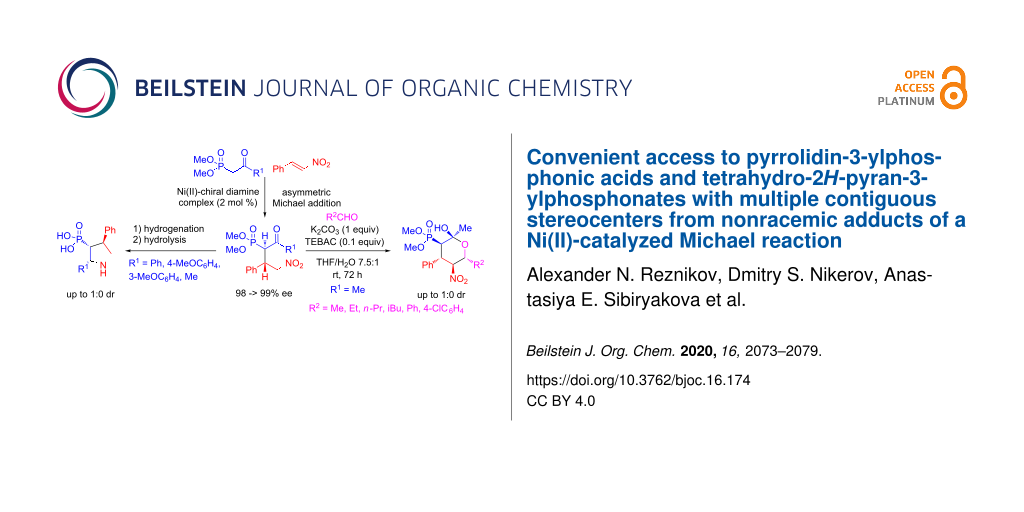
Abstract
A new synthetic strategy toward nonracemic phosphoryl-substituted pyrrolidines and tetrahydropyranes with three and five contiguous stereocenters is presented. Readily available β-keto phosphonates react with conjugated nitroolefins in the presence of a chiral Ni(II) complex to give nitro keto phosphonates with two stereocenters with excellent enantioselectivity and moderate to high diastereoselectivity. These products were used for a reductive cyclization leading to pyrrolidin-3-ylphosphonic acid and for reactions with aldehydes yielding tetrahydropyranylphosphonates as individual stereoisomers. These nonracemic heterocycles containing phosphoryl moieties are useful for designing new pharmacologically active compounds.

Graphical Abstract
Introduction
Сhiral phosphonates [1,2] and phosphoryl-substituted heterocycles [2-4] have received significant attention in recent years due to their wide range of biological activity. For example, SF-2312 (1) is a natural antibiotic – an enolase inhibitor produced by the actinomycete Micromonospora [5]. Dipeptide analogs with phosphonoproline 2 and piperidine-2-phosphonic acid 3 are potent inhibitors of dipeptidyl peptidase IV [6,7]. Oxygen-containing heterocycles containing a phosphoryl group are also of interest in the development of new drugs. It is known that phosphorylated carbohydrate analogues 4 and 5 are neuraminidase inhibitors (Figure 1) [8,9].
![[1860-5397-16-174-1]](/bjoc/content/figures/1860-5397-16-174-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Pharmacologically active nonracemic phosphonates with heterocyclic moieties.
Figure 1: Pharmacologically active nonracemic phosphonates with heterocyclic moieties.
These circumstances create an interest in discovering synthetic routes for obtaining nonracemic phosphoryl-substituted heterocycles. Thus, in recent years, effective methods for the asymmetric synthesis of chiral phosphonates containing octahydroindole [10], tetrahydroquinoline [11], tetrahydroisoquinoline [11,12], β-carboline [13], morpholine [14], and isoindoline [15] moieties have been developed. Obtained in an enantiomerically pure form, phosphonoproline and its analogues [16-19] were among the first compounds in this series. Surprisingly, the methods for obtaining chiral pyrrolidin-3-ylphosphonic acids, tetrahydropyranylphosphonic acids and their esters have experienced minimal development [20-22]. The use of atom-efficient methods of asymmetric catalysis for the synthesis of such compounds is extremely relevant. The chiral metal-complexes-catalyzed [23-26] or organocatalyzed [27-32] Michael reaction is one of the most important synthetic tools for the asymmetric formation of a C–C bond. Moreover, the generation of the first stereocenter of a given configuration during the catalytic process leads to the possibility of creating enantiomerically pure compounds with several stereocenters through the directed regulation of diastereoselectivity in subsequent transformations, which can be realized as stepwise or cascade processes. Due to this reason, the Michael reaction was successfully used over the last decade to synthesize a wide range of heterocycles [33-44]. 2-Oxo-4-nitrophosphonates [45,46], the Michael adducts of β-keto phosphonates and nitroolefins, can be considered as versatile reagents for the synthesis of various phosphoryl-substituted heterocycles. The present work reports on the synthesis of nonracemic phosphoryl-substituted pyrrolidines and tetrahydropyranes via Ni(II)-catalyzed asymmetric Michael addition of β-keto phosphonates to conjugated nitroolefins.
Results and Discussion
For the synthesis of nonracemic polysubstituted pyrrolidin-3-ylphosphonic acids and tetrahydropyranylphosphonates, we assumed that the 4-nitro-2-oxophosphonates – the Michael adducts of β-keto phosphonates and nitroolefins – are suitable precursors, considering that the Ni(II)-catalyzed Michael addition was carried out not only enantioselectively, but also diastereoselectively, such has been previously described [45]. These considerations prompted us to explore further applications of 4-nitro-2-oxophosphonates containing two contiguous stereocenters for the synthesis of the target compounds. Initially, 4-nitro-2-oxophosphonates 6a–f (Figure 2) were synthesized similar to the reported procedure [45,46] from corresponding β-keto phosphonates and nitroolefins in the presence of 2 mol % of the Ni(II) complex with dr 11:1 to 1:0 and 98 –> 99% ee (see Supporting Information File 1).
![[1860-5397-16-174-2]](/bjoc/content/figures/1860-5397-16-174-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Starting nonracemic 4-nitro-2-oxophosphonates.
Figure 2: Starting nonracemic 4-nitro-2-oxophosphonates.
A search for acceptable hydrogenation conditions was carried out using compound 6a. Hydrogenation in the presence of Raney nickel or Pd/C in various solvents leads to a mixture of products, among which 8 and 9 are identified (Scheme 1).
![[1860-5397-16-174-i1]](/bjoc/content/inline/1860-5397-16-174-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Intermolecular N-methylation of reduction product 7.
Scheme 1: Intermolecular N-methylation of reduction product 7.
A satisfactory yield of the desired product 7 (78% by 31P NMR of reaction mixture) was achieved when the hydrogenation was carried out in acetic acid in the presence of Pd/C. However, phosphonate 7 was unstable: already during its isolation, it was partially transformed into compounds 8 and 9. It can be assumed that N-alkylation by the phosphoryl group occurs [47-49].
To avoid this undesirable process, the product 7 was formylated before its isolation. The formylated product 10a may be purified by chromatography without decomposition. Subsequent acid hydrolysis of compound 10a leads to pyrrolidinylphosphonic acid hydrochloride 11a (Scheme 2).
![[1860-5397-16-174-i2]](/bjoc/content/inline/1860-5397-16-174-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of pyrrolidinyl phosphonic acids 11a–d.
Scheme 2: Synthesis of pyrrolidinyl phosphonic acids 11a–d.
Similarly, pyrrolidinylphosphonic acids 11b–d were obtained from the corresponding phosphonates 6b,c,e (Scheme 2).
An X-ray diffraction study [50] of the formyl derivative 10a showed its (2R,3R,4S)-configuration (Figure 3). The absolute configuration of the other phosphonates 10b–d and 11b–d is assumed by analogy.
![[1860-5397-16-174-3]](/bjoc/content/figures/1860-5397-16-174-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: ORTEP diagram of (2R,3R,4S)-10a.
Figure 3: ORTEP diagram of (2R,3R,4S)-10a.
Hydrogenation of phosphonates 6d,f with o-anisyl and bulky adamantan-1-yl substituents at the carbonyl leads to a mixture of unidentified products.
At the next stage of this work, the Henry/acetalization reaction with phosphonate 6e was studied. To optimize the reaction conditions various bases and solvents were used. The reaction of phosphonate 6e with propionic aldehyde 12a was used as a model reaction (see Table 1).
Table 1: Optimization of the conditions of Henry/acetalyzation reaction with phosphonate 6ea.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-174-i5.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Base | Phase transfer catalyst | Solvent | Yield of 13a (%) | drb |
| 1 | PhNMe2 | – | THF | 0 | – |
| 2 | Et3N | – | THF | 0c | – |
| 3 | DBU | – | THF | 0c | – |
| 4 | K3PO4 | TEBAC | THF | 0 | – |
| 5 | K2CO3 | TEBAC | THF | 0 | – |
| 6 | KF | TEBAC | THF | 0c | – |
| 7 | Cs2CO3 | – | THF | 0c | – |
| 8 | K2CO3 | TEBAC | THF/H2O 1:1 | 0c | – |
| 9 | K2CO3 | TEBAC | THF/H2O 7.5:1 | 34 | 1:0 |
| 10 | K2CO3 | 18-crown-6 | CH3CN | 30 | 11:1 |
| 11 | K2CO3 | 18-crown-6 | THF | 17 | 16:1 |
| 12 | K2CO3 | 18-crown-6 | CH2Cl2 | 12 | 10:1 |
aReaction conditions: phosphonate 6e (1.58 mmol), aldehyde 12a (15.8 mmol), solvent (17 mL), base (1.58 mmol), phase transfer catalyst (0.158 mmol), rt, 72 h; bdetermined by 31P NMR; cfull phosphonate 6e conversion was observed and unidentified products were formed.
Initially, the catalytic ability of organic bases was studied for the Henry/acetalyzation reaction by using 0.1–1.0 equiv of catalyst in THF as a solvent. Unfortunately, no product formation was observed in the presence of N,N-dimethylaniline (Table 1, entry 1) and unidentified products were formed when stronger bases such as triethylamine or DBU were used (Table 1, entries 2 and 3). Later on, inorganic bases in combination with a phase transfer agent such as benzyltriethylammonium chloride (TEBAC) were tried as catalysts for the same reaction. Similarly, no products were detected in the reaction even after 72 h in the presence of 1 equiv of potassium phosphate or carbonate with TEBAC (0.1 equiv, Table 1, entries 4 and 5). If potassium fluoride or cesium carbonate were used as a base, the initial phosphonate 6e was consumed in a few hours, but unidentified products were formed (Table 1, entries 6 and 7). After this, the reaction was carried out in an aqueous–organic medium. Fortunately, an individual diastereomer of Henry/acetalyzation product 13a was obtained when K2CO3 in combination with TEBAC was used as a catalyst. A THF/water ratio of 7.5:1 is important in order to get the best yield (Table 1, entry 9). Lower yields and dr values for compound 13a were obtained when 18-crown-6 was used as the phase-transfer catalyst (Table 1, entries 10–12).
With the optimized reaction conditions in hand, the transformation was studied with a series of phosphonates and aldehydes. As shown in Scheme 3, the reaction of phosphonate 6e gave moderate yields with various aliphatic and aromatic aldehydes 12a–f. The presence of a bulkier (compared to methyl) group at the carbonyl in the phosphonates 6a–d,f leads to no reaction.
![[1860-5397-16-174-i3]](/bjoc/content/inline/1860-5397-16-174-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of tetrahydropyranylphosphonates 13a–f via diastereoselective Henry/acetalyzation reaction.
Scheme 3: Synthesis of tetrahydropyranylphosphonates 13a–f via diastereoselective Henry/acetalyzation reactio...
Tetrahydropyranols 13a–f were obtained with good diastereomeric purity. In the 1H NMR spectra of 13a–f signals of methine groups with characteristic spin–spin coupling constants for axially located protons (12.1, 12.3 and 12.5 Hz) were observed.
The structure and absolute (2S,3R,4S,5S,6R)-configuration of the tetrahydropyranol 13b have been confirmed by single-crystal X-ray crystallographic analysis (Figure 4) [51].
![[1860-5397-16-174-4]](/bjoc/content/figures/1860-5397-16-174-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: ORTEP diagram of (2S,3R,4S,5S,6R)-13b.
Figure 4: ORTEP diagram of (2S,3R,4S,5S,6R)-13b.
All hydrogen atoms and the hydroxy group occupy an axial position, while the remaining substituents are in equatorial positions. This is consistent with the 1H NMR data for other similar tetrahydropyranols 13b–f, therefore, their configuration was assumed by analogy.
Dehydration of compound 13b in the presence of p-toluenesulfonic acid gives dihydropyran 14 as a single stereoisomer (Scheme 4).
![[1860-5397-16-174-i4]](/bjoc/content/inline/1860-5397-16-174-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of (3,4-dihydro-2H-pyran-5-yl)phosphonate 14.
Scheme 4: Synthesis of (3,4-dihydro-2H-pyran-5-yl)phosphonate 14.
Conclusion
In summary, a convenient protocol for the synthesis of phosphoryl-substituted pyrrolidines with three contiguous stereocenters via asymmetric addition of β-keto phosphonates to nitroolefins and subsequent reductive cyclization of enantioenriched, diastereomerically pure Michael adducts was developed. The present study also demonstrates that the diastereoselective cascade Henry/acetalyzation reaction with keto nitro phosphonates and aldehydes leading to tetrahydropyranylphosphonates can be efficiently performed within an aqueous-organic medium with K2CO3/TEBAC as the catalyst, and the corresponding products can be obtained with high diastereomeric purity. These methods enable the formation of highly enantioenriched phosphoryl-substituted heterocycles from readily available β-keto phosphonates using an inexpensive Ni(II) complex as the catalyst in the key step.
Funding
The authors gratefully acknowledge the support of this work by the Russian Science Foundation (project 18-13-00447). Spectral and chromatographic analysis was performed by joint use center «Research of physical and chemical properties of substances and materials» with financial support of the Russian Ministry of Science and Higher Education for a basic part of a research project 0778-2020-0005. The X-ray studies were performed in the frame of the development program of the Moscow State University.
References
-
Horsman, G. P.; Zechel, D. L. Chem. Rev. 2017, 117, 5704–5783. doi:10.1021/acs.chemrev.6b00536
Return to citation in text: [1] -
Mucha, A.; Kafarski, P.; Berlicki, Ł. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f
Return to citation in text: [1] [2] -
Chen, L.; Liu, X.-Y.; Zou, Y.-X. Adv. Synth. Catal. 2020, 362, 1724–1818. doi:10.1002/adsc.201901540
Return to citation in text: [1] -
Moonen, K.; Laureyn, I.; Stevens, C. V. Chem. Rev. 2004, 104, 6177–6216. doi:10.1021/cr030451c
Return to citation in text: [1] -
Hanaya, T.; Itoh, C. Heterocycles 2011, 82, 1675–1683. doi:10.3987/com-10-s(e)80
Return to citation in text: [1] -
Belyaev, A.; Zhang, X.; Augustyns, K.; Lambeir, A.-M.; De Meester, I.; Vedernikova, I.; Scharpé, S.; Haemers, A. J. Med. Chem. 1999, 42, 1041–1052. doi:10.1021/jm981033g
Return to citation in text: [1] -
Boduszek, B.; Oleksyszyn, J.; Kam, C.-M.; Selzler, J.; Smith, R. E.; Powers, J. C. J. Med. Chem. 1994, 37, 3969–3976. doi:10.1021/jm00049a016
Return to citation in text: [1] -
Shie, J.-J.; Fang, J.-M.; Lai, P.-T.; Wen, W.-H.; Wang, S.-Y.; Cheng, Y.-S. E.; Tsai, K.-C.; Yang, A.-S.; Wong, C.-H. J. Am. Chem. Soc. 2011, 133, 17959–17965. doi:10.1021/ja207892q
Return to citation in text: [1] -
Wallimann, K.; Vasella, A. Helv. Chim. Acta 1990, 73, 1359–1372. doi:10.1002/hlca.19900730523
Return to citation in text: [1] -
Viveros-Ceballos, J. L.; Martínez-Toto, E. I.; Eustaquio-Armenta, C.; Cativiela, C.; Ordóñez, M. Eur. J. Org. Chem. 2017, 6781–6787. doi:10.1002/ejoc.201701330
Return to citation in text: [1] -
Ordóñez, M.; Arizpe, A.; Sayago, F. J.; Jiménez, A. I.; Cativiela, C. Molecules 2016, 21, 1140. doi:10.3390/molecules21091140
Return to citation in text: [1] [2] -
Viveros-Ceballos, J. L.; Ordóñez, M.; Sayago, F. J.; Jiménez, A. I.; Cativiela, C. Eur. J. Org. Chem. 2016, 2711–2719. doi:10.1002/ejoc.201600313
Return to citation in text: [1] -
Viveros-Ceballos, J. L.; Sayago, F. J.; Cativiela, C.; Ordóñez, M. Eur. J. Org. Chem. 2015, 1084–1091. doi:10.1002/ejoc.201403418
Return to citation in text: [1] -
Bonilla-Landa, I.; Viveros-Ceballos, J. L.; Ordóñez, M. Tetrahedron: Asymmetry 2014, 25, 485–487. doi:10.1016/j.tetasy.2014.02.014
Return to citation in text: [1] -
Viveros-Ceballos, J. L.; Cativiela, C.; Ordóñez, M. Tetrahedron: Asymmetry 2011, 22, 1479–1484. doi:10.1016/j.tetasy.2011.08.003
Return to citation in text: [1] -
Hirata, S.; Kuriyama, M.; Onomura, O. Tetrahedron 2011, 67, 9411–9416. doi:10.1016/j.tet.2011.09.080
Return to citation in text: [1] -
Kaboudin, B.; Kato, J.-y.; Aoyama, H.; Yokomatsu, T. Tetrahedron: Asymmetry 2013, 24, 1562–1566. doi:10.1016/j.tetasy.2013.10.013
Return to citation in text: [1] -
Amedjkouh, M.; Westerlund, K. Tetrahedron Lett. 2004, 45, 5175–5177. doi:10.1016/j.tetlet.2004.04.009
Return to citation in text: [1] -
Kaname, M.; Arakawa, Y.; Yoshifuji, S. Tetrahedron Lett. 2001, 42, 2713–2716. doi:10.1016/s0040-4039(01)00283-0
Return to citation in text: [1] -
Markitanov, Y. N.; Timoshenko, V. M.; Shermolovich, Y. G.; Mykhalchuk, V. L.; Grafova, I. A.; Grafov, A. V. Chem. Heterocycl. Compd. 2016, 52, 503–506. doi:10.1007/s10593-016-1916-5
Return to citation in text: [1] -
Yan, L.; Hale, J. J.; Lynch, C. L.; Budhu, R.; Gentry, A.; Mills, S. G.; Hajdu, R.; Keohane, C. A.; Rosenbach, M. J.; Milligan, J. A.; Shei, G.-J.; Chrebet, G.; Bergstrom, J.; Card, D.; Rosen, H.; Mandala, S. M. Bioorg. Med. Chem. Lett. 2004, 14, 4861–4866. doi:10.1016/j.bmcl.2004.07.049
Return to citation in text: [1] -
Paulsen, H.; Greve, W. Chem. Ber. 1973, 106, 2114–2123. doi:10.1002/cber.19731060705
Return to citation in text: [1] -
Zheng, K.; Liu, X.; Feng, X. Chem. Rev. 2018, 118, 7586–7656. doi:10.1021/acs.chemrev.7b00692
Return to citation in text: [1] -
Reznikov, A. N.; Klimochkin, Y. N. Synthesis 2020, 52, 781–795. doi:10.1055/s-0039-1690044
Return to citation in text: [1] -
Pellissier, H. Adv. Synth. Catal. 2015, 357, 2745–2780. doi:10.1002/adsc.201500512
Return to citation in text: [1] -
Mauduit, M.; Baslé, O.; Clavier, H.; Crévisy, C.; Denicourt-Nowicki, A. Metal-Catalyzed Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes. In Comprehensive Organic Synthesis II, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier: Oxford, U.K., 2014; Vol. 4, pp 189–341. doi:10.1016/b978-0-08-097742-3.00406-7
Return to citation in text: [1] -
Pellissier, H. Curr. Org. Chem. 2018, 22, 323–344. doi:10.2174/1385272821666170818160915
Return to citation in text: [1] -
Maria Faísca Phillips, A. Curr. Org. Synth. 2016, 13, 687–725. doi:10.2174/1570179412666150914200843
Return to citation in text: [1] -
Heravi, M. M.; Hajiabbasi, P.; Hamidi, H. Curr. Org. Chem. 2014, 18, 489–511. doi:10.2174/13852728113176660149
Return to citation in text: [1] -
Somanathan, R.; Chavez, D.; Servin, F. A.; Romero, J. A.; Navarrete, A.; Parra-Hake, M.; Aguirre, G.; Anaya de Parrodi, C.; Gonzalez, J. Curr. Org. Chem. 2012, 16, 2440–2461. doi:10.2174/138527212803520128
Return to citation in text: [1] -
Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h
Return to citation in text: [1] -
Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023
Return to citation in text: [1] -
Pellissier, H. Adv. Synth. Catal. 2016, 358, 2194–2259. doi:10.1002/adsc.201600462
Return to citation in text: [1] -
Bugaut, X.; Bonne, D.; Coquerel, Y.; Rodriguez, J.; Constantieux, T. Curr. Org. Chem. 2013, 17, 1920–1928. doi:10.2174/13852728113179990090
Return to citation in text: [1] -
Gasperi, T.; Miceli, M.; Campagne, J.-M.; Marcia de Figueiredo, R. Molecules 2017, 22, 1636. doi:10.3390/molecules22101636
Return to citation in text: [1] -
Lu, L.-Q.; Chen, J.-R.; Xiao, W.-J. Acc. Chem. Res. 2012, 45, 1278–1293. doi:10.1021/ar200338s
Return to citation in text: [1] -
Urbanietz, G.; Atodiresei, I.; Enders, D. Synthesis 2014, 46, 1261–1269. doi:10.1055/s-0033-1340826
Return to citation in text: [1] -
Mondal, B.; Pan, S. C. Synlett 2018, 29, 576–580. doi:10.1055/s-0036-1591736
Return to citation in text: [1] -
Corbett, M. T.; Xu, Q.; Johnson, J. S. Org. Lett. 2014, 16, 2362–2365. doi:10.1021/ol500679w
Return to citation in text: [1] -
Guo, S.; Xie, Y.; Hu, X.; Huang, H. Org. Lett. 2011, 13, 5596–5599. doi:10.1021/ol2023196
Return to citation in text: [1] -
Nakamura, A.; Lectard, S.; Hashizume, D.; Hamashima, Y.; Sodeoka, M. J. Am. Chem. Soc. 2010, 132, 4036–4037. doi:10.1021/ja909457b
Return to citation in text: [1] -
Ma, H.; Liu, K.; Zhang, F.-G.; Zhu, C.-L.; Nie, J.; Ma, J.-A. J. Org. Chem. 2010, 75, 1402–1409. doi:10.1021/jo901991v
Return to citation in text: [1] -
Xu, Y.; Matsunaga, S.; Shibasaki, M. Org. Lett. 2010, 12, 3246–3249. doi:10.1021/ol101185p
Return to citation in text: [1] -
Barnes, D. M.; Ji, J.; Fickes, M. G.; Fitzgerald, M. A.; King, S. A.; Morton, H. E.; Plagge, F. A.; Preskill, M.; Wagaw, S. H.; Wittenberger, S. J.; Zhang, J. J. Am. Chem. Soc. 2002, 124, 13097–13105. doi:10.1021/ja026788y
Return to citation in text: [1] -
Reznikov, A. N.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Tetrahedron: Asymmetry 2015, 26, 1050–1057. doi:10.1016/j.tetasy.2015.08.003
Return to citation in text: [1] [2] [3] -
Reznikov, A. N.; Sibiryakova, A. E.; Klimochkin, Y. N. Russ. J. Gen. Chem. 2014, 84, 2280–2282. doi:10.1134/s1070363214110437
Return to citation in text: [1] [2] -
Budzisz, E.; Pastuszko, S. Tetrahedron 1999, 55, 4815–4824. doi:10.1016/s0040-4020(99)00153-2
Return to citation in text: [1] -
Gray, M. D. M.; Smith, D. J. H. Tetrahedron Lett. 1980, 21, 859–860. doi:10.1016/s0040-4039(00)71526-7
Return to citation in text: [1] -
Vassileva, V.; Georgiev, E. M.; Troev, K.; Roundhill, D. M. Phosphorus, Sulfur Silicon Relat. Elem. 1994, 92, 101–107. doi:10.1080/10426509408021462
Return to citation in text: [1] -
Reznikov, A. N.; Nikerov, D. S.; Sibiryakova, A. E.; Rybakov, V. B.; Golovin, E. V.; Klimochkin, Yu. N. CCDC 1919294: Experimental Crystal Structure Determination, 2020. doi:10.5517/ccdc.csd.cc22f5qf
Return to citation in text: [1] -
Reznikov, A. N.; Nikerov, D. S.; Sibiryakova, A. E.; Rybakov, V. B.; Golovin, E. V.; Klimochkin, Yu. N. CCDC 1846644: Experimental Crystal Structure Determination, 2018. doi:10.5517/ccdc.csd.cc1zzl5q
Return to citation in text: [1]
| 45. | Reznikov, A. N.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Tetrahedron: Asymmetry 2015, 26, 1050–1057. doi:10.1016/j.tetasy.2015.08.003 |
| 46. | Reznikov, A. N.; Sibiryakova, A. E.; Klimochkin, Y. N. Russ. J. Gen. Chem. 2014, 84, 2280–2282. doi:10.1134/s1070363214110437 |
| 45. | Reznikov, A. N.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Tetrahedron: Asymmetry 2015, 26, 1050–1057. doi:10.1016/j.tetasy.2015.08.003 |
| 46. | Reznikov, A. N.; Sibiryakova, A. E.; Klimochkin, Y. N. Russ. J. Gen. Chem. 2014, 84, 2280–2282. doi:10.1134/s1070363214110437 |
| 45. | Reznikov, A. N.; Sibiryakova, A. E.; Rybakov, V. B.; Klimochkin, Y. N. Tetrahedron: Asymmetry 2015, 26, 1050–1057. doi:10.1016/j.tetasy.2015.08.003 |
| 1. | Horsman, G. P.; Zechel, D. L. Chem. Rev. 2017, 117, 5704–5783. doi:10.1021/acs.chemrev.6b00536 |
| 2. | Mucha, A.; Kafarski, P.; Berlicki, Ł. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 8. | Shie, J.-J.; Fang, J.-M.; Lai, P.-T.; Wen, W.-H.; Wang, S.-Y.; Cheng, Y.-S. E.; Tsai, K.-C.; Yang, A.-S.; Wong, C.-H. J. Am. Chem. Soc. 2011, 133, 17959–17965. doi:10.1021/ja207892q |
| 9. | Wallimann, K.; Vasella, A. Helv. Chim. Acta 1990, 73, 1359–1372. doi:10.1002/hlca.19900730523 |
| 27. | Pellissier, H. Curr. Org. Chem. 2018, 22, 323–344. doi:10.2174/1385272821666170818160915 |
| 28. | Maria Faísca Phillips, A. Curr. Org. Synth. 2016, 13, 687–725. doi:10.2174/1570179412666150914200843 |
| 29. | Heravi, M. M.; Hajiabbasi, P.; Hamidi, H. Curr. Org. Chem. 2014, 18, 489–511. doi:10.2174/13852728113176660149 |
| 30. | Somanathan, R.; Chavez, D.; Servin, F. A.; Romero, J. A.; Navarrete, A.; Parra-Hake, M.; Aguirre, G.; Anaya de Parrodi, C.; Gonzalez, J. Curr. Org. Chem. 2012, 16, 2440–2461. doi:10.2174/138527212803520128 |
| 31. | Zhang, Y.; Wang, W. Catal. Sci. Technol. 2012, 2, 42–53. doi:10.1039/c1cy00334h |
| 32. | Almaşi, D.; Alonso, D. A.; Nájera, C. Tetrahedron: Asymmetry 2007, 18, 299–365. doi:10.1016/j.tetasy.2007.01.023 |
| 6. | Belyaev, A.; Zhang, X.; Augustyns, K.; Lambeir, A.-M.; De Meester, I.; Vedernikova, I.; Scharpé, S.; Haemers, A. J. Med. Chem. 1999, 42, 1041–1052. doi:10.1021/jm981033g |
| 7. | Boduszek, B.; Oleksyszyn, J.; Kam, C.-M.; Selzler, J.; Smith, R. E.; Powers, J. C. J. Med. Chem. 1994, 37, 3969–3976. doi:10.1021/jm00049a016 |
| 33. | Pellissier, H. Adv. Synth. Catal. 2016, 358, 2194–2259. doi:10.1002/adsc.201600462 |
| 34. | Bugaut, X.; Bonne, D.; Coquerel, Y.; Rodriguez, J.; Constantieux, T. Curr. Org. Chem. 2013, 17, 1920–1928. doi:10.2174/13852728113179990090 |
| 35. | Gasperi, T.; Miceli, M.; Campagne, J.-M.; Marcia de Figueiredo, R. Molecules 2017, 22, 1636. doi:10.3390/molecules22101636 |
| 36. | Lu, L.-Q.; Chen, J.-R.; Xiao, W.-J. Acc. Chem. Res. 2012, 45, 1278–1293. doi:10.1021/ar200338s |
| 37. | Urbanietz, G.; Atodiresei, I.; Enders, D. Synthesis 2014, 46, 1261–1269. doi:10.1055/s-0033-1340826 |
| 38. | Mondal, B.; Pan, S. C. Synlett 2018, 29, 576–580. doi:10.1055/s-0036-1591736 |
| 39. | Corbett, M. T.; Xu, Q.; Johnson, J. S. Org. Lett. 2014, 16, 2362–2365. doi:10.1021/ol500679w |
| 40. | Guo, S.; Xie, Y.; Hu, X.; Huang, H. Org. Lett. 2011, 13, 5596–5599. doi:10.1021/ol2023196 |
| 41. | Nakamura, A.; Lectard, S.; Hashizume, D.; Hamashima, Y.; Sodeoka, M. J. Am. Chem. Soc. 2010, 132, 4036–4037. doi:10.1021/ja909457b |
| 42. | Ma, H.; Liu, K.; Zhang, F.-G.; Zhu, C.-L.; Nie, J.; Ma, J.-A. J. Org. Chem. 2010, 75, 1402–1409. doi:10.1021/jo901991v |
| 43. | Xu, Y.; Matsunaga, S.; Shibasaki, M. Org. Lett. 2010, 12, 3246–3249. doi:10.1021/ol101185p |
| 44. | Barnes, D. M.; Ji, J.; Fickes, M. G.; Fitzgerald, M. A.; King, S. A.; Morton, H. E.; Plagge, F. A.; Preskill, M.; Wagaw, S. H.; Wittenberger, S. J.; Zhang, J. J. Am. Chem. Soc. 2002, 124, 13097–13105. doi:10.1021/ja026788y |
| 5. | Hanaya, T.; Itoh, C. Heterocycles 2011, 82, 1675–1683. doi:10.3987/com-10-s(e)80 |
| 20. | Markitanov, Y. N.; Timoshenko, V. M.; Shermolovich, Y. G.; Mykhalchuk, V. L.; Grafova, I. A.; Grafov, A. V. Chem. Heterocycl. Compd. 2016, 52, 503–506. doi:10.1007/s10593-016-1916-5 |
| 21. | Yan, L.; Hale, J. J.; Lynch, C. L.; Budhu, R.; Gentry, A.; Mills, S. G.; Hajdu, R.; Keohane, C. A.; Rosenbach, M. J.; Milligan, J. A.; Shei, G.-J.; Chrebet, G.; Bergstrom, J.; Card, D.; Rosen, H.; Mandala, S. M. Bioorg. Med. Chem. Lett. 2004, 14, 4861–4866. doi:10.1016/j.bmcl.2004.07.049 |
| 22. | Paulsen, H.; Greve, W. Chem. Ber. 1973, 106, 2114–2123. doi:10.1002/cber.19731060705 |
| 2. | Mucha, A.; Kafarski, P.; Berlicki, Ł. J. Med. Chem. 2011, 54, 5955–5980. doi:10.1021/jm200587f |
| 3. | Chen, L.; Liu, X.-Y.; Zou, Y.-X. Adv. Synth. Catal. 2020, 362, 1724–1818. doi:10.1002/adsc.201901540 |
| 4. | Moonen, K.; Laureyn, I.; Stevens, C. V. Chem. Rev. 2004, 104, 6177–6216. doi:10.1021/cr030451c |
| 23. | Zheng, K.; Liu, X.; Feng, X. Chem. Rev. 2018, 118, 7586–7656. doi:10.1021/acs.chemrev.7b00692 |
| 24. | Reznikov, A. N.; Klimochkin, Y. N. Synthesis 2020, 52, 781–795. doi:10.1055/s-0039-1690044 |
| 25. | Pellissier, H. Adv. Synth. Catal. 2015, 357, 2745–2780. doi:10.1002/adsc.201500512 |
| 26. | Mauduit, M.; Baslé, O.; Clavier, H.; Crévisy, C.; Denicourt-Nowicki, A. Metal-Catalyzed Asymmetric Nucleophilic Addition to Electron-Deficient Alkenes. In Comprehensive Organic Synthesis II, 2nd ed.; Knochel, P.; Molander, G. A., Eds.; Elsevier: Oxford, U.K., 2014; Vol. 4, pp 189–341. doi:10.1016/b978-0-08-097742-3.00406-7 |
| 13. | Viveros-Ceballos, J. L.; Sayago, F. J.; Cativiela, C.; Ordóñez, M. Eur. J. Org. Chem. 2015, 1084–1091. doi:10.1002/ejoc.201403418 |
| 15. | Viveros-Ceballos, J. L.; Cativiela, C.; Ordóñez, M. Tetrahedron: Asymmetry 2011, 22, 1479–1484. doi:10.1016/j.tetasy.2011.08.003 |
| 51. | Reznikov, A. N.; Nikerov, D. S.; Sibiryakova, A. E.; Rybakov, V. B.; Golovin, E. V.; Klimochkin, Yu. N. CCDC 1846644: Experimental Crystal Structure Determination, 2018. doi:10.5517/ccdc.csd.cc1zzl5q |
| 11. | Ordóñez, M.; Arizpe, A.; Sayago, F. J.; Jiménez, A. I.; Cativiela, C. Molecules 2016, 21, 1140. doi:10.3390/molecules21091140 |
| 12. | Viveros-Ceballos, J. L.; Ordóñez, M.; Sayago, F. J.; Jiménez, A. I.; Cativiela, C. Eur. J. Org. Chem. 2016, 2711–2719. doi:10.1002/ejoc.201600313 |
| 16. | Hirata, S.; Kuriyama, M.; Onomura, O. Tetrahedron 2011, 67, 9411–9416. doi:10.1016/j.tet.2011.09.080 |
| 17. | Kaboudin, B.; Kato, J.-y.; Aoyama, H.; Yokomatsu, T. Tetrahedron: Asymmetry 2013, 24, 1562–1566. doi:10.1016/j.tetasy.2013.10.013 |
| 18. | Amedjkouh, M.; Westerlund, K. Tetrahedron Lett. 2004, 45, 5175–5177. doi:10.1016/j.tetlet.2004.04.009 |
| 19. | Kaname, M.; Arakawa, Y.; Yoshifuji, S. Tetrahedron Lett. 2001, 42, 2713–2716. doi:10.1016/s0040-4039(01)00283-0 |
| 11. | Ordóñez, M.; Arizpe, A.; Sayago, F. J.; Jiménez, A. I.; Cativiela, C. Molecules 2016, 21, 1140. doi:10.3390/molecules21091140 |
| 47. | Budzisz, E.; Pastuszko, S. Tetrahedron 1999, 55, 4815–4824. doi:10.1016/s0040-4020(99)00153-2 |
| 48. | Gray, M. D. M.; Smith, D. J. H. Tetrahedron Lett. 1980, 21, 859–860. doi:10.1016/s0040-4039(00)71526-7 |
| 49. | Vassileva, V.; Georgiev, E. M.; Troev, K.; Roundhill, D. M. Phosphorus, Sulfur Silicon Relat. Elem. 1994, 92, 101–107. doi:10.1080/10426509408021462 |
| 10. | Viveros-Ceballos, J. L.; Martínez-Toto, E. I.; Eustaquio-Armenta, C.; Cativiela, C.; Ordóñez, M. Eur. J. Org. Chem. 2017, 6781–6787. doi:10.1002/ejoc.201701330 |
| 14. | Bonilla-Landa, I.; Viveros-Ceballos, J. L.; Ordóñez, M. Tetrahedron: Asymmetry 2014, 25, 485–487. doi:10.1016/j.tetasy.2014.02.014 |
| 50. | Reznikov, A. N.; Nikerov, D. S.; Sibiryakova, A. E.; Rybakov, V. B.; Golovin, E. V.; Klimochkin, Yu. N. CCDC 1919294: Experimental Crystal Structure Determination, 2020. doi:10.5517/ccdc.csd.cc22f5qf |
© 2020 Reznikov et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)