singh, R.; Rawat, D. S. J. Heterocycl. Chem. 2009, 46, 69–73. doi:10.1002/jhet.32 |
| 69. | Guo, K.; Thompson, M. J.; Chen, B. J. Org. Chem. 2009, 74, 6999–7006. doi:10.1021/jo901232b |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 71. | Shinde, P. V.; Sonar, S. S.; Shingate, B. B.; Shingare, M. S. Tetrahedron Lett. 2010, 51, 1309–1312. doi:10.1016/j.tetlet.2009.12.146 |
| 72. | Srinivasula Reddy, L.; Ram Reddy, T.; Mohan, R. B.; Mahesh, A.; Lingappa, Y.; Gangi Reddy, N. C. Chem. Pharm. Bull. 2013, 61, 1114–1120. doi:10.1248/cpb.c13-00412 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 32. | Berlinck, R. G. S.; Burtoloso, A. C. B.; Kossuga, M. H. Nat. Prod. Rep. 2008, 25, 919–954. doi:10.1039/b507874c |
| 33. | Hu, E. H.; Sidler, D. R.; Dolling, U.-H. J. Org. Chem. 1998, 63, 3454–3457. doi:10.1021/jo970846u |
| 34. | Sakata, K.-I.; Someya, M.; Matsumoto, Y.; Tauchi, H.; Kai, M.; Toyota, M.; Takagi, M.; Hareyama, M.; Fukushima, M. Cancer Sci. 2011, 102, 1712–1716. doi:10.1111/j.1349-7006.2011.02004.x |
| 35. | Ramesh, B.; Bhalgat, C. M. Eur. J. Med. Chem. 2011, 46, 1882–1891. doi:10.1016/j.ejmech.2011.02.052 |
| 36. | Kaira, K.; Serizawa, M.; Koh, Y.; Miura, S.; Kaira, R.; Abe, M.; Nakagawa, K.; Ohde, Y.; Okumura, T.; Murakami, H.; Tsuya, A.; Nakamura, Y.; Naito, T.; Takahashi, T.; Kondo, H.; Nakajima, T.; Endo, M.; Yamamoto, N. Lung Cancer 2011, 74, 419–425. doi:10.1016/j.lungcan.2011.04.001 |
| 37. | Schroeder, P. E.; Hasinoff, B. B. Drug Metab. Dispos. 2005, 33, 1367–1372. doi:10.1124/dmd.105.005546 |
| 48. | Ranu, B. C.; Hajra, A.; Jana, U. J. Org. Chem. 2000, 65, 6270–6272. doi:10.1021/jo000711f |
| 49. | Lu, J.; Bai, Y. Synthesis 2002, 466–470. doi:10.1055/s-2002-20956 |
| 50. | Gangadasu, B.; Palaniappan, S.; Rao, V. J. Synlett 2004, 1285–1287. doi:10.1055/s-2004-822925 |
| 51. | Russowsky, D.; Lopes, F. A.; da Silva, V. S. S.; Canto, K. F. S.; Montes D'Oca, M. G.; Godoi, M. N. J. Braz. Chem. Soc. 2004, 15, 165–169. doi:10.1590/s0103-50532004000200002 |
| 52. | Tu, S.; Fang, F.; Zhu, S.; Li, T.; Zhang, X.; Zhuang, Q. Synlett 2004, 537–539. doi:10.1055/s-2004-815419 |
| 53. | Fazaeli, R.; Tangestaninejad, S.; Aliyan, H.; Moghadam, M. Appl. Catal., A 2006, 309, 44–51. doi:10.1016/j.apcata.2006.04.043 |
| 54. | Ahmed, B.; Khan, R. A.; Habibullah; Keshari, M. Tetrahedron Lett. 2009, 50, 2889–2892. doi:10.1016/j.tetlet.2009.03.177 |
| 55. | Phukan, M.; Kalita, M. K.; Borah, R. Green Chem. Lett. Rev. 2010, 3, 329–334. doi:10.1080/17518253.2010.487841 |
| 56. |
Zhang, X.; Gu, X.; Gao, Y.; Nie, S.; Lu, H. Appl. Organomet. Chem. 2017, 31, e3590. doi:10.1002/aoc.3590
And references therein. |
| 57. | Chitra, S.; Pandiarajan, K. Tetrahedron Lett. 2009, 50, 2222–2224. doi:10.1016/j.tetlet.2009.02.162 |
| 58. | Han, B.; Han, R.-F.; Ren, Y.-W.; Duan, X.-Y.; Xu, Y.-C.; Zhang, W. Tetrahedron 2011, 67, 5615–5620. doi:10.1016/j.tet.2011.05.105 |
| 48. | Ranu, B. C.; Hajra, A.; Jana, U. J. Org. Chem. 2000, 65, 6270–6272. doi:10.1021/jo000711f |
| 31. |
Costanzo, P.; Nardi, M.; Oliverio, M. Eur. J. Org. Chem. 2020, 3954–3964. doi:10.1002/ejoc.201901923
And references therein. |
| 59. | Shen, Z.-L.; Xu, X.-P.; Ji, S.-J. J. Org. Chem. 2010, 75, 1162–1167. doi:10.1021/jo902394y |
| 60. | Sheik Mansoor, S.; Syed Shafi, S.; Zaheer Ahmed, S. Arabian J. Chem. 2016, 9, S846–S851. doi:10.1016/j.arabjc.2011.09.018 |
| 61. | Pandey, J.; Anand, N.; Tripathi, R. P. Tetrahedron 2009, 65, 9350–9356. doi:10.1016/j.tet.2009.09.002 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 22. | Ravi, V.; Ramu, E.; Vijay, K.; Srinivas Rao, A. Chem. Pharm. Bull. 2007, 55, 1254–1257. doi:10.1248/cpb.55.1254 |
| 23. | Wan, J.-P.; Gan, S.-F.; Wu, J.-M.; Pan, Y. Green Chem. 2009, 11, 1633–1637. doi:10.1039/b914286j |
| 24. | Sharma, S. D.; Konwar, D. Synth. Commun. 2009, 39, 980–991. doi:10.1080/00397910802448440 |
| 25. | Reddy, L. S.; Reddy, N. C. G.; Reddy, T. R.; Lingappa, Y.; Mohan, R. B. J. Korean Chem. Soc. 2011, 55, 304–307. doi:10.5012/jkcs.2011.55.2.304 |
| 26. | Chebolu, R.; Kommi, D. N.; Kumar, D.; Bollineni, N.; Chakraborti, A. K. J. Org. Chem. 2012, 77, 10158–10167. doi:10.1021/jo301793z |
| 27. |
Zhang, L.-J.; Xia, J.; Zhou, Y.-Q.; Wang, H.; Wang, S.-W. Synth. Commun. 2012, 42, 328–336. doi:10.1080/00397911.2010.524337
And references therein. |
| 28. | Kumar, D.; Kommi, D. N.; Chebolu, R.; Garg, S. K.; Kumar, R.; Chakraborti, A. K. RSC Adv. 2013, 3, 91–98. doi:10.1039/c2ra21994h |
| 29. |
Senthilkumar, S.; Kumarraja, M. Tetrahedron Lett. 2014, 55, 1971–1974. doi:10.1016/j.tetlet.2014.01.140
And references therein. |
| 30. |
Herrera Cano, N.; Uranga, J. G.; Nardi, M.; Procopio, A.; Wunderlin, D. A.; Santiago, A. N. Beilstein J. Org. Chem. 2016, 12, 2410–2419. doi:10.3762/bjoc.12.235
And references therein. |
| 48. | Ranu, B. C.; Hajra, A.; Jana, U. J. Org. Chem. 2000, 65, 6270–6272. doi:10.1021/jo000711f |
| 20. |
Mariappan, G.; Hazarika, R.; Alam, F.; Karki, R.; Patangia, U.; Nath, S. Arabian J. Chem. 2015, 8, 715–719. doi:10.1016/j.arabjc.2011.11.008
And referenes therein. |
| 21. |
Salahuddin; Shaharyar, M.; Mazumder, A.; Ahsan, M. J. Arabian J. Chem. 2014, 7, 418–424. doi:10.1016/j.arabjc.2013.02.001
And references therein. |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 39. | Fu, N.-Y.; Yuan, Y.-F.; Cao, Z.; Wang, S.-W.; Wang, J.-T.; Peppe, C. Tetrahedron 2002, 58, 4801–4807. doi:10.1016/s0040-4020(02)00455-6 |
| 40. | Rodríguez-Domínguez, J. C.; Bernardi, D.; Kirsch, G. Tetrahedron Lett. 2007, 48, 5777–5780. doi:10.1016/j.tetlet.2007.06.104 |
| 41. | Ahmed, N.; van Lier, J. E. Tetrahedron Lett. 2007, 48, 5407–5409. doi:10.1016/j.tetlet.2007.06.005 |
| 42. | Chen, X.-H.; Xu, X.-Y.; Liu, H.; Cun, L.-F.; Gong, L.-Z. J. Am. Chem. Soc. 2006, 128, 14802–14803. doi:10.1021/ja065267y |
| 43. | Ryabukhin, S. V.; Plaskon, A. S.; Ostapchuk, E. N.; Volochnyuk, D. M.; Shishkin, O. V.; Shivanyuk, A. N.; Tolmachev, A. A. Org. Lett. 2007, 9, 4215–4218. doi:10.1021/ol701782v |
| 44. | Li, N.; Chen, X.-H.; Song, J.; Luo, S.-W.; Fan, W.; Gong, L.-Z. J. Am. Chem. Soc. 2009, 131, 15301–15310. doi:10.1021/ja905320q |
| 45. |
Guggilapu, S. D.; Prajapati, S. K.; Nagarsenkar, A.; Lalita, G.; Vegi, G. M. N.; Babu, B. N. New J. Chem. 2016, 40, 838–843. doi:10.1039/c5nj02444g
And references therein. |
| 46. |
Barbero, M.; Cadamuro, S.; Dughera, S. Green Chem. 2017, 19, 1529–1535. doi:10.1039/c6gc03274e
And references therein. |
| 47. | Oliverio, M.; Costanzo, P.; Nardi, M.; Rivalta, I.; Procopio, A. ACS Sustainable Chem. Eng. 2014, 2, 1228–1233. doi:10.1021/sc5000682 |
| 47. | Oliverio, M.; Costanzo, P.; Nardi, M.; Rivalta, I.; Procopio, A. ACS Sustainable Chem. Eng. 2014, 2, 1228–1233. doi:10.1021/sc5000682 |
| 78. | Shawky, A.; El-Sheikh, S. M.; Rashed, M. N.; Abdo, S. M.; El-Dosoqy, T. I. J. Environ. Chem. Eng. 2019, 7, 103174. doi:10.1016/j.jece.2019.103174 |
| 73. | Tamaddon, F.; Tayefi, M.; Hosseini, E.; Zare, E. J. Mol. Catal. A: Chem. 2013, 366, 36–42. doi:10.1016/j.molcata.2012.08.027 |
| 76. | Gunasekaran, S.; Anbalagan, G.; Pandi, S. J. Raman Spectrosc. 2006, 37, 892–899. doi:10.1002/jrs.1518 |
| 77. | Xu, B.; Poduska, K. M. Phys. Chem. Chem. Phys. 2014, 16, 17634–17639. doi:10.1039/c4cp01772b |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 38. | Ma, Y.; Qian, C.; Wang, L.; Yang, M. J. Org. Chem. 2000, 65, 3864–3868. doi:10.1021/jo9919052 |
| 85. |
Yadav, A. K.; Singh, P. RSC Adv. 2015, 5, 67583–67609. doi:10.1039/c5ra13043c
And references therein. |
| 86. | Debure, M.; Andreazza, P.; Canizarès, A.; Grangeon, S.; Lerouge, C.; Mack, P.; Madé, B.; Simon, P.; Veron, E.; Warmont, F.; Vayer, M. ACS Earth Space Chem. 2017, 1, 442–454. doi:10.1021/acsearthspacechem.7b00073 |
| 76. | Gunasekaran, S.; Anbalagan, G.; Pandi, S. J. Raman Spectrosc. 2006, 37, 892–899. doi:10.1002/jrs.1518 |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 76. | Gunasekaran, S.; Anbalagan, G.; Pandi, S. J. Raman Spectrosc. 2006, 37, 892–899. doi:10.1002/jrs.1518 |
| 77. | Xu, B.; Poduska, K. M. Phys. Chem. Chem. Phys. 2014, 16, 17634–17639. doi:10.1039/c4cp01772b |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 82. | Ramasesha, K.; De Marco, L.; Mandal, A.; Tokmakoff, A. Nat. Chem. 2013, 5, 935–940. doi:10.1038/nchem.1757 |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 83. | Shalaby, N. H.; Elsalamony, R. A.; El Naggar, A. M. A. New J. Chem. 2018, 42, 9177–9186. doi:10.1039/c8nj01479e |
| 84. | Lin, Y.-F.; Chen, H.-W.; Chang, C.-C.; Hung, W.-C.; Chiou, C.-S. J. Chem. Technol. Biotechnol. 2011, 86, 1449–1456. doi:10.1002/jctb.2665 |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 79. | Darezereshki, E.; Ranjbar, M.; Bakhtiari, F. J. Alloys Compd. 2010, 502, 257–260. doi:10.1016/j.jallcom.2010.04.163 |
| 80. | Shao, M.; Ning, F.; Zhao, J.; Wei, M.; Evans, D. G.; Duan, X. J. Am. Chem. Soc. 2012, 134, 1071–1077. doi:10.1021/ja2086323 |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
| 76. | Gunasekaran, S.; Anbalagan, G.; Pandi, S. J. Raman Spectrosc. 2006, 37, 892–899. doi:10.1002/jrs.1518 |
| 77. | Xu, B.; Poduska, K. M. Phys. Chem. Chem. Phys. 2014, 16, 17634–17639. doi:10.1039/c4cp01772b |
| 81. | White, W. B.; Karr, C., Jr., Eds. Infrared and Raman Spectroscopy of Lunar and Terrestrial Minerals; Academic Press: New York, NY, USA, 1975. |
| 70. | Banerjee, S.; Sereda, G. Tetrahedron Lett. 2009, 50, 6959–6962. doi:10.1016/j.tetlet.2009.09.137 |
© 2020 Godugu et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)