Abstract
3-Aryl-1-(trifluoromethyl)prop-2-yn-1-iminium triflate salts represent a novel, highly reactive class of acetylenic iminium salts. Herein we present several reactions which are based on the electron-poor acetylenic bond and on the high electrophilicity of the CF3-substituted iminium group. These salts were found to be highly reactive dienophiles in Diels–Alder reactions with cyclopentadiene, 2,3-dimethylbutadiene and even anthracene. At higher temperature, the cycloadducts undergo an intramolecular SE(Ar) reaction leading to condensed carbocycles incorporating a 1-(trifluoromethyl)-1-(dimethylamine)indene ring system. With styrenes and some substituted styrenes, cascade reactions take place, which likely include cyclobutene and several cationic intermediates and mainly yield 2-(1-phenylvinyl)indenes. In a similar reaction cascade, a fulvene derivative was obtained with 1,4-diphenylbutadiene as the substrate.

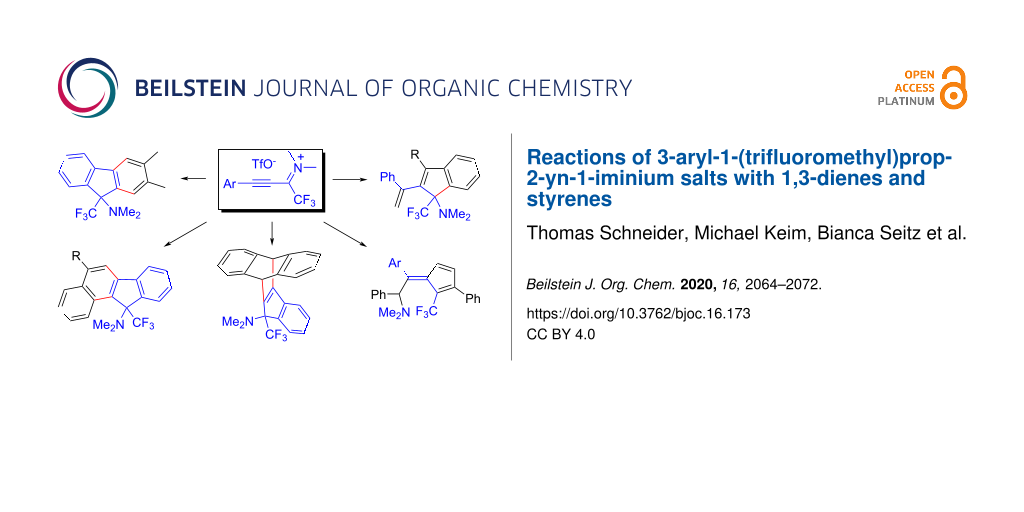
Graphical Abstract
Introduction
In recent years, a trifluoromethyl substituent has been included quite often in the design of compounds which were developed for applications in various fields, such as biological and medicinal chemistry, agrochemicals, transition metal ligands, and materials science [1-5]. The particular characteristics of the C‒F bond [6,7] are the basis for the electronic and steric properties of the CF3 group, such as a strong electron-withdrawing (−I) effect, the accumulation of negative charge density in a relatively small volume and the low polarizability of the fluoro atoms. These and other substituent effects can modulate the conformational, physicochemical and electronic properties of a molecule.
Two major strategies exist to introduce a CF3 group into a target molecule: formation of a carbon‒ or heteroatom‒CF3 bond [8,9] and the use of preformed CF3-substituted building blocks. During our studies on acetylenic iminium salts, among the numerous CF3-substituted building blocks α-(trifluoromethyl)iminium salts RCH(CF3)=N+R’2 X− attracted our attention, because a) the CF3 group should significantly increase the electrophilicity of the iminium functional group and b) these salts are known to react with a variety of nucleophiles to afford products containing a C(CF3)NR2 moiety. In particular C(CF3)NHR and C(CF3)NH2 groups are of interest as pharmacophores in the design of bioactive compounds [10-13].
Simple α-(trifluoromethyl)iminium salts (RCH(CF3)=N+Me2 X−, R= H, CF3) with weakly nucleophilic or non-nucleophilic anions can be isolated [14,15], but in organic synthesis, they are most often generated in situ from suitable precursors and are directly exposed to diverse nucleophiles. Such transformations have been achieved using CH2(CF3)NR2 as the iminium ion precursors [15], trifluoroacetaldehyde hemiaminals [16-18], N,O-acetals [19] and N,N-acetals [20], 2-(trifluoromethyl)-1,3-oxazolidines [21], and N-(tert-butylsulfinyl)trifluoroacetaldimine [11,22]. Other synthetic approaches to α-CF3-substituted amines include nucleophilic trifluoromethylation strategies [17], such as the reaction of trifluoroacetaldehyde hemiaminals with enolizable carbonyl compounds in the presence of a strong base [23], the reaction of aldiminium salts with (trifluoromethyl)trimethylsilane/Lewis base [24], and the preparation of secondary α-(trifluoromethyl)propargylamines from imines CF3CH=NR and lithium acetylides [25]. By a photoredox-catalytic process, primary α-(trifluoromethyl)-α-(4-pyridyl)benzylamines were obtained from α-(trifluoromethyl)-benzaldoximes and 4-cyanopyridine [26].
We have recently introduced a new class of acetylenic iminium salts, namely 1-(trifluoromethyl)prop-2-yn-1-iminium triflates R‒C≡C‒C(CF3)=N+Me2 TfO− [27]. As a first synthetic application, we have reported the phospha-Michael addition providing 3-(triphenylphosphonio)-1-(trifluoromethyl)-1-(dimethylamino)allenes, which were subsequently transformed into α-(trifluoromethyl)pyrroles. In the present paper, we consider the reactivity of the electrophilic (“electron-poor”) acetylenic bond toward 1,3-dienes, and show how the expected [4 + 2] or [2 + 2] cycloaddition products can enter subsequent cascade reactions toward carbocycles which incorporate a C(CF3)NMe2 structural unit.
Results and Discussion
The Diels–Alder reaction of 1-CF3-substituted propyn-1-iminium salt 1a with cyclopentadiene was carried out in order to assess the dienophilic reactivity of the cation. High conversion into cycloaddition product 2 was observed already within one hour at 0 °C. Because of its high hydrolytic lability, adduct 2 was not isolated but directly converted into the norbornadienyl trifluoromethyl ketone 3 (Scheme 1). The smooth [4 + 2] cycloaddition of 1a as compared to comparably harsh thermal conditions of other propyne ketiminium salts with an internal acetylenic bond reveals the activating influence of the CF3 group, which has both an electronic (electron-withdrawing) and steric (e.g., CF3 vs a phenyl substituent [28]) component. Moreover, a comparison with the thermal conditions of the Diels–Alder reaction of 4-phenyl-1,1,1-trifluorobut-3-yn-2-one and cyclopentadiene [29] confirms the expected accelerating effect of the iminium activation.
![[1860-5397-16-173-i1]](/bjoc/content/inline/1860-5397-16-173-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Diels–Alder reaction of propyn-1-iminium salt 1a compared with the reported [29] reaction of 4-phenyl-1,1,1-trifluorobut-3-yn-2-one.
Scheme 1: Diels–Alder reaction of propyn-1-iminium salt 1a compared with the reported [29] reaction of 4-phenyl-1...
The Diels–Alder reaction of alkyne 1a with 2,3-dimethylbutadiene also occurred under very mild conditions and yielded the iminium-substituted 1,4-cyclohexadiene 4-Ch (Scheme 2), which, due to its high sensitivity toward moisture, was not purified but was further converted in two steps into cyclohexadienyl ketone 5-Ch by intentional hydrolysis followed by dehydrogenating aromatization leading to biphenyl-2-yl trifluoromethyl ketone 6. The latter product was more effectively prepared in a one-pot cycloaddition/hydrolysis/aromatization sequence. 1H NMR spectra of unpurified 1,4-cyclohexadien-1-iminium salt 4-Ch and 1,4-cyclohexadien-1-yl ketone 5-Ch indicated the presence of a minor byproduct. In the case of 5-Ch, obtained as an oil, the two components could not be separated by column chromatography; however, the 1H NMR spectrum suggested the cyclobutene structure 5-Cb for the byproduct. Thus, signals of the two methyl groups (δ 1.63, 1.78 ppm) and two terminal olefinic protons (δ 4.88 ppm, m) are observed, and the ring-CH2 protons appear as an AB spin system (δ 2.75 and 2.92 ppm, 2J = 15.2 Hz). Obviously, 4-Cb and 5-Cb result from a formal [2 + 2] cycloaddition of 1a and 2,3-dimethylbutadiene (DMBD), the regioselectivity of which is as expected for a highly asynchronous transition state with effective stabilization of the positive charge or a two-step ionic process (vide infra). The high electrophilic power of the 1-CF3-substituted propyn-1-iminium ion presumably renders an ionic [2 + 2] cycloaddition pathway competitive with the Diels–Alder reaction. The few reported examples of cyclobutene formation from alkynes and unactivated 1,3-dienes include the sensitized photocycloaddition of phenylacetylene and DMBD [30] and the gold(I)-catalyzed reaction of phenylacetylene and DMBD or isoprene [31]. On the other hand, the 1,1-diphenylpropargyl cation was found to react with 2,4-dimethyl-1,3-pentadiene to afford a product derived from an initial [4 + 2] cycloaddition [32].
![[1860-5397-16-173-i2]](/bjoc/content/inline/1860-5397-16-173-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Sequential Diels–Alder/intramolecular SE(Ar) reaction of propyn-1-iminium triflates 1a,b. Conditions: a) CH3CN, aqueous K2CO3, 15 min; b) 1. 1a + 2,3-dimethylbutadiene, dry CH2Cl2, 0 °C, 30 min, then rt, 2 h; 2. o-chloranil, CH2Cl2, rt, 20 h; 3. K2CO3, CH3CN/H2O, rt, 2 h; c) o-chloranil, dry CH2Cl2, rt, 22 h, then K2CO3, H2O; d) 1. dry CH3CN, rt, 1 h; 2. 50 °C, 22 h; 3. o-chloranil, CH2Cl2, rt, 12 h.
Scheme 2: Sequential Diels–Alder/intramolecular SE(Ar) reaction of propyn-1-iminium triflates 1a,b. Condition...
When the Diels–Alder reaction of 1a with DMBD was carried out at room temperature instead of 0 °C, 19F NMR monitoring of the reaction’s progress indicated the appearance of a second product beside the 1,4-cyclohexadien-1-iminium salt 4-Ch. Further investigations revealed that the new product was the dihydrofluorene 7, resulting from 4-Ch by an intramolecular electrophilic aromatic substitution with the reactive (trifluoromethyl)iminium group as the electrophile (Scheme 2). The thermal conversion of 4-Ch into 7 was optimized and finally allowed the preparation of the latter from 1a in a one-pot, two-step, temperature-dependent Diels–Alder reaction/intramolecular SE(Ar) reaction sequence in good yield. Dehydrogenation of 7 with chloranil then provided 9-(dimethylamino)-9-(trifluoromethyl)fluorene 8.
In an analogous reaction sequence, 11H-benzo[a]fluorene derivative 9 was obtained from 3-(2-naphthyl)propyn-1-iminium salt 1b and 2,3-dimethylbutadiene (Scheme 2) in a one-pot three-step sequence. On the other hand, the successful thermal iminium-ion induced SE(Ar) reactions shown in Scheme 2 were not applicable to the iminium-substituted norbornadiene 2, which suffered undefined polymerization on moderate heating in various solvents.
In contrast to so far unknown 9‑amino-9-(trifluoromethyl)-9H-fluorenes, compounds containing a 9-(trifluoromethyl)-9H-fluoren-9‑ol partial structure can be found in the patent literature [33-35], where they have been claimed for their pyruvate dehydrogenase kinase (PDHK) inhibitory activity.
The remarkable dienophilic reactivity of 1-CF3-substituted propyn-1-iminium salts is also exemplified by the Diels–Alder reaction of 1a with anthracene (Scheme 3). After 12 h at room temperature, a 91% conversion was observed, and subsequent moderate heating gave the cycloaddition product 10 in 95% yield. The subsequent iminium-induced electrophilic cyclization required extended heating in refluxing toluene and finally furnished the neutral polycycle 11 in good yield. The paddlewheel-shaped structure of 11 was established by an XRD structure determination and is shown in Figure 1.
![[1860-5397-16-173-i3]](/bjoc/content/inline/1860-5397-16-173-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Diels–Alder reaction of 1a and anthracene followed by an intramolecular SE(Ar) reaction.
Scheme 3: Diels–Alder reaction of 1a and anthracene followed by an intramolecular SE(Ar) reaction.
![[1860-5397-16-173-1]](/bjoc/content/figures/1860-5397-16-173-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Solid-state molecular structure of 11 (ORTEP plot).
Figure 1: Solid-state molecular structure of 11 (ORTEP plot).
The reactivity of 1a in the Diels–Alder reaction with anthracene may be compared with that of other dienophiles. Thus, other propyn-1-iminium salts with an internal C,C triple bond and not containing a CF3 substituent (toluene, 120 °C, several hours [28]), DMAD (no solvent, 170–180 °C, 1 h [36]) and hexafluoro-2-butyne (200 °C, 2 h [37]) react only under harsher conditions, whereas terminal propyn-1-iminium salts (CH2Cl2, rt, 2‒4 h [38]) and tetracyanoethylene (rt, 12 h [39]) were found to react equally well or even faster than 1a. Although these comparisons are only qualitative, they suggest that 1-CF3-substituted propyn-1-iminium salts have a high electrophilicity power and therefore, are candidates for polar Diels–Alder reactions [40,41].
The thermal cyclization of Diels–Alder adducts as shown in Scheme 2 and Scheme 3 appear to be the first intramolecular SE(Ar) reactions of an α-(trifluoromethyl)iminium functional group. Analogous intermolecular reactions of CF3-substituted iminium groups with electron-rich (hetero)aromatics are known [18,24]. Furthermore, 1-(trifluoromethyl)indenes have recently been generated by cationic cyclization of β-aryl trifluoromethyl enones under superacid conditions [42-44].
Styrenes are also known to behave as dienes in [4 + 2] cycloaddition reactions [45]. Thus, while styrene and maleic anhydride react only at elevated temperatures, with the more electrophilic (methoxycarbonyl)maleic anhydride two 2:1 adducts are already formed at room temperature: one by two consecutive Diels–Alder reactions, the other one by a Diels–Alder/ene reaction sequence [46]. Taking into account the presumably high electrophilic character of the 1-(trifluoromethyl)propyn-1-iminium ion, for its reaction with styrenes we could not exclude a priori an initial electrophilic addition at the olefinic bond (with formation of a benzyl cation intermediate).
The reaction of propyn-1-iminium salt 1a with styrene in acetonitrile was considered first and was monitored by 19F NMR spectroscopy. Whereas no reaction appeared to occur at room temperature, a slow transformation into two fluorine-containing products was observed at 70 °C, which after neutralization and work-up were identified by their NMR and analytical data as 2-(1-phenylvinyl)indene 12a and a small amount of benzo[a]fluorene 13a (δF = −69.74 and −69.20 ppm, respectively).
The results obtained with styrenes bearing a substituent at the olefinic bond provide useful information with respect to the reaction mechanism. Thus, the reaction of 1a with α-methylstyrene or 1,1-diphenylethene proceeded at a faster rate than with styrene and yielded 3-methyl- and 3-phenyl-2-vinylindenes 12b and 12c (structure confirmed by an XRD analysis, see Figure 2) in high yields. Benzo[a]fluorenes were found to a minor extent (13c) or not at all (13b) (Scheme 4). A remarkable stereochemical aspect accompanied the reaction of 1a with (E)-1-phenyl-1-propene leading to 2-((E)-1-phenylprop-1-enyl)indene 12d, where trans(Ph,Me)→cis isomerization at the olefinic bond has occurred. The E-configuration was assigned based on NOESY NMR experiments and confirmed by an X-ray structure determination (see Figure 3).
![[1860-5397-16-173-i4]](/bjoc/content/inline/1860-5397-16-173-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Reactions of propyn-1-iminium salt 1a with styrenes.
Scheme 4: Reactions of propyn-1-iminium salt 1a with styrenes.
![[1860-5397-16-173-2]](/bjoc/content/figures/1860-5397-16-173-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Solid-state molecular structure of 12c (ORTEP plot).
Figure 2: Solid-state molecular structure of 12c (ORTEP plot).
![[1860-5397-16-173-3]](/bjoc/content/figures/1860-5397-16-173-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Solid-state molecular structure of 12d (ORTEP plot). Both the R and the S enantiomer are present in the acentric unit cell of the crystal (space group P21, Z = 4).
Figure 3: Solid-state molecular structure of 12d (ORTEP plot). Both the R and the S enantiomer are present in...
A mechanistic scheme for the formation of indenes 12 and benzo[a]fluorenes 13 is proposed in Scheme 5. The electrophilic propyn-1-iminium ion 1a adds chemoselectively (by conjugate addition) and regioselectively (Markovnikov-type addition) at the olefinic bond of the styrene to form the resonance-stabilized benzyl cation 14 which can add intramolecularly to the nucleophilic central carbon atom of the aminoallene moiety, either by an electrophilic 1,4-cyclization yielding a cyclobutene 15 or by an 1,6-cyclization yielding a dihydronaphthalene 16. Formally speaking, 15 results from a [2 + 2] cycloaddition and 16 from a [4 + 2] cycloaddition (Diels–Alder reaction). Under the reaction conditions, cyclobutene 15 undergoes a fast electrocyclic ring opening leading to a butadiene 17, which is finally transformed into 2-(1-phenylvinyl)indene 12 through an intramolecular iminium ion-induced 1,5-cyclization. The same cyclization type together with oxidative aromatization converts dihydronaphthalenes 16 into benzo[a]fluorenes 13. The stereochemistry of 12d can be explained by a stereoselective formation of trans-3,4-disubstituted cyclobutene 15d and subsequent conrotatory electrocyclic ring-opening, from which Z(1,2),E(3,4)-configured butadiene intermediate 17d results.
![[1860-5397-16-173-i5]](/bjoc/content/inline/1860-5397-16-173-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: A mechanistic proposal for the reaction of alkyne 1a with styrenes.
Scheme 5: A mechanistic proposal for the reaction of alkyne 1a with styrenes.
The intermediacy of a cyclobutene 15 in the mechanistic scenario of Scheme 5 is corroborated by the isolation of cyclobutene 18 from the reaction of 1a with 1,2-dihydronaphthalene, a cyclic styrene derivative (Scheme 6; compare also the cyclobutene byproduct in Scheme 2). The structure of 18 was derived from its 1H and 13C NMR chemical shifts; a NOE NMR experiment indicated the vicinity of the phenyl ring and the CH2CH part or the molecule in line with the expected orientation of the cycloaddition. Cis-annelated cyclobutene 18 (1H NMR: 3JH,H = 3.8 Hz for the angular protons) and the iminium-substituted primary cycloadduct are not expected to undergo a fast ring opening under moderate thermal conditions, because the orbital-symmetry-allowed concerted conrotatory process [47,48] would create a strained cis,trans-dihydrobenzo[8]annulene ring system.
![[1860-5397-16-173-i6]](/bjoc/content/inline/1860-5397-16-173-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Reaction of alkyne 1a with 1,2-dihydronaphthalene.
Scheme 6: Reaction of alkyne 1a with 1,2-dihydronaphthalene.
Cyclobutenes resulting from a [2 + 2] cycloaddition of electrophilic alkynes and alkenes under moderate thermal conditions have been isolated also from the reaction of CF3-free propyniminium salts with cyclic enol ethers [49] and of other very electrophilic alkynes (i.e., Lewis acid-activated acetylenic esters [50], 1-(trifluoroacetyl)-3-haloacetylenes [44] and 4-chloro-2-oxobut-3-ynoic esters [51-53] with unactivated alkenes (including cyclohexene [51], which did not react with 1a). For the 3-arylpropyn-1-iminium ions 1, a charge delocalization can be assumed, which is described by the resonance structure of a 1-aryl-3-aminoallenyl cation, hence their electronic structure shows a certain analogy to the triphenylpropargyl/triphenylallenyl cation. It has been reported that this cation reacts with cyclopentadiene in two different ways: concerted [4 + 2] cycloaddition and a stepwise [2 + 2] cycloaddition via an allenyl-cyclopentenyl cation (which could be trapped with OH−) [54,55].
Styrene structural moieties are also present in (E,E)-1,4-diphenylbuta-1,3-diene. Therefore, it was of interest to know whether it would react with propyn-1-iminium salts 1 as a styrene or a 1,3-diene. With 3-(4-bromophenyl)propyn-1-iminium salt 1c in acetonitrile, no reaction was observed at 20 °C, but within two hours at 45 °C, an unclean reaction took place, which became evident by a multitude of 19F NMR signals. Assuming that some of the signals represented products that would easily undergo further thermal reactions, the solution was additionally heated at 70 °C for 40 hours. The resulting reaction mixture still contained several products, of which only the major fluorine-containing component (δF = −46.76 ppm, a value quite different from those of products 12 and 13) could finally be isolated in modest yield and was identified by an X-ray structure analysis as the CF3-substituted fulvene 19 (Scheme 7).
![[1860-5397-16-173-i7]](/bjoc/content/inline/1860-5397-16-173-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis and solid-state molecular structure (ORTEP plot) of pentafulvene 19; selected bond distances (Å), see molecule plot for atom numbering: C8‒C7 1.370(2), C7‒C17 1.481(2), C17‒C18 1.343(2), C18‒C5 1.471(2), C5‒C6 1.374(2), C6‒C7 1.496(2).
Scheme 7: Synthesis and solid-state molecular structure (ORTEP plot) of pentafulvene 19; selected bond distan...
A reaction pathway leading to fulvene 19 is proposed in Scheme 8. It begins with the formal [2 + 2] cycloaddition of 1c and the diene component, which is probably a two-step process as shown in Scheme 5. Cyclobutene 20 is prone to a thermally induced conrotatory electrocyclic ring-opening, which yields iminium-substituted triene 21. In a similar reaction, an α-phenyliminium salt structurally analogous to 21 could be isolated [56]. A cationic 1,5-cylization converts 21 into cyclopentene 22, from which fulvene 19 is formed by deprotonation and a formal 1,4-shift of the NMe2 group. The details of this rearrangement are not known, an N,N-dimethyldihydropyrrolium intermediate may be involved.
![[1860-5397-16-173-i8]](/bjoc/content/inline/1860-5397-16-173-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Proposed mechanistic pathway leading to fulvene 19.
Scheme 8: Proposed mechanistic pathway leading to fulvene 19.
Conclusion
This study has uncovered new applications of 3-aryl-1-(trifluoromethyl)prop-2-yn-1-iminium ions as CF3-substituted C3 building blocks. They are not only very electrophilic dienophiles in Diels–Alder reactions with normal electron demand (HOMOdiene‒LUMOdienophile controlled, in the language of FMO theory), but also represent powerful 1,3-biselectrophiles. Thus, Diels–Alder reactions followed by an intramolecular SE(Ar) reaction of the α-(trifluoromethyl)iminium functional group were achieved as a two-step one-pot synthesis. On the other hand, an electrophilic (Markownikow type) addition of the propyn-1-iminium ion via its C3-position to the olefinic bond of styrenes initiated a reaction cascade which was again terminated by the already mentioned cyclization step through intramolecular electrophilic aromatic substitution, resulting in the formation of 2-(1-phenylvinyl)-1-(trifluoromethyl)-1-(dimethylamino)indenes as the major products. Various other synthetic applications of these reactive propyn-1-iminium salts will be reported in due course.
Supporting Information
| Supporting Information File 1: Experimental procedures, NMR (1H, 13C, 19F) and IR spectra of synthesized compounds. | ||
| Format: PDF | Size: 4.0 MB | Download |
| Supporting Information File 2: Crystal and structure refinement data for compounds 11, 12c, 12d and 19. | ||
| Format: PDF | Size: 281.0 KB | Download |
References
-
Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351
Return to citation in text: [1] -
Haufe, G.; Leroux, F., Eds. Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals; Academic Press: Oxford, U.K., 2019.
Return to citation in text: [1] -
Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, U.K., 2009. doi:10.1002/9781444312096
Return to citation in text: [1] -
Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, NJ, USA, 2008.
Return to citation in text: [1] -
Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Hunter, L. Beilstein J. Org. Chem. 2010, 6, No. 38. doi:10.3762/bjoc.6.38
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Alonso, C.; Martínez de Marigorta, E.; Rubiales, G.; Palacios, F. Chem. Rev. 2015, 115, 1847–1935. doi:10.1021/cr500368h
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Mei, H.; Xie, C.; Han, J.; Soloshonok, V. A. Eur. J. Org. Chem. 2016, 5917–5932. doi:10.1002/ejoc.201600578
Return to citation in text: [1] [2] -
Gauthier, J. Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L. T.; Falgueyret, J.-P.; Kimmel, D. B.; Lamontagne, S.; Léger, S.; LeRiche, T.; Li, C. S.; Massé, F.; McKay, D. J.; Nicoll-Griffith, D. A.; Oballa, R. M.; Palmer, J. T.; Percival, M. D.; Riendeau, D.; Robichaud, J.; Rodan, G. A.; Rodan, S. B.; Seto, C.; Thérien, M.; Truong, V.-L.; Venuti, M. C.; Wesolowski, G.; Young, R. N.; Zamboni, R.; Black, W. C. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. doi:10.1016/j.bmcl.2007.12.047
Return to citation in text: [1] -
Sewald, N.; Seymour, L. C.; Burger, K.; Osipov, S. N.; Kolomiets, A. F.; Fokin, A. V. Tetrahedron: Asymmetry 1994, 5, 1051–1060. doi:10.1016/0957-4166(94)80055-3
Return to citation in text: [1] -
Henle, H.; Geisel, M.; Mews, R. J. Fluorine Chem. 1984, 26, 133–148. doi:10.1016/s0022-1139(00)80917-3
Return to citation in text: [1] -
Ates, C.; Janousek, Z.; Viehe, H. G. Tetrahedron Lett. 1993, 34, 5711–5714. doi:10.1016/s0040-4039(00)73840-8
Return to citation in text: [1] [2] -
Billard, T.; Langlois, B. R. J. Org. Chem. 2002, 67, 997–1000. doi:10.1021/jo016265t
Return to citation in text: [1] -
Langlois, B. R.; Billard, T. Synthesis 2003, 185–194. doi:10.1055/s-2003-36812
Return to citation in text: [1] [2] -
Gong, Y.; Kato, K. J. Fluorine Chem. 2002, 116, 103–107. doi:10.1016/s0022-1139(02)00044-1
Return to citation in text: [1] [2] -
Fuchigami, T.; Ichikawa, S. J. Org. Chem. 1994, 59, 607–615. doi:10.1021/jo00082a018
Return to citation in text: [1] -
Xu, Y.; Dolbier, W. R., Jr. J. Org. Chem. 2000, 65, 2134–2137. doi:10.1021/jo991750y
Return to citation in text: [1] -
Lebouvier, N.; Laroche, C.; Huguenot, F.; Brigaud, T. Tetrahedron Lett. 2002, 43, 2827–2830. doi:10.1016/s0040-4039(02)00330-1
Return to citation in text: [1] -
Wu, L.; Xie, C.; Zhou, J.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. J. Fluorine Chem. 2015, 170, 57–65. doi:10.1016/j.jfluchem.2015.01.001
Return to citation in text: [1] -
Blond, G.; Billard, T.; Langlois, B. R. J. Org. Chem. 2001, 66, 4826–4830. doi:10.1021/jo015587u
Return to citation in text: [1] -
Levin, V. V.; Kozlov, M. A.; Song, Y.-H.; Dilman, A. D.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Tetrahedron Lett. 2008, 49, 3108–3111. doi:10.1016/j.tetlet.2008.03.043
Return to citation in text: [1] [2] -
Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2005, 46, 2219–2221. doi:10.1016/j.tetlet.2005.02.030
Return to citation in text: [1] -
Nicastri, M. C.; Lehnherr, D.; Lam, Y.-h.; DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2020, 142, 987–998. doi:10.1021/jacs.9b10871
Return to citation in text: [1] -
Schneider, T.; Seitz, B.; Schiwek, M.; Maas, G. J. Fluorine Chem. 2020, 235, 109567. doi:10.1016/j.jfluchem.2020.109567
Return to citation in text: [1] -
Nikolai, J.; Schlegel, J.; Regitz, M.; Maas, G. Synthesis 2002, 497–504. doi:10.1055/s-2002-20964
Return to citation in text: [1] [2] -
Zenova, A. Y.; Borisenko, A. A.; Platonov, V. V.; Proskurnina, M. V.; Zefirov, N. S. Russ. J. Org. Chem. 1996, 32, 951–954.
Zh. Org. Khim. 1996, 32, 992–995.
Return to citation in text: [1] [2] -
Akbulut, N.; Hartsough, D.; Kim, J.-I.; Schuster, G. B. J. Org. Chem. 1989, 54, 2549–2556. doi:10.1021/jo00272a017
Return to citation in text: [1] -
de Orbe, M. E.; Echavarren, A. M. Eur. J. Org. Chem. 2018, 2740–2752. doi:10.1002/ejoc.201800170
Return to citation in text: [1] -
Gassman, P. G.; Singleton, D. A. Tetrahedron Lett. 1987, 28, 5969–5972. doi:10.1016/s0040-4039(00)96839-4
Return to citation in text: [1] -
Motomura, T.; Nagamori, H.; Suzawa, K.; Ito, H.; Morita, T.; Kobayashi, S.; Shinkai, H. Fluorene Compound and Use Thereof for Medical Purposes. WO Patent WO2010041748, April 15, 2010.
Return to citation in text: [1] -
Motomura, T.; Matsuo, T.; Shomi, G.; Inoue, M. Pyrazole Alcohol Compounds and Pharmaceutical Use Thereof. U.S. Patent US20150025120, Jan 20, 2015.
Return to citation in text: [1] -
Motomura, T.; Matsuo, T.; Shomi, G. Fluorene-Amide Compounds and Pharamacceutical Use Thereof. U.S. Patent US20150018403, Jan 15, 2015.
Return to citation in text: [1] -
Bouffard, J.; Eaton, R. F.; Müller, P.; Swager, T. M. J. Org. Chem. 2007, 72, 10166–10180. doi:10.1021/jo702000d
Return to citation in text: [1] -
Krespan, C. G.; McKusick, B. C.; Cairns, T. L. J. Am. Chem. Soc. 1961, 83, 3428–3432. doi:10.1021/ja01477a018
Return to citation in text: [1] -
Keim, M.; Kratzer, P.; Derksen, H.; Isakov, D.; Maas, G. Eur. J. Org. Chem. 2019, 826–844. doi:10.1002/ejoc.201801511
Return to citation in text: [1] -
Roberts, R. M. G. J. Organomet. Chem. 1990, 388, 181–186. doi:10.1016/0022-328x(90)85360-b
Return to citation in text: [1] -
Domingo, L. R.; Aurell, M. J.; Pérez, P.; Contreras, R. Tetrahedron 2002, 58, 4417–4423. doi:10.1016/s0040-4020(02)00410-6
Return to citation in text: [1] -
Domingo, L. R.; Sáez, J. A. Org. Biomol. Chem. 2009, 7, 3576–3583. doi:10.1039/b909611f
Return to citation in text: [1] -
Kazakova, A. N.; Iakovenko, R. O.; Boyarskaya, I. A.; Ivanov, A. Y.; Avdontceva, M. S.; Zolotarev, A. A.; Panikorovsky, T. L.; Starova, G. L.; Nenajdenko, V. G.; Vasilyev, A. V. Org. Chem. Front. 2017, 4, 255–265. doi:10.1039/c6qo00643d
Return to citation in text: [1] -
Iakovenko, R. O.; Kazakova, A. N.; Boyarskaya, I. A.; Gurzhiy, V. V.; Avdontceva, M. S.; Panikorovsky, T. L.; Muzalevskiy, V. M.; Nenajdenko, V. G.; Vasilyev, A. V. Eur. J. Org. Chem. 2017, 5632–5643. doi:10.1002/ejoc.201701085
Return to citation in text: [1] -
Politanskaya, L. V.; Selivanova, G. A.; Panteleeva, E. V.; Tretyakov, E. V.; Platonov, V. E.; Nikul’shin, P. V.; Vinogradov, A. S.; Zonov, Y. V.; Karpov, V. M.; Mezhenkova, T. V.; Vasilyev, A. V.; Koldobskii, A. B.; Shilova, O. S.; Morozova, S. M.; Burgart, Y. V.; Shchegolkov, E. V.; Saloutin, V. I.; Sokolov, V. B.; Aksinenko, A. Y.; Nenajdenko, V. G.; Moskalik, M. Y.; Astakhova, V. V.; Shainyan, B. A.; Tabolin, A. A.; Ioffe, S. L.; Muzalevskiy, V. M.; Balenkova, E. S.; Shastin, A. V.; Tyutyunov, A. A.; Boiko, V. E.; Igumnov, S. M.; Dilman, A. D.; Adonin, N. Y.; Bardin, V. V.; Masoud, S. M.; Vorobyeva, D. V.; Osipov, S. N.; Nosova, E. V.; Lipunova, G. N.; ACharushin, V. N.; APrima, D. O.; Makarov, A. G.; Zibarev, A. V.; Trofimov, B. A.; Sobenina, L. N.; Belyaeva, K. V.; Sosnovskikh, V. Y.; Obydennov, D. L.; Usachev, S. A. Russ. Chem. Rev. 2019, 88, 425–569. doi:10.1070/rcr4871
Return to citation in text: [1] [2] -
Wagner-Jauregg, T. Synthesis 1980, 769–798. doi:10.1055/s-1980-29206
Return to citation in text: [1] -
Hall, H. K., Jr.; Nogues, P.; Rhoades, J. W.; Sentman, R. C.; Detar, M. J. Org. Chem. 1982, 47, 1451–1455. doi:10.1021/jo00347a014
Return to citation in text: [1] -
Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811
Angew. Chem. 1969, 81, 797–870. doi:10.1002/ange.19690812102
Return to citation in text: [1] -
Dolbier, W. R., Jr.; Koroniak, H.; Houk, K. N.; Sheu, C. Acc. Chem. Res. 1996, 29, 471–477. doi:10.1021/ar9501986
Return to citation in text: [1] -
Kratzer, P.; Gerster, H.; Maas, G. Synthesis 2015, 47, 2805–2818. doi:10.1055/s-0034-1380223
Return to citation in text: [1] -
Snider, B. B.; Roush, D. M.; Rodini, D. J.; Gonzalez, D.; Spindell, D. J. Org. Chem. 1980, 45, 2773–2785. doi:10.1021/jo01302a007
Return to citation in text: [1] -
Koldobskii, A. B.; Solodova, E. V.; Godovikov, I. A.; Kalinin, V. N. Tetrahedron 2008, 64, 9555–9560. doi:10.1016/j.tet.2008.07.066
Return to citation in text: [1] [2] -
Koldobskii, A. B.; Tsvetkov, N. P.; Verteletskii, P. V.; Godovikov, I. A.; Kalinin, V. N. Russ. Chem. Bull. 2009, 58, 1431–1437. doi:10.1007/s11172-009-0191-3
Return to citation in text: [1] -
Koldobskii, A. B.; Shilova, O. S.; Artyushin, O. I.; Kagramanov, N. D.; Moiseev, S. K. J. Fluorine Chem. 2020, 231, 109463. doi:10.1016/j.jfluchem.2020.109463
Return to citation in text: [1] -
Bäuml, E.; Mayr, H. Chem. Ber. 1985, 118, 683–693. doi:10.1002/cber.19851180228
Return to citation in text: [1] -
Bäuml, E.; Mayr, H. Chem. Ber. 1985, 118, 694–703. doi:10.1002/cber.19851180229
Return to citation in text: [1] -
Keim, M. Terminale Propiniminium-Salze und CF3-haltige 3-Trifloxypropeniminium-Salze als reaktive C3-Bausteine zum Aufbau neuartig funktionalisierter Carbo- und Heterocyclen. Ph.D. Thesis, Ulm University, Ulm, Germany, 2018.
Return to citation in text: [1]
| 38. | Keim, M.; Kratzer, P.; Derksen, H.; Isakov, D.; Maas, G. Eur. J. Org. Chem. 2019, 826–844. doi:10.1002/ejoc.201801511 |
| 39. | Roberts, R. M. G. J. Organomet. Chem. 1990, 388, 181–186. doi:10.1016/0022-328x(90)85360-b |
| 40. | Domingo, L. R.; Aurell, M. J.; Pérez, P.; Contreras, R. Tetrahedron 2002, 58, 4417–4423. doi:10.1016/s0040-4020(02)00410-6 |
| 41. | Domingo, L. R.; Sáez, J. A. Org. Biomol. Chem. 2009, 7, 3576–3583. doi:10.1039/b909611f |
| 1. | Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2013. doi:10.1002/9783527651351 |
| 2. | Haufe, G.; Leroux, F., Eds. Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals; Academic Press: Oxford, U.K., 2019. |
| 3. | Ojima, I., Ed. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Chichester, U.K., 2009. doi:10.1002/9781444312096 |
| 4. | Bégué, J.-P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, NJ, USA, 2008. |
| 5. | Böhm, H.-J.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. ChemBioChem 2004, 5, 637–643. doi:10.1002/cbic.200301023 |
| 14. | Henle, H.; Geisel, M.; Mews, R. J. Fluorine Chem. 1984, 26, 133–148. doi:10.1016/s0022-1139(00)80917-3 |
| 15. | Ates, C.; Janousek, Z.; Viehe, H. G. Tetrahedron Lett. 1993, 34, 5711–5714. doi:10.1016/s0040-4039(00)73840-8 |
| 25. | Magueur, G.; Crousse, B.; Bonnet-Delpon, D. Tetrahedron Lett. 2005, 46, 2219–2221. doi:10.1016/j.tetlet.2005.02.030 |
| 50. | Snider, B. B.; Roush, D. M.; Rodini, D. J.; Gonzalez, D.; Spindell, D. J. Org. Chem. 1980, 45, 2773–2785. doi:10.1021/jo01302a007 |
| 10. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 11. | Mei, H.; Xie, C.; Han, J.; Soloshonok, V. A. Eur. J. Org. Chem. 2016, 5917–5932. doi:10.1002/ejoc.201600578 |
| 12. | Gauthier, J. Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L. T.; Falgueyret, J.-P.; Kimmel, D. B.; Lamontagne, S.; Léger, S.; LeRiche, T.; Li, C. S.; Massé, F.; McKay, D. J.; Nicoll-Griffith, D. A.; Oballa, R. M.; Palmer, J. T.; Percival, M. D.; Riendeau, D.; Robichaud, J.; Rodan, G. A.; Rodan, S. B.; Seto, C.; Thérien, M.; Truong, V.-L.; Venuti, M. C.; Wesolowski, G.; Young, R. N.; Zamboni, R.; Black, W. C. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. doi:10.1016/j.bmcl.2007.12.047 |
| 13. | Sewald, N.; Seymour, L. C.; Burger, K.; Osipov, S. N.; Kolomiets, A. F.; Fokin, A. V. Tetrahedron: Asymmetry 1994, 5, 1051–1060. doi:10.1016/0957-4166(94)80055-3 |
| 26. | Nicastri, M. C.; Lehnherr, D.; Lam, Y.-h.; DiRocco, D. A.; Rovis, T. J. Am. Chem. Soc. 2020, 142, 987–998. doi:10.1021/jacs.9b10871 |
| 44. | Politanskaya, L. V.; Selivanova, G. A.; Panteleeva, E. V.; Tretyakov, E. V.; Platonov, V. E.; Nikul’shin, P. V.; Vinogradov, A. S.; Zonov, Y. V.; Karpov, V. M.; Mezhenkova, T. V.; Vasilyev, A. V.; Koldobskii, A. B.; Shilova, O. S.; Morozova, S. M.; Burgart, Y. V.; Shchegolkov, E. V.; Saloutin, V. I.; Sokolov, V. B.; Aksinenko, A. Y.; Nenajdenko, V. G.; Moskalik, M. Y.; Astakhova, V. V.; Shainyan, B. A.; Tabolin, A. A.; Ioffe, S. L.; Muzalevskiy, V. M.; Balenkova, E. S.; Shastin, A. V.; Tyutyunov, A. A.; Boiko, V. E.; Igumnov, S. M.; Dilman, A. D.; Adonin, N. Y.; Bardin, V. V.; Masoud, S. M.; Vorobyeva, D. V.; Osipov, S. N.; Nosova, E. V.; Lipunova, G. N.; ACharushin, V. N.; APrima, D. O.; Makarov, A. G.; Zibarev, A. V.; Trofimov, B. A.; Sobenina, L. N.; Belyaeva, K. V.; Sosnovskikh, V. Y.; Obydennov, D. L.; Usachev, S. A. Russ. Chem. Rev. 2019, 88, 425–569. doi:10.1070/rcr4871 |
| 8. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 9. | Alonso, C.; Martínez de Marigorta, E.; Rubiales, G.; Palacios, F. Chem. Rev. 2015, 115, 1847–1935. doi:10.1021/cr500368h |
| 23. | Blond, G.; Billard, T.; Langlois, B. R. J. Org. Chem. 2001, 66, 4826–4830. doi:10.1021/jo015587u |
| 47. |
Woodward, R. B.; Hoffmann, R. Angew. Chem., Int. Ed. Engl. 1969, 8, 781–853. doi:10.1002/anie.196907811
Angew. Chem. 1969, 81, 797–870. doi:10.1002/ange.19690812102 |
| 48. | Dolbier, W. R., Jr.; Koroniak, H.; Houk, K. N.; Sheu, C. Acc. Chem. Res. 1996, 29, 471–477. doi:10.1021/ar9501986 |
| 6. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 7. | Hunter, L. Beilstein J. Org. Chem. 2010, 6, No. 38. doi:10.3762/bjoc.6.38 |
| 24. | Levin, V. V.; Kozlov, M. A.; Song, Y.-H.; Dilman, A. D.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Tetrahedron Lett. 2008, 49, 3108–3111. doi:10.1016/j.tetlet.2008.03.043 |
| 49. | Kratzer, P.; Gerster, H.; Maas, G. Synthesis 2015, 47, 2805–2818. doi:10.1055/s-0034-1380223 |
| 20. | Xu, Y.; Dolbier, W. R., Jr. J. Org. Chem. 2000, 65, 2134–2137. doi:10.1021/jo991750y |
| 11. | Mei, H.; Xie, C.; Han, J.; Soloshonok, V. A. Eur. J. Org. Chem. 2016, 5917–5932. doi:10.1002/ejoc.201600578 |
| 22. | Wu, L.; Xie, C.; Zhou, J.; Mei, H.; Soloshonok, V. A.; Han, J.; Pan, Y. J. Fluorine Chem. 2015, 170, 57–65. doi:10.1016/j.jfluchem.2015.01.001 |
| 19. | Fuchigami, T.; Ichikawa, S. J. Org. Chem. 1994, 59, 607–615. doi:10.1021/jo00082a018 |
| 17. | Langlois, B. R.; Billard, T. Synthesis 2003, 185–194. doi:10.1055/s-2003-36812 |
| 46. | Hall, H. K., Jr.; Nogues, P.; Rhoades, J. W.; Sentman, R. C.; Detar, M. J. Org. Chem. 1982, 47, 1451–1455. doi:10.1021/jo00347a014 |
| 16. | Billard, T.; Langlois, B. R. J. Org. Chem. 2002, 67, 997–1000. doi:10.1021/jo016265t |
| 17. | Langlois, B. R.; Billard, T. Synthesis 2003, 185–194. doi:10.1055/s-2003-36812 |
| 18. | Gong, Y.; Kato, K. J. Fluorine Chem. 2002, 116, 103–107. doi:10.1016/s0022-1139(02)00044-1 |
| 18. | Gong, Y.; Kato, K. J. Fluorine Chem. 2002, 116, 103–107. doi:10.1016/s0022-1139(02)00044-1 |
| 24. | Levin, V. V.; Kozlov, M. A.; Song, Y.-H.; Dilman, A. D.; Belyakov, P. A.; Struchkova, M. I.; Tartakovsky, V. A. Tetrahedron Lett. 2008, 49, 3108–3111. doi:10.1016/j.tetlet.2008.03.043 |
| 15. | Ates, C.; Janousek, Z.; Viehe, H. G. Tetrahedron Lett. 1993, 34, 5711–5714. doi:10.1016/s0040-4039(00)73840-8 |
| 21. | Lebouvier, N.; Laroche, C.; Huguenot, F.; Brigaud, T. Tetrahedron Lett. 2002, 43, 2827–2830. doi:10.1016/s0040-4039(02)00330-1 |
| 42. | Kazakova, A. N.; Iakovenko, R. O.; Boyarskaya, I. A.; Ivanov, A. Y.; Avdontceva, M. S.; Zolotarev, A. A.; Panikorovsky, T. L.; Starova, G. L.; Nenajdenko, V. G.; Vasilyev, A. V. Org. Chem. Front. 2017, 4, 255–265. doi:10.1039/c6qo00643d |
| 43. | Iakovenko, R. O.; Kazakova, A. N.; Boyarskaya, I. A.; Gurzhiy, V. V.; Avdontceva, M. S.; Panikorovsky, T. L.; Muzalevskiy, V. M.; Nenajdenko, V. G.; Vasilyev, A. V. Eur. J. Org. Chem. 2017, 5632–5643. doi:10.1002/ejoc.201701085 |
| 44. | Politanskaya, L. V.; Selivanova, G. A.; Panteleeva, E. V.; Tretyakov, E. V.; Platonov, V. E.; Nikul’shin, P. V.; Vinogradov, A. S.; Zonov, Y. V.; Karpov, V. M.; Mezhenkova, T. V.; Vasilyev, A. V.; Koldobskii, A. B.; Shilova, O. S.; Morozova, S. M.; Burgart, Y. V.; Shchegolkov, E. V.; Saloutin, V. I.; Sokolov, V. B.; Aksinenko, A. Y.; Nenajdenko, V. G.; Moskalik, M. Y.; Astakhova, V. V.; Shainyan, B. A.; Tabolin, A. A.; Ioffe, S. L.; Muzalevskiy, V. M.; Balenkova, E. S.; Shastin, A. V.; Tyutyunov, A. A.; Boiko, V. E.; Igumnov, S. M.; Dilman, A. D.; Adonin, N. Y.; Bardin, V. V.; Masoud, S. M.; Vorobyeva, D. V.; Osipov, S. N.; Nosova, E. V.; Lipunova, G. N.; ACharushin, V. N.; APrima, D. O.; Makarov, A. G.; Zibarev, A. V.; Trofimov, B. A.; Sobenina, L. N.; Belyaeva, K. V.; Sosnovskikh, V. Y.; Obydennov, D. L.; Usachev, S. A. Russ. Chem. Rev. 2019, 88, 425–569. doi:10.1070/rcr4871 |
| 29. |
Zenova, A. Y.; Borisenko, A. A.; Platonov, V. V.; Proskurnina, M. V.; Zefirov, N. S. Russ. J. Org. Chem. 1996, 32, 951–954.
Zh. Org. Khim. 1996, 32, 992–995. |
| 27. | Schneider, T.; Seitz, B.; Schiwek, M.; Maas, G. J. Fluorine Chem. 2020, 235, 109567. doi:10.1016/j.jfluchem.2020.109567 |
| 51. | Koldobskii, A. B.; Solodova, E. V.; Godovikov, I. A.; Kalinin, V. N. Tetrahedron 2008, 64, 9555–9560. doi:10.1016/j.tet.2008.07.066 |
| 52. | Koldobskii, A. B.; Tsvetkov, N. P.; Verteletskii, P. V.; Godovikov, I. A.; Kalinin, V. N. Russ. Chem. Bull. 2009, 58, 1431–1437. doi:10.1007/s11172-009-0191-3 |
| 53. | Koldobskii, A. B.; Shilova, O. S.; Artyushin, O. I.; Kagramanov, N. D.; Moiseev, S. K. J. Fluorine Chem. 2020, 231, 109463. doi:10.1016/j.jfluchem.2020.109463 |
| 28. | Nikolai, J.; Schlegel, J.; Regitz, M.; Maas, G. Synthesis 2002, 497–504. doi:10.1055/s-2002-20964 |
| 51. | Koldobskii, A. B.; Solodova, E. V.; Godovikov, I. A.; Kalinin, V. N. Tetrahedron 2008, 64, 9555–9560. doi:10.1016/j.tet.2008.07.066 |
| 54. | Bäuml, E.; Mayr, H. Chem. Ber. 1985, 118, 683–693. doi:10.1002/cber.19851180228 |
| 55. | Bäuml, E.; Mayr, H. Chem. Ber. 1985, 118, 694–703. doi:10.1002/cber.19851180229 |
| 36. | Bouffard, J.; Eaton, R. F.; Müller, P.; Swager, T. M. J. Org. Chem. 2007, 72, 10166–10180. doi:10.1021/jo702000d |
| 37. | Krespan, C. G.; McKusick, B. C.; Cairns, T. L. J. Am. Chem. Soc. 1961, 83, 3428–3432. doi:10.1021/ja01477a018 |
| 33. | Motomura, T.; Nagamori, H.; Suzawa, K.; Ito, H.; Morita, T.; Kobayashi, S.; Shinkai, H. Fluorene Compound and Use Thereof for Medical Purposes. WO Patent WO2010041748, April 15, 2010. |
| 34. | Motomura, T.; Matsuo, T.; Shomi, G.; Inoue, M. Pyrazole Alcohol Compounds and Pharmaceutical Use Thereof. U.S. Patent US20150025120, Jan 20, 2015. |
| 35. | Motomura, T.; Matsuo, T.; Shomi, G. Fluorene-Amide Compounds and Pharamacceutical Use Thereof. U.S. Patent US20150018403, Jan 15, 2015. |
| 28. | Nikolai, J.; Schlegel, J.; Regitz, M.; Maas, G. Synthesis 2002, 497–504. doi:10.1055/s-2002-20964 |
| 31. | de Orbe, M. E.; Echavarren, A. M. Eur. J. Org. Chem. 2018, 2740–2752. doi:10.1002/ejoc.201800170 |
| 32. | Gassman, P. G.; Singleton, D. A. Tetrahedron Lett. 1987, 28, 5969–5972. doi:10.1016/s0040-4039(00)96839-4 |
| 29. |
Zenova, A. Y.; Borisenko, A. A.; Platonov, V. V.; Proskurnina, M. V.; Zefirov, N. S. Russ. J. Org. Chem. 1996, 32, 951–954.
Zh. Org. Khim. 1996, 32, 992–995. |
| 56. | Keim, M. Terminale Propiniminium-Salze und CF3-haltige 3-Trifloxypropeniminium-Salze als reaktive C3-Bausteine zum Aufbau neuartig funktionalisierter Carbo- und Heterocyclen. Ph.D. Thesis, Ulm University, Ulm, Germany, 2018. |
| 30. | Akbulut, N.; Hartsough, D.; Kim, J.-I.; Schuster, G. B. J. Org. Chem. 1989, 54, 2549–2556. doi:10.1021/jo00272a017 |
© 2020 Schneider et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)