

Abstract
We report that phenacyl azides are key compounds for a regiodivergent synthesis of valuable, functionalized imidazole (32–98% yield) and pyrimidine derivatives (45–88% yield), with a broad substrate scope, when using deep eutectic solvents [choline chloride (ChCl)/glycerol (1:2 mol/mol) and ChCl/urea (1:2 mol/mol)] as environmentally benign and non-innocent reaction media, by modulating the temperature (25 or 80 °C) in the presence or absence of bases (Et3N).
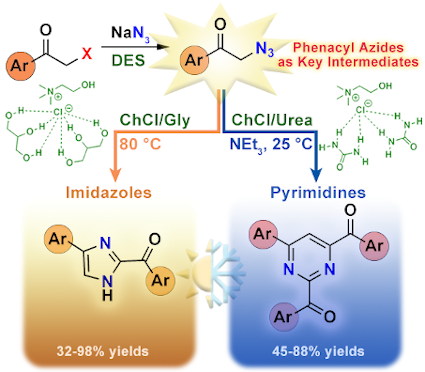
Graphical Abstract
Introduction
In a world with dwindling petroleum resources, the setting up of more and more sustainable routes for the preparation of heterocyclic compounds is an ongoing synthetic endeavor as these scaffolds are ubiquitous motifs in many biologically active compounds and pharmaceuticals. In this context, in the last decades, the so-called deep eutectic solvents (DESs) have received an increasing attention due to their biodegradability, high thermal stability, non-flammability, and low volatility. These are mixtures usually obtained from the combination of 2 or 3 safe, inexpensive and nature-inspired components able to engage in reciprocal hydrogen-bond interactions, and that form fluids at a specified mixing ratio at the desired temperature. Owing to the broad tunability of their physicochemical properties and the ability to act not only as solvents but also as catalysts and reagents, DESs have progressively replaced toxic and volatile organic solvents (VOCs) in countless heterocyclodehydration processes and multicomponent reactions (MCRs) [1-3].
As part of our ongoing research in DES chemistry, we reported recently on the preparation of valuable heterocycles by (a) nucleophilic substitution (tetrahydrofuran derivatives) [4], (b) heterocyclodehydration reactions (2-aminoimidazoles, 2-pyrazinones, benzoxazines, thiophenes) [5-8], (c) carbon–sulfur bond-forming reactions [9], (d) directed ortho-metalation and nucleophilic acyl substitution strategies [10], (e) Pd-catalyzed aminocarbonylation of aryl iodides, Suzuki–Miyaura and Sonogashira cross-coupling reactions [11-13], (f) Cu-catalyzed C–N coupling reactions [14], and (g) heterogeneous “click” cycloaddition reactions [15] using DESs as environmentally responsible and non-innocent reaction media. Telescoped, one-pot transformations of phenacyl halides to symmetrical 2,5-disubstituted pyrazines (A), through phenacyl azides as intermediates, were also found to take place smoothly using such neoteric solvents (Scheme 1, path a) [16].
![[1860-5397-16-158-i1]](/bjoc/content/inline/1860-5397-16-158-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: One-pot synthesis of 2,5-diarylpyrazines (A) (path a) or 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles (B) (path b), or 2,4-diaroyl-6-arylpyrimidines (C) (path c) in DES from phenacyl azides (rt = room temperature).
Scheme 1: One-pot synthesis of 2,5-diarylpyrazines (A) (path a) or 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles (B) ...
Among nitrogen-containing heterocyles, imidazoles and pyrimidines are important structural scaffolds commonly found in natural products [17,18], light-emitting devices [19,20], and pharmacologically active compounds as anticancer, anti-inflammatory, antitubercular, antihypertensive, antihistaminic, anti-obesity, antiviral, and other medicinal agents [21-27]. Herein, we wish to report that either 2-aroylimidazoles (B) (Scheme 1, path b) or 2,4-diaroylpyrimidines (C) (Scheme 1, path c) can regioselectively be prepared from the same phenacyl azide as starting material by properly selecting the nature of the eutectic mixture and the temperature, in the presence or absence of bases.
To the best of our knowledge, while there are a few reports for the synthesis of 2-aroylimidazoles (a) through the condensation of α-azido ketones in iPrOH in the presence of potassium ethylxanthate as a catalyst [28], (b) by exploiting the reaction of arylglyoxals with an excess amount of ammonium acetate in water [29], (c) by the cathodic reduction of 2-azido-1-phenylethanone in a DMF/LiClO4 medium [30], (d) by radical chain reactions of α-azido ketones with tributyltin hydride [31], or (e) by a modified Radziszewski’s synthesis when using phenylglyoxals, benzaldeydes, and ammonium acetate as ammonia source in acetic acid or methylene chloride or N,N-dimethylformamide as the solvent [32], there are no adequate studies covering the preparation of aroylpyrimidines. In 1995, Yamamoto et al. reported the direct introduction of acyl groups into pyrimidine rings by reacting trimethylstannyl derivatives with acylformyl chlorides in dry benzene under a N2 stream [33].
Results and Discussion
During our studies on the synthesis of symmetrical pyrazines, we observed that phenacyl bromide (1a, 1.5 mmol) could be almost quantitatively converted into phenacyl azide (2a, 97% yield), within 4 h, when treated with NaN3 (1.65 mmol) as the azide source in a choline chloride (ChCl)/glycerol (Gly) (1:2 mol/mol) eutectic mixture at room temperature (rt, 25 °C) (Scheme 2) [16]. By warming the mixture up to 50 °C, the yield of 2a dropped to 89% after 1 h, and we noticed in the crude the appearance of an additional product, which was detected as a fluorescent spot on TLC, whose amount increased by increasing the temperature. After 4 h warming at 50 °C, we were able to isolate by column chromatography on silica gel a product which was characterized as 2-benzoyl-(4 or 5)-phenyl-(1H)-imidazole (3a/3a', Scheme 2). This adduct was formed as a mixture of two tautomers (3a and 3a'; 3a/3a' ratio: 57:43, Supporting Information File 1) [28,29] in an overall 16% yield, the remaining being mainly 2a, which was isolated in 75% yield (Scheme 2). We thus became interested in improving the yield of 3a/3a'.
![[1860-5397-16-158-i2]](/bjoc/content/inline/1860-5397-16-158-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Transformation of phenacyl bromide (1a) in ChCl/Gly into phenacyl azide (2a) and 2-benzoyl-(4 or 5)-phenyl-(1H)-imidazole (3a and 3a') depending on the temperature.
Scheme 2: Transformation of phenacyl bromide (1a) in ChCl/Gly into phenacyl azide (2a) and 2-benzoyl-(4 or 5)...
After careful evaluation of the reaction parameters (temperature and time) and the amount of NaN3 used, we found that the treatment of phenacyl chloride (1b) with NaN3 (1.5 equiv) in ChCl/Gly at 80 °C gave the best results, as it provided the adducts 3a/3a' in an overall 88% yield after 12 h reaction time (Table 1, entries 1–4). Of note, imidazoles 3a/3a' were found to precipitate directly from the aforementioned eutectic mixture during the reaction, and thus they could be isolated by simple decantation or centrifugation and washing with a few drops of EtOAc or Et2O. This procedure left azide 2a in solution. The latter could be quantified (10% yield) by simple dilution with an equal volume mixture of water and EtOAc, followed by the separation of the organic layer from water, and removal of the volatiles under reduced pressure.
Table 1: Optimization of the reaction conditions for the synthesis of 3a/3a'.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-158-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | T (°C) | t (h) | NaN3 (equiv) | 2a yield (%)b | 3a/3a' yield (%)b |
| 1 | 80 | 4 | 1.2 | 37 | 55 |
| 2 | 100 | 16 | 1.2 | 7 | 60c |
| 3 | 80 | 16 | 0.9 | 68 | 32 |
| 4 | 80 | 12 | 1.5 | 10 | 88 |
aReaction conditions: 1b (0.5 mmol), ChCl/Gly (1.0 g); byield refers to products isolated after column chromatography on silica gel; c15% of 2,4-dibenzoyl-6-phenylpyrimidine was also isolated (see main text and Table 2).
To examine the scope and limitation of this transformation, various functionalized phenacyl halides (1c–i) were tested as substrates. As can be seen from the results compiled in Scheme 3, the reaction is amenable to “neutral” (Me), electron-withdrawing (Cl, Br, CN) and electron-donating (MeO, OH) substituents as the desired imidazoles 3b/3b'–3h were isolated in 78–98% yield. The presence of additional halogen groups such as chlorine and bromine in 3c/3c' and 3d allows further downstream diversification by cross-coupling reactions. A 4-fluoro-substituted phenacyl chloride as well as a bromomethyl 2-naphthyl ketone proved to be competent substrates as well, thus furnishing the corresponding imidazoles 3i/3i' and 3j in 67–87% yield. Conversely, a phenacyl halide decorated with an additional phenyl group at the para-position delivered the expected adduct 3k/3k' in 32% yield even by prolonging the reaction time to 16 h, most probably because of the poor solubility in the eutectic mixture and/or the higher thermal stability towards the loss of N2 (vide infra) of the corresponding phenacyl azide 2k as it is formed. The latter was indeed isolated as the main product (68% yield, Scheme 3).
![[1860-5397-16-158-i3]](/bjoc/content/inline/1860-5397-16-158-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles 3. Scope of the reaction. Typical conditions: 1 (0.5 mmol), NaN3 (0.75 mmol), ChCl/Gly (1.0 g), 80 °C, 4–16 h; yield refers to products isolated after column chromatography on silica gel; only one tautomer has been depicted for simplicity; imidazoles 3c/3c', 3i/3i', 3j, 3k/3k' were found to precipitate as they formed, and were isolated by filtration/centrifugation and washing with a few drops of AcOEt or Et2O; synthesis of imidazoles 3k/3k': 10% (w/w) EtOH was added to the eutectic mixture to improve the solubility of 2k.
Scheme 3: Synthesis of 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles 3. Scope of the reaction. Typical conditions: 1 ...
A plausible mechanism for the formation of the 2-aroylimidazoles 3/3' is depicted in Scheme 4. A key intermediate may be the in situ generated α-imino ketone 5. The latter is known to undergo dimerization to give 6 followed by dehydration. The two tautomers 3/3' would most probably originate by two competitive pathways (a and b; Scheme 4) via a [1,5]-hydrogen shift [28,29]. Hydrogen-bond catalysis promoted by DES components may be playing an important role in favoring the loss of nitrogen under mild conditions from a putative azide tautomer 4, the latter deriving from the corresponding α-phenacyl azide precursor 2 via an acid-catalyzed enolization process [34]. The pyrolysis of α-azido ketones in conventional VOCs (trichlorobenzene) is known to take place under harsh conditions, which are based on heating the mixture between 180 °C and 240 °C [35].
![[1860-5397-16-158-i4]](/bjoc/content/inline/1860-5397-16-158-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the formation of 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles 3/3' from α-phenacyl azides 2 in ChCl/Gly.
Scheme 4: Proposed mechanism for the formation of 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles 3/3' from α-phenacyl ...
The investigation of the thermal stability of phenacyl azides 2 led to the fortuitous discovery of the competitive formation of a different heterocycle. Indeed, while phenacyl azide (2a) was stable per se in a ChCl/Gly mixture after stirring at rt for 12 h, a new fluorescent spot was detected on TLC plate in the presence of an excess of Et3N (3 equiv). After column chromatography on silica gel, we were able to isolate a new product that was characterized as 2,4-dibenzoyl-6-phenylpyrimidine (7a, 55% yield) in addition to 3a/3a' (26% yield), the remaining being starting material only (conversion: 81%, Table 2, entry 1). The employment of ChCl/urea (1:2 mol/mol) as the eutectic mixture provided 7a in 57% yield and 3a/3a' in 43% yield, but with full conversion (Table 2, entry 2). By lowering the equivalents of Et3N up to 1, or alternatively using K2CO3 (3 equiv) as a base, in ChCl/urea, the yield of 7a dropped down up to 33% and that of 3a/3a' up to 25% (Table 2, entries 3–5). By changing the solvent to pure Gly, 7a and 3a/3a' now formed in 52 and 37% yield, respectively (Table 2, entry 6). When Et3N (3 equiv) was alternatively used as the sole solvent, the formation of 7a was suppressed dramatically (27% yield, Table 2, entry 7) [36]. This last result is consistent with a synergistic cooperation of the basic ChCl/urea eutectic mixture [37] with Et3N in promoting the transformation at rt. α-Azido ketones are, indeed, known to be highly base sensitive and to undergo a base-promoted loss of nitrogen to form α-imino ketones upon protonation [38].
Table 2: Investigation of the reaction conditions in the synthesis of 7a.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-158-i8.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent | 7a yield (%)b | 3a/3a' yield (%)b | conversion yield (%) |
| 1 | ChCl/Glyc | 55 | 26 | 81 |
| 2 | ChCl/ureac | 57 | 43 | full |
| 3 | ChCl/uread | 41 | 33 | 74 |
| 4 | ChCl/ureae | 37 | 25 | 62 |
| 5 | ChCl/ureaf | 33 | 31 | 66 |
| 6 | Glyc | 52 | 37 | 89 |
| 7 | Et3Nc | 27 | – | 27 |
aReaction conditions: 2a (0.3 mmol), solvent (0.5 g); byield refers to products isolated after column chromatography on silica gel; cEt3N: 3 equiv; dEt3N: 2 equiv; eEt3N: 1 equiv; fK2CO3: 3 equiv.
A plausible mechanism for the formation of pyrimidine derivative 7a from 2a, in competition with imidazoles 3a/3a', is depicted in Scheme 5.
![[1860-5397-16-158-i5]](/bjoc/content/inline/1860-5397-16-158-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Proposed mechanism for the formation of 2-benzoyl-(4 or 5)-phenyl-(1H)-imidazoles 3a/3a' and 2,4-dibenzoyl-6-phenylpyrimidine (7a) from α-phenacyl azide (2a) in ChCl/urea in the presence of Et3N.
Scheme 5: Proposed mechanism for the formation of 2-benzoyl-(4 or 5)-phenyl-(1H)-imidazoles 3a/3a' and 2,4-di...
The key intermediate 5a, formed by elimination of nitrogen from the enol-azide 4a, would undergo either dimerization to give 6a, and then imidazoles 3a/3a' after cyclization and dehydration (see Scheme 5), or trimerization to afford adduct 8a further to an additional attack of 5a to the internal imino group of 6a and dehydration. Consecutive tautomerization, followed by an intramolecular nucleophilic attack to the terminal imino group of 9a, provides cyclized adduct 10a, and finally pyrimidine derivative 7a by aromatization/elimination of NH3. To the best of our knowledge, this is the first one-pot synthesis of functionalized pyrimidines using phenacyl azides as the sole starting material [39]. Variation of the phenacyl azides was next investigated in the preparation of various pyrimidines 7 (Scheme 6).
![[1860-5397-16-158-i6]](/bjoc/content/inline/1860-5397-16-158-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Scope of the synthesis of 2,4-diaroyl-6-arylpyrimidines 7. Typical conditions: 2 (0.3 mmol), Et3N (0.9 mmol), ChCl/urea (0.5 g), rt, 4 h; yield refers to products isolated after column chromatography on silica gel; synthesis of pyrimidine derivatives 7b and 7d, reaction time: 12 h.
Scheme 6: Scope of the synthesis of 2,4-diaroyl-6-arylpyrimidines 7. Typical conditions: 2 (0.3 mmol), Et3N (...
The results shown in Scheme 6 demonstrate that this protocol allows the use of phenacyl azides as starting material also for the preparation of a variety of 2,4,6-trisubstituted pyrimidines. The cyclotrimerization of phenacyl azides containing an alkyl group (4-Me, 2b), halides (4-Cl, 4-Br and 4-F, 2c, d, and 2i) or strong electron-withdrawing groups (2-CF3, 4-CF3, 2l, m) occurs smoothly at rt generally within 4 h in a ChCl/urea eutectic mixture, thereby providing the desired pyrimidines 7b–h in 45–88% yield. Variable amounts of difunctionalized imidazoles 3/3' (20–40%), deriving from a dimerization process, also formed competitively in some cases under the aforementioned conditions. The latter, however, could be easily isolated by column chromatography. The presence of strong electron-donating groups like MeO was not tolerated. Indeed, phenacyl azides 2f, and 2g remained unreacted when stirred in ChCl/urea at rt even for 12 h.
Conclusion
In summary, we have shown that phenacyl halides can be straightforwardly converted, via phenacyl azides, into valuable 2-aroyl-(4 or 5)-aryl-(1H)-imidazoles when a solution in ChCl/Gly (1:2) is heated to 80 °C for 4–16 h in the presence of NaN3 (1.5 equiv). In most cases, their isolation can be performed by a very gentle procedure: decantation/centrifugation as soon as they form from the above eutectic mixture. The reaction proceeds in very good yields (67–98%) when phenacyl azides are soluble in the eutectic mixture and is applicable to a range of substrates. Phenacyl azides, in turn, can also be competitively converted into 2,4-diaroyl-6-arylpyrimidines (45–88% yield) via an unprecedented cyclotrimerization reaction the key intermediate α-imino ketone undergoes when a solution in ChCl/urea is stirred at rt for 4 h in the presence of Et3N (3 equiv) as a base. These cyclizations proved to be relatively sensitive to the electronic properties of the starting phenacyl azides as they did not take place in the presence of strong electron-donating groups like MeO. Studies to expand even more the scope and the selectivity of such DES-promoted heterocyclization reactions and to elucidate their mechanism are in progress.
Experimental
General methods
Deep eutectic solvents [choline chloride (ChCl)/glycerol (Gly) (1:2 mol/mol); choline chloride (ChCl)/urea (1:2 mol/mol)] were prepared by heating under stirring up to 80 °C for 10–15 min the corresponding individual components until a clear solution was obtained. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury 300 or on a Bruker 400 or 600 MHz spectrometer and chemical shifts are reported in parts per million (δ); CDCl3, (CD3)2CO and (CD3)2SO were used as solvents. GC–MS analyses were performed on a HP 6890 gas chromatograph, Series II by using a HP1 column (methyl siloxane; 30 m × 0.32 mm × 0.25 μm film thickness) equipped with a mass-selective detector operating at 70 eV. Analytical thin-layer chromatography (TLC) was carried out on pre-coated 0.25 mm thick plates of Kieselgel 60 F254; visualization was accomplished by UV light (254 nm) or by spraying with a solution of 5% (w/v) ammonium molybdate and 0.2% (w/v) cerium(III) sulfate in 100 mL 17.6% (w/v) aq. sulfuric acid and heating to 473 K until blue spots appeared. Column chromatography was conducted by using silica gel 60 with a particle size distribution of 40–63 μm and 230–400 ASTM, using hexane/EtOAc mixtures as the eluent. High-resolution mass spectrometry (HRMS) analyses were performed using a Bruker microTOF QII mass spectrometer equipped with an electrospray ion source (ESI). NaN3, 2-haloketones, and all other reagents, unless otherwise specified, were purchased from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA), and used without any further purification.
Experimental procedures
General procedure for the synthesis of 2-azido ketones 2d, 2e, 2f, 2k, 2l and 2m
α-Haloketone 1 (1.5 mmol) and NaN3 (107 mg, 1.65 mmol) were sequentially added to the ChCl/Gly (1:2 mol/mol, 2.0 g) eutectic mixture. The reaction mixture was stirred at 25 °C under air for 3–12 h until complete consumption of the starting material (monitored by thin-layer chromatography). Then, water was added, and the mixture was extracted with EtOAc (3 × 10 mL). The collected organic layers were dried using anhydrous Na2SO4, filtered, and the volatile evaporated under reduced pressure to afford the crude product, which was purified by column chromatography on silica gel (hexane/EtOAc 5:1–4:1) to afford the desired α-azido ketone 2. Characterization data of the isolated 2-azido ketones are provided in Supporting Information File 1.
Synthesis of 2-benzoyl-4-phenyl-1H-imidazole (3a) and 2-benzoyl-5-phenyl-1H-imidazole (3a'). Typical procedure
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-158-i9.svg?max-width=637&scale=1.18182)
Sodium azide (0.75 mmol) was added to a solution of 2-chloroacetophenone (1a, 0.5 mmol) in ChCl/Gly (1:2 mol/mol, 1.0 g) under air and with vigorous stirring. The mixture was then warmed to 80 °C. After 12 h, 10 mL of water were added and the solid 3a was recovered by decantation (or filtration) from the reaction mixture and washed with a few drops of EtOAc or Et2O. The solution was extracted with EtOAc (3 × 10 mL). The combined organic phases were dried over anhydrous Na2SO4 and the solvent was concentrated in vacuo. The addition of Et2O to the crude mixture allowed the further precipitation of 3a, which was finally recovered in 88% yield. Characterization data of the isolated imidazoles are provided in Supporting Information File 1.
Synthesis of 2,4-dibenzoyl-6-phenylpyrimidine (7a). Typical procedure
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-158-i10.svg?max-width=637&scale=1.18182)
Et3N (0.930 mmol, 0.130 mL) was added to a solution of 2-azidoacetophenone (2a, 0.31 mmol, 0.05 g) in ChCl/ urea (1:2 mol/mol, 0.5 g) eutectic mixture at 25 °C, under air and with vigorous stirring. The reaction was monitored by TLC (hexane/EtOAc 7:3). After 4 h, 5 mL of water were added and the reaction mixture was extracted with EtOAc (3 × 5 mL). The combined organic phases were dried over anhydrous Na2SO4 and the solvent was concentrated under reduced pressure. Purification by column chromatography on silica gel (hexane/EtOAc 9:1) provided pyrimidine 7a in 57% yield. A mixture of imidazoles 3a/3a' (43% yield) was also isolated. Characterization data of the isolated pyrimidines are provided in Supporting Information File 1.
Supporting Information
| Supporting Information File 1: Compound characterization data and NMR spectra. | ||
| Format: PDF | Size: 2.4 MB | Download |
Acknowledgements
The authors are also indebted to Dr. Francesco Lavolpe, Dr. Walter Barella, Dr. Giuseppe Dilauro, and Dr. Francesco Messa for their contribution to the experimental work.
Funding
This work was carried out under the framework of the national PRIN project “Unlocking Sustainable Technologies Through Nature-inspired Solvents (NATUREChem) (grant number: 2017A5HXFC_002) financially supported by the University of Bari “A. Moro” (codes: SFARMA.RicercaLocale.Vitale Fondi Ateneo 17-18, PernaF.18 FondiAteneo15-16, VitaleP.18 FondiAteneo15-16), the University of Salento, the Interuniversity Consortium C.I.N.M.P.I.S., and the Ministero dell’Università e della Ricerca (MIUR-PRIN).
References
-
Alonso, D. A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Eur. J. Org. Chem. 2016, 612–632. doi:10.1002/ejoc.201501197
Return to citation in text: [1] -
Ramón, D. J.; Guillena, G., Eds. Deep Eutectic Solvents: Synthesis, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2019. doi:10.1002/9783527818488
Return to citation in text: [1] -
Perna, F. M.; Vitale, P.; Capriati, V. Curr. Opin. Green Sustainable Chem. 2020, 21, 27–33. doi:10.1016/j.cogsc.2019.09.004
Return to citation in text: [1] -
Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F. M.; Salomone, A.; Florio, S.; Capriati, V. Chem. Sci. 2016, 7, 1192–1199. doi:10.1039/c5sc03436a
Return to citation in text: [1] -
Capua, M.; Perrone, S.; Perna, F. M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. Molecules 2016, 21, 924. doi:10.3390/molecules21070924
Return to citation in text: [1] -
Mancuso, R.; Maner, A.; Cicco, L.; Perna, F. M.; Capriati, V.; Gabriele, B. Tetrahedron 2016, 72, 4239–4244. doi:10.1016/j.tet.2016.05.062
Return to citation in text: [1] -
Perrone, S.; Capua, M.; Messa, F.; Salomone, A.; Troisi, L. Tetrahedron 2017, 73, 6193–6198. doi:10.1016/j.tet.2017.09.013
Return to citation in text: [1] -
Behalo, M. S.; Bloise, E.; Mele, G.; Salomone, A.; Messa, F.; Carbone, L.; Mazzetto, S. E.; Lomonaco, D. J. Heterocycl. Chem. 2020, 57, 768–773. doi:10.1002/jhet.3818
Return to citation in text: [1] -
Dilauro, G.; Cicco, L.; Perna, F. M.; Vitale, P.; Capriati, V. C. R. Chim. 2017, 20, 617–623. doi:10.1016/j.crci.2017.01.008
Return to citation in text: [1] -
Ghinato, S.; Dilauro, G.; Perna, F. M.; Capriati, V.; Blangetti, M.; Prandi, C. Chem. Commun. 2019, 55, 7741–7744. doi:10.1039/c9cc03927a
Return to citation in text: [1] -
Messa, F.; Perrone, S.; Capua, M.; Tolomeo, F.; Troisi, L.; Capriati, V.; Salomone, A. Chem. Commun. 2018, 54, 8100–8103. doi:10.1039/c8cc03858a
Return to citation in text: [1] -
Dilauro, G.; García, S. M.; Tagarelli, D.; Vitale, P.; Perna, F. M.; Capriati, V. ChemSusChem 2018, 11, 3495–3501. doi:10.1002/cssc.201801382
Return to citation in text: [1] -
Messa, F.; Dilauro, G.; Perna, F. M.; Vitale, P.; Capriati, V.; Salomone, A. ChemCatChem 2020, 12, 1979–1984. doi:10.1002/cctc.201902380
Return to citation in text: [1] -
Quivelli, A. F.; Vitale, P.; Perna, F. M.; Capriati, V. Front. Chem. (Lausanne, Switz.) 2019, 7, 723. doi:10.3389/fchem.2019.00723
Return to citation in text: [1] -
Vitale, P.; Lavolpe, F.; Valerio, F.; Di Biase, M.; Perna, F. M.; Messina, E.; Agrimi, G.; Pisano, I.; Capriati, V. React. Chem. Eng. 2020, 5, 859–864. doi:10.1039/d0re00067a
Return to citation in text: [1] -
Vitale, P.; Cicco, L.; Messa, F.; Perna, F. M.; Salomone, A.; Capriati, V. Eur. J. Org. Chem. 2019, 5557–5562. doi:10.1002/ejoc.201900722
Return to citation in text: [1] [2] -
De Luca, L. Curr. Med. Chem. 2006, 13, 1–23.
Return to citation in text: [1] -
Lagoja, I. M. Chem. Biodiversity 2005, 2, 1–50. doi:10.1002/cbdv.200490173
Return to citation in text: [1] -
Han, H.-B.; Tu, Z.-L.; Wu, Z.-G.; Zheng, Y.-X. Dyes Pigm. 2019, 160, 863–871. doi:10.1016/j.dyepig.2018.09.017
Return to citation in text: [1] -
Chen, W.-C.; Zhu, Z.-L.; Lee, C.-S. Adv. Opt. Mater. 2018, 6, 1800258. doi:10.1002/adom.201800258
Return to citation in text: [1] -
Zhang, L.; Peng, X.-M.; Damu, G. L. V.; Geng, R.-X.; Zhou, C.-H. Med. Res. Rev. 2014, 34, 340–437. doi:10.1002/med.21290
Return to citation in text: [1] -
Sharma, A.; Kumar, V.; Kharb, R.; Kumar, S.; Chander Sharma, P.; Pal Pathak, D. Curr. Pharm. Des. 2016, 22, 3265–3301. doi:10.2174/1381612822666160226144333
Return to citation in text: [1] -
Kuzu, B.; Tan, M.; Taslimi, P.; Gülçin, İ.; Taşpınar, M.; Menges, N. Bioorg. Chem. 2019, 86, 187–196. doi:10.1016/j.bioorg.2019.01.044
Return to citation in text: [1] -
Agarwal, A.; Srivastava, K.; Puri, S. K.; Chauhan, P. M. S. Bioorg. Med. Chem. 2005, 13, 4645–4650. doi:10.1016/j.bmc.2005.04.061
Return to citation in text: [1] -
Shipe, W. D.; Sharik, S. S.; Barrow, J. C.; McGaughey, G. B.; Theberge, C. R.; Uslaner, J. M.; Yan, Y.; Renger, J. J.; Smith, S. M.; Coleman, P. J.; Cox, C. D. J. Med. Chem. 2015, 58, 7888–7894. doi:10.1021/acs.jmedchem.5b00983
Return to citation in text: [1] -
Kaur, R.; Kaur, P.; Sharma, S.; Singh, G.; Mehndiratta, S.; Bedi, P. M. S.; Nepali, K. Recent Pat. Anti-Cancer Drug Discovery 2015, 10, 23–71. doi:10.2174/1574892809666140917104502
Return to citation in text: [1] -
Farag, A. K.; Elkamhawy, A.; Londhe, A. M.; Lee, K.-T.; Pae, A. N.; Roh, E. J. Eur. J. Med. Chem. 2017, 141, 657–675. doi:10.1016/j.ejmech.2017.10.003
Return to citation in text: [1] -
Chen, J.; Chen, W.; Yu, Y.; Zhang, G. Tetrahedron Lett. 2013, 54, 1572–1575. doi:10.1016/j.tetlet.2013.01.042
Return to citation in text: [1] [2] [3] -
Khalili, B.; Tondro, T.; Hashemi, M. M. Tetrahedron 2009, 65, 6882–6887. doi:10.1016/j.tet.2009.06.082
Return to citation in text: [1] [2] [3] -
Batanero, B.; Escudero, J.; Barba, F. Org. Lett. 1999, 1, 1521–1522. doi:10.1021/ol990200j
Return to citation in text: [1] -
Benati, L.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Strazzari, S.; Zanardi, G.; Calestani, G. Tetrahedron 2002, 58, 3485–3492. doi:10.1016/s0040-4020(02)00302-2
Return to citation in text: [1] -
Zuliani, V.; Cocconcelli, G.; Fantini, M.; Ghiron, C.; Rivara, M. J. Org. Chem. 2007, 72, 4551–4553. doi:10.1021/jo070187d
Return to citation in text: [1] -
Yamamoto, Y.; Ouchi, H.; Tanaka, T.; Morita, Y. Heterocycles 1995, 41, 1275–1290. doi:10.3987/com-95-7055
Return to citation in text: [1] -
Skulcova, A.; Russ, A.; Jablonsky, M.; Sima, J. BioResources 2018, 13, 5042–5051.
Return to citation in text: [1] -
Boyer, J. H.; Straw, D. J. Am. Chem. Soc. 1952, 74, 4506–4508. doi:10.1021/ja01138a011
Return to citation in text: [1] -
Patonay, T.; Juhász-Tóth, É.; Bényei, A. Eur. J. Org. Chem. 2002, 285–295. doi:10.1002/1099-0690(20021)2002:2<285::aid-ejoc285>3.0.co;2-j
Return to citation in text: [1] -
Mjalli, F. S.; Ahmed, O. U. Korean J. Chem. Eng. 2016, 33, 337–343. doi:10.1007/s11814-015-0134-7
Return to citation in text: [1] -
Reddy, C. N.; Sathish, M.; Adhikary, S.; Nanubolu, J. B.; Alarifi, A.; Maurya, R. A.; Kamal, A. Org. Biomol. Chem. 2017, 15, 2730–2733. doi:10.1039/c7ob00299h
Return to citation in text: [1] -
Hu, M.; Wu, J.; Zhang, Y.; Qiu, F.; Yu, Y. Tetrahedron 2011, 67, 2676–2680. doi:10.1016/j.tet.2011.01.062
Return to citation in text: [1]
| 1. | Alonso, D. A.; Baeza, A.; Chinchilla, R.; Guillena, G.; Pastor, I. M.; Ramón, D. J. Eur. J. Org. Chem. 2016, 612–632. doi:10.1002/ejoc.201501197 |
| 2. | Ramón, D. J.; Guillena, G., Eds. Deep Eutectic Solvents: Synthesis, Properties, and Applications; Wiley-VCH: Weinheim, Germany, 2019. doi:10.1002/9783527818488 |
| 3. | Perna, F. M.; Vitale, P.; Capriati, V. Curr. Opin. Green Sustainable Chem. 2020, 21, 27–33. doi:10.1016/j.cogsc.2019.09.004 |
| 10. | Ghinato, S.; Dilauro, G.; Perna, F. M.; Capriati, V.; Blangetti, M.; Prandi, C. Chem. Commun. 2019, 55, 7741–7744. doi:10.1039/c9cc03927a |
| 30. | Batanero, B.; Escudero, J.; Barba, F. Org. Lett. 1999, 1, 1521–1522. doi:10.1021/ol990200j |
| 9. | Dilauro, G.; Cicco, L.; Perna, F. M.; Vitale, P.; Capriati, V. C. R. Chim. 2017, 20, 617–623. doi:10.1016/j.crci.2017.01.008 |
| 31. | Benati, L.; Leardini, R.; Minozzi, M.; Nanni, D.; Spagnolo, P.; Strazzari, S.; Zanardi, G.; Calestani, G. Tetrahedron 2002, 58, 3485–3492. doi:10.1016/s0040-4020(02)00302-2 |
| 5. | Capua, M.; Perrone, S.; Perna, F. M.; Vitale, P.; Troisi, L.; Salomone, A.; Capriati, V. Molecules 2016, 21, 924. doi:10.3390/molecules21070924 |
| 6. | Mancuso, R.; Maner, A.; Cicco, L.; Perna, F. M.; Capriati, V.; Gabriele, B. Tetrahedron 2016, 72, 4239–4244. doi:10.1016/j.tet.2016.05.062 |
| 7. | Perrone, S.; Capua, M.; Messa, F.; Salomone, A.; Troisi, L. Tetrahedron 2017, 73, 6193–6198. doi:10.1016/j.tet.2017.09.013 |
| 8. | Behalo, M. S.; Bloise, E.; Mele, G.; Salomone, A.; Messa, F.; Carbone, L.; Mazzetto, S. E.; Lomonaco, D. J. Heterocycl. Chem. 2020, 57, 768–773. doi:10.1002/jhet.3818 |
| 28. | Chen, J.; Chen, W.; Yu, Y.; Zhang, G. Tetrahedron Lett. 2013, 54, 1572–1575. doi:10.1016/j.tetlet.2013.01.042 |
| 4. | Cicco, L.; Sblendorio, S.; Mansueto, R.; Perna, F. M.; Salomone, A.; Florio, S.; Capriati, V. Chem. Sci. 2016, 7, 1192–1199. doi:10.1039/c5sc03436a |
| 29. | Khalili, B.; Tondro, T.; Hashemi, M. M. Tetrahedron 2009, 65, 6882–6887. doi:10.1016/j.tet.2009.06.082 |
| 16. | Vitale, P.; Cicco, L.; Messa, F.; Perna, F. M.; Salomone, A.; Capriati, V. Eur. J. Org. Chem. 2019, 5557–5562. doi:10.1002/ejoc.201900722 |
| 19. | Han, H.-B.; Tu, Z.-L.; Wu, Z.-G.; Zheng, Y.-X. Dyes Pigm. 2019, 160, 863–871. doi:10.1016/j.dyepig.2018.09.017 |
| 20. | Chen, W.-C.; Zhu, Z.-L.; Lee, C.-S. Adv. Opt. Mater. 2018, 6, 1800258. doi:10.1002/adom.201800258 |
| 15. | Vitale, P.; Lavolpe, F.; Valerio, F.; Di Biase, M.; Perna, F. M.; Messina, E.; Agrimi, G.; Pisano, I.; Capriati, V. React. Chem. Eng. 2020, 5, 859–864. doi:10.1039/d0re00067a |
| 21. | Zhang, L.; Peng, X.-M.; Damu, G. L. V.; Geng, R.-X.; Zhou, C.-H. Med. Res. Rev. 2014, 34, 340–437. doi:10.1002/med.21290 |
| 22. | Sharma, A.; Kumar, V.; Kharb, R.; Kumar, S.; Chander Sharma, P.; Pal Pathak, D. Curr. Pharm. Des. 2016, 22, 3265–3301. doi:10.2174/1381612822666160226144333 |
| 23. | Kuzu, B.; Tan, M.; Taslimi, P.; Gülçin, İ.; Taşpınar, M.; Menges, N. Bioorg. Chem. 2019, 86, 187–196. doi:10.1016/j.bioorg.2019.01.044 |
| 24. | Agarwal, A.; Srivastava, K.; Puri, S. K.; Chauhan, P. M. S. Bioorg. Med. Chem. 2005, 13, 4645–4650. doi:10.1016/j.bmc.2005.04.061 |
| 25. | Shipe, W. D.; Sharik, S. S.; Barrow, J. C.; McGaughey, G. B.; Theberge, C. R.; Uslaner, J. M.; Yan, Y.; Renger, J. J.; Smith, S. M.; Coleman, P. J.; Cox, C. D. J. Med. Chem. 2015, 58, 7888–7894. doi:10.1021/acs.jmedchem.5b00983 |
| 26. | Kaur, R.; Kaur, P.; Sharma, S.; Singh, G.; Mehndiratta, S.; Bedi, P. M. S.; Nepali, K. Recent Pat. Anti-Cancer Drug Discovery 2015, 10, 23–71. doi:10.2174/1574892809666140917104502 |
| 27. | Farag, A. K.; Elkamhawy, A.; Londhe, A. M.; Lee, K.-T.; Pae, A. N.; Roh, E. J. Eur. J. Med. Chem. 2017, 141, 657–675. doi:10.1016/j.ejmech.2017.10.003 |
| 14. | Quivelli, A. F.; Vitale, P.; Perna, F. M.; Capriati, V. Front. Chem. (Lausanne, Switz.) 2019, 7, 723. doi:10.3389/fchem.2019.00723 |
| 11. | Messa, F.; Perrone, S.; Capua, M.; Tolomeo, F.; Troisi, L.; Capriati, V.; Salomone, A. Chem. Commun. 2018, 54, 8100–8103. doi:10.1039/c8cc03858a |
| 12. | Dilauro, G.; García, S. M.; Tagarelli, D.; Vitale, P.; Perna, F. M.; Capriati, V. ChemSusChem 2018, 11, 3495–3501. doi:10.1002/cssc.201801382 |
| 13. | Messa, F.; Dilauro, G.; Perna, F. M.; Vitale, P.; Capriati, V.; Salomone, A. ChemCatChem 2020, 12, 1979–1984. doi:10.1002/cctc.201902380 |
| 17. | De Luca, L. Curr. Med. Chem. 2006, 13, 1–23. |
| 18. | Lagoja, I. M. Chem. Biodiversity 2005, 2, 1–50. doi:10.1002/cbdv.200490173 |
| 16. | Vitale, P.; Cicco, L.; Messa, F.; Perna, F. M.; Salomone, A.; Capriati, V. Eur. J. Org. Chem. 2019, 5557–5562. doi:10.1002/ejoc.201900722 |
| 32. | Zuliani, V.; Cocconcelli, G.; Fantini, M.; Ghiron, C.; Rivara, M. J. Org. Chem. 2007, 72, 4551–4553. doi:10.1021/jo070187d |
| 33. | Yamamoto, Y.; Ouchi, H.; Tanaka, T.; Morita, Y. Heterocycles 1995, 41, 1275–1290. doi:10.3987/com-95-7055 |
| 38. | Reddy, C. N.; Sathish, M.; Adhikary, S.; Nanubolu, J. B.; Alarifi, A.; Maurya, R. A.; Kamal, A. Org. Biomol. Chem. 2017, 15, 2730–2733. doi:10.1039/c7ob00299h |
| 39. | Hu, M.; Wu, J.; Zhang, Y.; Qiu, F.; Yu, Y. Tetrahedron 2011, 67, 2676–2680. doi:10.1016/j.tet.2011.01.062 |
| 36. | Patonay, T.; Juhász-Tóth, É.; Bényei, A. Eur. J. Org. Chem. 2002, 285–295. doi:10.1002/1099-0690(20021)2002:2<285::aid-ejoc285>3.0.co;2-j |
| 37. | Mjalli, F. S.; Ahmed, O. U. Korean J. Chem. Eng. 2016, 33, 337–343. doi:10.1007/s11814-015-0134-7 |
| 34. | Skulcova, A.; Russ, A.; Jablonsky, M.; Sima, J. BioResources 2018, 13, 5042–5051. |
| 35. | Boyer, J. H.; Straw, D. J. Am. Chem. Soc. 1952, 74, 4506–4508. doi:10.1021/ja01138a011 |
| 28. | Chen, J.; Chen, W.; Yu, Y.; Zhang, G. Tetrahedron Lett. 2013, 54, 1572–1575. doi:10.1016/j.tetlet.2013.01.042 |
| 29. | Khalili, B.; Tondro, T.; Hashemi, M. M. Tetrahedron 2009, 65, 6882–6887. doi:10.1016/j.tet.2009.06.082 |
| 28. | Chen, J.; Chen, W.; Yu, Y.; Zhang, G. Tetrahedron Lett. 2013, 54, 1572–1575. doi:10.1016/j.tetlet.2013.01.042 |
| 29. | Khalili, B.; Tondro, T.; Hashemi, M. M. Tetrahedron 2009, 65, 6882–6887. doi:10.1016/j.tet.2009.06.082 |
© 2020 Vitale et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)
