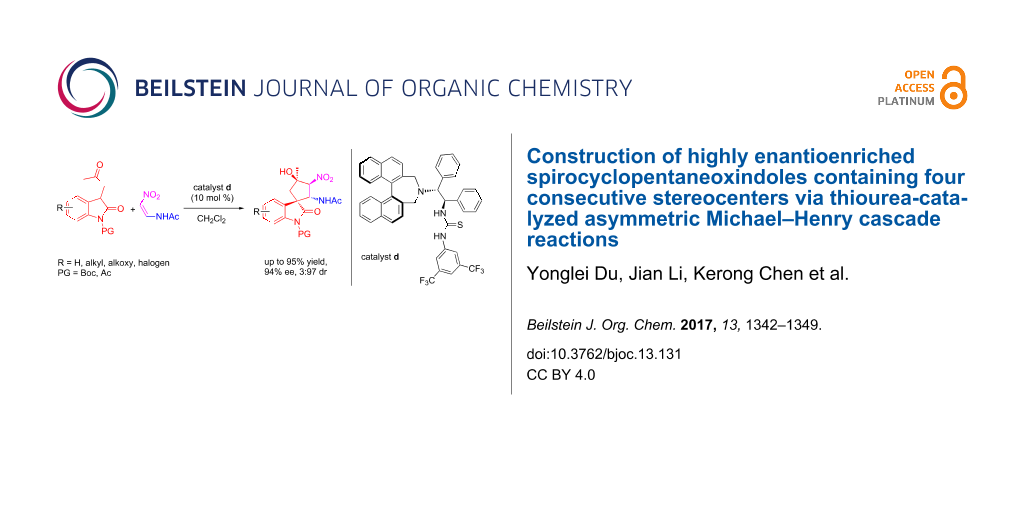
Abstract
The thiourea-catalyzed asymmetric synthesis of highly enantioenriched spirocyclopentaneoxindoles containing chiral amide functional groups using simple 3-substituted oxindoles and nitrovinylacetamide as starting materials was achieved successfully. This protocol features operational simplicity, high atom economy, and high catalytic asymmetry, thus representing a versatile approach to the synthesis of highly enantioenriched spirocyclopentaneoxindoles.

Graphical Abstract
Introduction
The spirocyclic oxindole core represents an important scaffold that is encountered frequently in many biologically active molecules and natural products (Figure 1) [1-19]. Despite many advances in asymmetric synthesis in the construction of heterocyclic spirooxindoles in the past decade [2,4,11,20-22], the development of general and practical strategies to obtain saturated spirocyclopentaneoxindoles containing multiple contiguous stereocenters remains challenging [23-26]. The medicinal properties of these frameworks mean that fast enrichment of spirooxindoles bearing diverse functional groups is of considerable importance.
![[1860-5397-13-131-1]](/bjoc/content/figures/1860-5397-13-131-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representative spirooxindole natural products.
Figure 1: Representative spirooxindole natural products.
Recently, an increasing number of asymmetric catalysis strategies with chiral transition metals [27-33], organocatalysts such as secondary amines [34-36], nucleophilic phosphines [26,37-44], tertiary amines [45], N-heterocyclic carbenes (NHCs) [46-48], and cinchona alkaloid derivatives [25,28,49,50] have been used to construct successfully spirooxindole privileged scaffolds. However, most of them were devoted to exploring new catalyst systems to improve the reaction efficiency and selectivity [51-64], and studies extending the reaction scope using functionalized nitrovinylacetamides are rare. Chiral thioureas [28,38,39] have evolved as powerful hydrogen bonding catalysts for the asymmetric synthesis of spirocyclopentaneoxindoles, which have been demonstrated as acceptable but still considerably limited. Organocatalytic iminium–enamine cascade reactions [35] (Scheme 1, reaction 1A) and Michael–Henry cascade reactions [25] (Scheme 1, reaction 1B) reported by Barbas III's group involve the cyclization between α,β-unsaturated aldehydes and nitrostyrenes with 3-substituted oxindoles to generate the corresponding CHO- or NO2-substituted spirooxindole derivatives with good enantiomeric excess (ee) values, respectively. However, the utility of the reaction is limited to α,β-unsaturated aldehydes with aromatic/alkane substitutions and nitroolefins with aromatic substitutions. Additionally, Shao’s group (Scheme 1, reaction 1C) developed a one-pot thiourea-catalyzed Michael addition/intramolecular silyl nitronate-olefin cycloaddition (ISOC)/fragmentation sequence to produce highly enantioenriched spirocyclopentaneoxindoles containing an oxime functional group from easily accessible 3-allyl-substituted oxindoles and nitroolefins, which has received wide attention because of its high efficiency in constructing functionalized spirocyclopentaneoxindoles. However, lower temperatures are required (−30 °C) [50]. Therefore, it is highly desirable to develop novel and efficient methods to access directly various spirocycles. In our continuous endeavor to develop effective strategies to construct biologically active spirocyclic oxindoles [65-68], we have built successfully interesting spirooxindoles via an NHC-catalyzed [4 + 2] annulation involving an oxidative γ-carbon activation of common α,β-unsaturated aldehydes [68]. Herein, we report another effective asymmetric catalytic synthesis of saturated spirocyclopentaneoxindoles containing four consecutive stereocenters with 3-substituted oxindoles and nitrovinylacetamide using a bifunctional thiourea catalyst in good yields (up to 95%) with excellent diastereoselectivity (up to 3:97) and enantioselectivity (up to 94%) (Scheme 1, reaction 1D).
![[1860-5397-13-131-i1]](/bjoc/content/inline/1860-5397-13-131-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Construction of spirocyclopentaneoxindole scaffolds.
Scheme 1: Construction of spirocyclopentaneoxindole scaffolds.
Results and Discussion
To establish the optimal experimental conditions for the synthesis of spirocyclopentaneoxindoles, we chose 3-substituted oxindole 1a and nitrovinylacetamide (2a) as the model substrates, and the results are summarized in Table 1. Initially, a variety of organocatalysts (a–f) were investigated in CH2Cl2 at −20 °C for 12 h to evaluate their ability to promote the transformation (Table 1, entries 1–6). When cinchona alkaloid-derived catalyst a and quinine-derived amine catalyst b were tested, however, poor yields or ee values were obtained, respectively (Table 1, entries 1 and 2). Further experiments showed that a bifunctional thiourea catalyst d was the most efficient for the synthesis of spirocyclic oxindole derivatives in good yields (80%) with excellent diastereoselectivity (8:92 dr), and moderate enantioselectivity (83% ee, Table 1, entries 3–6). Therefore, catalyst d was chosen as the optimal catalyst for further investigation (Table 1, entry 4). The reaction temperature was investigated, however elevated temperatures are detrimental (Table 1, entries 4, 12, 13). Different solvents, such as methanol, toluene, acetone, ether and chloroform were further screened. The results suggested that changing the solvent had an adverse effect on the ee value, and CH2Cl2 remained the best choice for this transformation (Table 1, entries 4, 7–11). Subsequently, we investigated some additives, for example, p-MeC6H4SO3H, trimethylsilyl chloride (TMSCl) and trifluoroacetic acid (TFA) for this catalytic system to increase the ee value of the target product; however, the use of the additives proved ineffective (Table 1, entries 15–17).
Table 1: Optimization for the reaction conditionsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-13-131-i4.svg?max-width=637&scale=1.0)
|
||||||
| entry | catalyst | solvent | additive | yield(%)b | drc | ee (%)d |
| 1 | a | CH2Cl2 | – | 37 | 5:95 | 99 |
| 2 | b | CH2Cl2 | – | 57 | 20:80 | 10 |
| 3 | c | CH2Cl2 | – | 6 | 16:84 | 6 |
| 4 | d | CH2Cl2 | – | 80 | 8:92 | 83 |
| 5 | e | CH2Cl2 | – | 21 | 4:96 | 29 |
| 6 | f | CH2Cl2 | – | 40 | – | – |
| 7 | d | CH3OH | – | – | – | 16 |
| 8 | d | toluene | – | – | – | 47 |
| 9 | d | acetone | – | – | – | 28 |
| 10 | d | Et2O | – | – | – | 41 |
| 11 | d | CHCl3 | – | – | – | 2 |
| 12e | d | CH2Cl2 | – | 56 | – | 4 |
| 13f | d | CH2Cl2 | – | trace | – | – |
| 14g | d | CH2Cl2 | – | 72 | – | 82 |
| 15h | d | CH2Cl2 | p-MeC6H4SO3H | – | – | 25 |
| 16h | d | CH2Cl2 | TMSCl | – | – | 13 |
| 17h | d | CH2Cl2 | TFA | – | – | 11 |
aReaction conditions: Unless noted, the reaction was carried out with 1a (0.11 mmol), 2a (0.1 mmol), catalyst a–f (0.01 mmol), solvent (2–3 mL), 12 h. bIsolated yield. cDetermined by 1H NMR spectroscopy of the crude mixture. dDetermined by chiral HPLC analysis of the major diastereomer. eTemperature (0 °C). fTemperature (rt). gSolvent (1 mL). hp-MeC6H4SO3H (0.01 mmol), TMSCl (0.01 mmol), TFA (0.01 mmol) as the additive. “–” represents: not determined.
With the optimized reaction conditions established, we next investigated the substrate scope of 3-substituted oxindoles in this transformation with nitroolefins (Scheme 2). In general, the diverse 3-substituted oxindoles 1a–j with electron-donating, electron-withdrawing, or halide groups could undergo a smooth reaction with nitrovinylacetamide (2a) in moderate yields, good diastereoselectivity, and general enantioselectivity (3a–j). For example, the protocol showed moderate yields (75–76%), excellent diastereoselectivity (9:91–3:97 dr) and good enantioselectivity (85–94% ee) for substrates containing 5-CH3 or 5-OCH3 groups (3b and 3d). Substrates carrying 5-F, 7-F, 5-Cl and 6-Cl afforded the corresponding products 3f–i in high yields (85–95%) with excellent diastereoselectivity (17:83–3:97 dr) and good enantioselectivity (76–93% ee). Surprisingly, introducing a bromo group into 6-position of the oxindole scaffold resulted in a low enantioselectivity (52% ee for 3j), although at a high yield (79%) and excellent diastereoselectivity (3:97 dr). To further extend the reaction scope, we attempted to exchange the N-Boc group of the 3-substituted oxindoles with other protecting groups, such as Bn, CH3 or an acetyl group. The results demonstrated that only an acetyl protecting group proved to be well tolerated, providing for the efficient synthesis of spirocyclopentaneoxindoles 3k–o with moderate yields (47–74%) and excellent diastereoselectivity (9:91–2:98 dr). However, replacing the N-Boc group with an N-acetyl group on spirooxindoles had a negative effect on the stereoselectivities of the target products, probably because the N-acetyl group decreases steric hindrance. In addition, substituents (CH3, Boc) at R′ and the free N-H substituted oxindole were investigated that did not give the corresponding target products (3p, 3q and 3r).
![[1860-5397-13-131-i2]](/bjoc/content/inline/1860-5397-13-131-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of enantioselective synthesis of spirooxindoles. Reaction conditions: catalyst d (0.01 mmol), oxindoles 1a–o (0.11 mmol) and nitroolefins 2a–c (0.1 mmol) in CH2Cl2 (3 mL), method A or method B. The ee values were determined by chiral HPLC analysis of major diastereomer. The dr values were determined by 1H NMR analysis. Method A: with 10 mol % of d as a catalyst, −20 °C, 12 h. Method B: with 10 mol % of d as a catalyst, −10 °C, 24 h.
Scheme 2: Scope of enantioselective synthesis of spirooxindoles. Reaction conditions: catalyst d (0.01 mmol),...
On the basis of the dual activation model proposed by Takemoto et al. [69], a plausible reaction mechanism was proposed in Scheme 3. The multifunctional organocatalyst d has a chiral scaffold including a thiourea moiety and an amino group. Both the 3-substituted oxindoles 1 and nitrovinylacetamide (2a) that participate in this reaction are activated simultaneously via multiple hydrogen bonds. In addition, the electrophilicity of the reacting carbon center of nitrovinylacetamide is likely enhanced by H-bonding, thereby enabling the Michael addition to construct a unique quaternary stereogenic center complex A which would cyclize concurrently via Henry reaction to give the product 3 and regenerates the catalyst d. The absolute configuration of 3g was determined by X-ray analysis (see Supporting Information File 1, Figure S1).
![[1860-5397-13-131-i3]](/bjoc/content/inline/1860-5397-13-131-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
We have developed a highly efficient and practical strategy for a single step construction of saturated spirocyclopentaneoxindoles containing four consecutive stereocenters and a unique quaternary stereogenic center, with good yields, and excellent diastereoselectivity and enantioselectivity using thiourea-catalyzed Michael–Henry cascade reactions. We anticipate that this reaction will simplify the synthesis of complex spirooxindoles containing multiple chiral centers with potential pharmacological properties.
Experimental
General procedure for the synthesis of products (3a–j): To a mixture of 1a–j (0.11 mmol) and 2a (0.1 mmol) in CH2Cl2 (3 mL) was added catalyst d (0.01 mmol). Then the mixture was stirred at −20 °C for 12 h. After completion of the reaction, the solvent was removed by evaporation. The crude product was purified by column chromatography on silica gel to afford the desired products 3a–j.
General procedure for the synthesis of products (3k–o): 3-Substituted oxindoles 1k–o (0.11 mmol) and nitrovinylacetamide (2a, 0.1 mmol) were dissolved in 3 mL CH2Cl2, the catalyst d (0.01 mmol) was added at −10 °C for 24 h. After nitrovinylacetamide (2a) was consumed completely, the solvent was removed under vacuum. The crude product was purified by column chromatography on silica gel to afford the desired products 3k–o.
Supporting Information
| Supporting Information File 1: General information, experimental details, characterization data and copies of 1H and 13C NMR spectra, and HPLC experimental data. | ||
| Format: PDF | Size: 4.1 MB | Download |
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (81620108027, 21632008, 91229204 and 81220108025) the Major Project of Chinese National Programs for Fundamental Research and Development (2015CB910304), and National S&T Major Projects (2014ZX09507002-001).
References
-
Sharma, N.; Li, Z.; Sharma, U. K.; Van der Eycken, E. V. Org. Lett. 2014, 16, 3884–3887. doi:10.1021/ol5019079
Return to citation in text: [1] -
Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/C2OB25184A
Return to citation in text: [1] [2] -
Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156–1171. doi:10.1021/ar2000343
Return to citation in text: [1] -
Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161
Return to citation in text: [1] [2] -
Rottmann, M.; McNamara, C.; Yeung, B. K. S.; Lee, M. C. S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D. M.; Dharia, N. V.; Tan, J.; Cohen, S. B.; Spencer, K. R.; González-Paez, G. E.; Lakshminarayana, S. B.; Goh, A.; Suwanarusk, R.; Jegla, T.; Schmitt, E. K.; Beck, H.-P.; Brun, R.; Nosten, F.; Renia, L.; Dartois, V.; Keller, T. H.; Fidock, D. A.; Winzeler, E. A.; Diagana, T. T. Science 2010, 329, 1175–1180. doi:10.1126/science.1193225
Return to citation in text: [1] -
Antonchick, A. P.; Gerding-Reimers, C.; Catarinella, M.; Schuermann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Nat. Chem. 2010, 2, 735–740. doi:10.1038/nchem.730
Return to citation in text: [1] -
Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R. S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; Bernard, D.; Zhang, J.; Lu, Y.; Gu, Q.; Shah, R. B.; Pienta, K. J.; Ling, X.; Kang, S.; Guo, M.; Sun, Y.; Yang, D.; Wang, S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3933–3938. doi:10.1073/pnas.0708917105
Return to citation in text: [1] -
Periyasami, G.; Raghunathan, R.; Surendiran, G.; Mathivanan, N. Bioorg. Med. Chem. Lett. 2008, 18, 2342–2345. doi:10.1016/j.bmcl.2008.02.065
Return to citation in text: [1] -
Mohr, J. T.; Krout, M. R.; Stoltz, B. M. Nature 2008, 455, 323–332. doi:10.1038/nature07370
Return to citation in text: [1] -
Fensome, A.; Adams, W. R.; Adams, A. L.; Berrodin, T. J.; Cohen, J.; Huselton, C.; Illenberger, A.; Kern, J. C.; Hudak, V. A.; Marella, M. A.; Melenski, E. G.; McComas, C. C.; Mugford, C. A.; Slayden, O. D.; Yudt, M.; Zhang, Z.; Zhang, P.; Zhu, Y.; Winneker, R. C.; Wrobel, J. E. J. Med. Chem. 2008, 51, 1861–1873. doi:10.1021/jm701080t
Return to citation in text: [1] -
Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342
Return to citation in text: [1] [2] -
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Mercado-Marin, E. V.; Garcia-Reynaga, P.; Romminger, S.; Pimenta, E. F.; Romney, D. K.; Lodewyk, M. W.; Williams, D. E.; Andersen, R. J.; Miller, S. J.; Tantillo, D. J.; Berlinck, R. G. S.; Sarpong, R. Nature 2014, 509, 318–324. doi:10.1038/nature13273
Return to citation in text: [1] -
Pimenta, E. F.; Vita-Marques, A. M.; Tininis, A.; Seleghim, M. H. R.; Sette, L. D.; Veloso, K.; Ferreira, A. G.; Williams, D. E.; Patrick, B. O.; Dalisay, D. S.; Andersen, R. J.; Berlinck, R. G. S. J. Nat. Prod. 2010, 73, 1821–1832. doi:10.1021/np100470h
Return to citation in text: [1] -
McIver, A. L.; Deiters, A. Org. Lett. 2010, 12, 1288–1291. doi:10.1021/ol100177u
Return to citation in text: [1] -
Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975
Return to citation in text: [1] -
Greshock, T. J.; Grubbs, A. W.; Jiao, P.; Wicklow, D. T.; Gloer, J. B.; Williams, R. M. Angew. Chem., Int. Ed. 2008, 47, 3573–3577. doi:10.1002/anie.200800106
Return to citation in text: [1] -
Mugishima, T.; Tsuda, M.; Kasai, Y.; Ishiyama, H.; Fukushi, E.; Kawabata, J.; Watanabe, M.; Akao, K.; Kobayashi, J. J. Org. Chem. 2005, 70, 9430–9435. doi:10.1021/jo051499o
Return to citation in text: [1] -
Bond, R. F.; Boeyens, J. C. A.; Holzapfel, C. W.; Steyn, P. S. J. Chem. Soc., Perkin Trans. 1 1979, 1751–1761. doi:10.1039/p19790001751
Return to citation in text: [1] -
Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r
Return to citation in text: [1] -
Zhou, B.; Yang, Y.; Shi, J.; Luo, Z.; Li, Y. J. Org. Chem. 2013, 78, 2897–2907. doi:10.1021/jo302655u
Return to citation in text: [1] -
Shi, Y.; Lin, A.; Mao, H.; Mao, Z.; Li, W.; Hu, H.; Zhu, C.; Cheng, Y. Chem. – Eur. J. 2013, 19, 1914–1918. doi:10.1002/chem.201202937
Return to citation in text: [1] -
Albertshofer, K.; Tan, B.; Barbas, C. F., III. Org. Lett. 2012, 14, 1834–1837. doi:10.1021/ol300441z
Return to citation in text: [1] [2] [3] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w
Return to citation in text: [1] [2] -
Sun, W.; Zhu, G.; Wu, C.; Hong, L.; Wang, R. Chem. – Eur. J. 2012, 18, 13959–13963. doi:10.1002/chem.201201976
Return to citation in text: [1] -
Li, Y.-M.; Li, X.; Peng, F.-Z.; Li, Z.-Q.; Wu, S.-T.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6200–6203. doi:10.1021/ol202624f
Return to citation in text: [1] [2] [3] -
Trost, B. M.; Cramer, N.; Silverman, S. M. J. Am. Chem. Soc. 2007, 129, 12396–12397. doi:10.1021/ja075335w
Return to citation in text: [1] -
Corkey, B. K.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 2764–2765. doi:10.1021/ja068723r
Return to citation in text: [1] -
Parthasarathy, K.; Praveen, C.; Jeyaveeran, J. C.; Prince, A. A. M. Bioorg. Med. Chem. Lett. 2016, 26, 4310–4317. doi:10.1016/j.bmcl.2016.07.036
Return to citation in text: [1] -
Arai, T.; Miyazaki, T.; Ogawa, H.; Masu, H. Org. Lett. 2016, 18, 5824–5827. doi:10.1021/acs.orglett.6b02783
Return to citation in text: [1] -
Dhara, K.; Mandal, T.; Das, J.; Dash, J. Angew. Chem., Int. Ed. 2015, 54, 15831–15835. doi:10.1002/anie.201508746
Return to citation in text: [1] -
Ding, L.-Z.; Zhong, T.-S.; Wu, H.; Wang, Y.-M. Eur. J. Org. Chem. 2014, 5139–5143. doi:10.1002/ejoc.201402687
Return to citation in text: [1] -
Albertshofer, K.; Anderson, K. E.; Barbas, C. F., III. Org. Lett. 2012, 14, 5968–5971. doi:10.1021/ol302876c
Return to citation in text: [1] [2] -
Zhang, S. L.; Xie, H.-X.; Zhu, J.; Li, H.; Zhang, X.-S.; Li, J.; Wang, W. Nat. Commun. 2011, 2, 211. doi:10.1038/ncomms1214
Return to citation in text: [1] -
Wang, Y.; Liu, L.; Zhang, T.; Zhong, N.-J.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2012, 77, 4143–4147. doi:10.1021/jo3002535
Return to citation in text: [1] -
Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r
Return to citation in text: [1] [2] -
Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094
Return to citation in text: [1] [2] -
Voituriez, A.; Pinto, N.; Neel, M.; Retailleau, P.; Marinetti, A. Chem. – Eur. J. 2010, 16, 12541–12544. doi:10.1002/chem.201001791
Return to citation in text: [1] -
Pinto, N.; Neel, M.; Panossian, A.; Retailleau, P.; Frison, G.; Voituriez, A.; Marinetti, A. Chem. – Eur. J. 2010, 16, 1033–1045. doi:10.1002/chem.200901893
Return to citation in text: [1] -
Zhang, J.; Cao, D.; Wang, H.; Zheng, C.; Zhao, G.; Shang, Y. J. Org. Chem. 2016, 81, 10558–10568. doi:10.1021/acs.joc.6b01553
Return to citation in text: [1] -
Zhang, X.-Y.; Shen, Z.; Hu, L.-L.; Wang, L.-J.; Lin, Y.-S.; Xie, J.-W.; Cui, H.-L. Tetrahedron Lett. 2016, 57, 3790–3794. doi:10.1016/j.tetlet.2016.07.035
Return to citation in text: [1] -
Voituriez, A.; Marinetti, A.; Gicquel, M. Synlett 2015, 26, 142–166. doi:10.1055/s-0034-1379251
Return to citation in text: [1] -
Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h
Return to citation in text: [1] -
Jiang, K.; Tiwari, B.; Chi, Y. R. Org. Lett. 2012, 14, 2382–2385. doi:10.1021/ol3008028
Return to citation in text: [1] -
Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415
Return to citation in text: [1] -
Menon, R. S.; Biju, A. T.; Nair, V. Chem. Soc. Rev. 2015, 44, 5040–5052. doi:10.1039/c5cs00162e
Return to citation in text: [1] -
Tan, B.; Candeias, N. R.; Barbas, C. F., III. Nat. Chem. 2011, 3, 473–477. doi:10.1038/nchem.1039
Return to citation in text: [1] -
Li, X.; Li, Y.-M.; Peng, F.-Z.; Wu, S.-T.; Li, Z.-Q.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6160–6163. doi:10.1021/ol2024955
Return to citation in text: [1] [2] -
Zhu, S.; Yu, S.; Ma, D. Angew. Chem., Int. Ed. 2008, 47, 545–548. doi:10.1002/anie.200704161
Return to citation in text: [1] -
Wiesner, M.; Revell, J. D.; Wennemers, H. Angew. Chem., Int. Ed. 2008, 47, 1871–1874. doi:10.1002/anie.200704972
Return to citation in text: [1] -
Wiesner, M.; Revell, J. D.; Tonazzi, S.; Wennemers, H. J. Am. Chem. Soc. 2008, 130, 5610–5611. doi:10.1021/ja801027s
Return to citation in text: [1] -
Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Angew. Chem., Int. Ed. 2008, 47, 4722–4724. doi:10.1002/anie.200801130
Return to citation in text: [1] -
García-García, P.; Ladépêche, A.; Halder, R.; List, B. Angew. Chem., Int. Ed. 2008, 47, 4719–4721. doi:10.1002/anie.200800847
Return to citation in text: [1] -
McCooey, S. H.; Connon, S. J. Org. Lett. 2007, 9, 599–602. doi:10.1021/ol0628006
Return to citation in text: [1] -
Xu, Y.; Zou, W.; Sundén, H.; Ibrahem, S.; Córdova, A. Adv. Synth. Catal. 2006, 348, 418–424. doi:10.1002/adsc.200505373
Return to citation in text: [1] -
Wang, J.; Li, H.; Lou, B.; Zu, L.; Guo, H.; Wang, W. Chem. – Eur. J. 2006, 12, 4321–4332. doi:10.1002/chem.200600115
Return to citation in text: [1] -
Palomo, C.; Vera, S.; Mielgo, A.; Gómez-Bengoa, E. Angew. Chem., Int. Ed. 2006, 45, 5984–5987. doi:10.1002/anie.200602207
Return to citation in text: [1] -
Mossé, S.; Alexakis, A. Org. Lett. 2006, 8, 3577–3580. doi:10.1021/ol0614727
Return to citation in text: [1] -
Luo, S.; Mi, X.; Zhang, L.; Liu, S.; Xu, H.; Cheng, J.-P. Angew. Chem., Int. Ed. 2006, 45, 3093–3097. doi:10.1002/anie.200600048
Return to citation in text: [1] -
Wang, W.; Wang, J.; Li, H. Angew. Chem., Int. Ed. 2005, 44, 1369–1371. doi:10.1002/anie.200461959
Return to citation in text: [1] -
Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. doi:10.1002/anie.200500599
Return to citation in text: [1] -
Andrey, O.; Alexakis, A.; Tomassini, A.; Bernardinelli, G. Adv. Synth. Catal. 2004, 346, 1147–1168. doi:10.1002/adsc.200404037
Return to citation in text: [1] -
Cai, H.; Zhou, Y.; Zhang, D.; Xu, J.; Liu, H. Chem. Commun. 2014, 50, 14771–14774. doi:10.1039/c4cc06000h
Return to citation in text: [1] -
Rong, X.; Yao, H.; Xia, W.; Du, Y.; Zhou, Y.; Liu, H. ACS Comb. Sci. 2016, 18, 220–224. doi:10.1021/acscombsci.5b00197
Return to citation in text: [1] -
Chen, X.; Chen, H.; Ji, X.; Jiang, H.; Yao, Z.-J.; Liu, H. Org. Lett. 2013, 15, 1846–1849. doi:10.1021/ol4004542
Return to citation in text: [1] -
Yao, H.; Zhou, Y.; Chen, X.; Zhang, P.; Xu, J.; Liu, H. J. Org. Chem. 2016, 81, 8888–8899. doi:10.1021/acs.joc.6b01596
Return to citation in text: [1] [2] -
Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119–125. doi:10.1021/ja044370p
Return to citation in text: [1]
| 69. | Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119–125. doi:10.1021/ja044370p |
| 1. | Sharma, N.; Li, Z.; Sharma, U. K.; Van der Eycken, E. V. Org. Lett. 2014, 16, 3884–3887. doi:10.1021/ol5019079 |
| 2. | Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/C2OB25184A |
| 3. | Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156–1171. doi:10.1021/ar2000343 |
| 4. | Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161 |
| 5. | Rottmann, M.; McNamara, C.; Yeung, B. K. S.; Lee, M. C. S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D. M.; Dharia, N. V.; Tan, J.; Cohen, S. B.; Spencer, K. R.; González-Paez, G. E.; Lakshminarayana, S. B.; Goh, A.; Suwanarusk, R.; Jegla, T.; Schmitt, E. K.; Beck, H.-P.; Brun, R.; Nosten, F.; Renia, L.; Dartois, V.; Keller, T. H.; Fidock, D. A.; Winzeler, E. A.; Diagana, T. T. Science 2010, 329, 1175–1180. doi:10.1126/science.1193225 |
| 6. | Antonchick, A. P.; Gerding-Reimers, C.; Catarinella, M.; Schuermann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Nat. Chem. 2010, 2, 735–740. doi:10.1038/nchem.730 |
| 7. | Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R. S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; Bernard, D.; Zhang, J.; Lu, Y.; Gu, Q.; Shah, R. B.; Pienta, K. J.; Ling, X.; Kang, S.; Guo, M.; Sun, Y.; Yang, D.; Wang, S. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3933–3938. doi:10.1073/pnas.0708917105 |
| 8. | Periyasami, G.; Raghunathan, R.; Surendiran, G.; Mathivanan, N. Bioorg. Med. Chem. Lett. 2008, 18, 2342–2345. doi:10.1016/j.bmcl.2008.02.065 |
| 9. | Mohr, J. T.; Krout, M. R.; Stoltz, B. M. Nature 2008, 455, 323–332. doi:10.1038/nature07370 |
| 10. | Fensome, A.; Adams, W. R.; Adams, A. L.; Berrodin, T. J.; Cohen, J.; Huselton, C.; Illenberger, A.; Kern, J. C.; Hudak, V. A.; Marella, M. A.; Melenski, E. G.; McComas, C. C.; Mugford, C. A.; Slayden, O. D.; Yudt, M.; Zhang, Z.; Zhang, P.; Zhu, Y.; Winneker, R. C.; Wrobel, J. E. J. Med. Chem. 2008, 51, 1861–1873. doi:10.1021/jm701080t |
| 11. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 12. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050 |
| 13. | Mercado-Marin, E. V.; Garcia-Reynaga, P.; Romminger, S.; Pimenta, E. F.; Romney, D. K.; Lodewyk, M. W.; Williams, D. E.; Andersen, R. J.; Miller, S. J.; Tantillo, D. J.; Berlinck, R. G. S.; Sarpong, R. Nature 2014, 509, 318–324. doi:10.1038/nature13273 |
| 14. | Pimenta, E. F.; Vita-Marques, A. M.; Tininis, A.; Seleghim, M. H. R.; Sette, L. D.; Veloso, K.; Ferreira, A. G.; Williams, D. E.; Patrick, B. O.; Dalisay, D. S.; Andersen, R. J.; Berlinck, R. G. S. J. Nat. Prod. 2010, 73, 1821–1832. doi:10.1021/np100470h |
| 15. | McIver, A. L.; Deiters, A. Org. Lett. 2010, 12, 1288–1291. doi:10.1021/ol100177u |
| 16. | Trost, B. M.; Brennan, M. K. Synthesis 2009, 3003–3025. doi:10.1055/s-0029-1216975 |
| 17. | Greshock, T. J.; Grubbs, A. W.; Jiao, P.; Wicklow, D. T.; Gloer, J. B.; Williams, R. M. Angew. Chem., Int. Ed. 2008, 47, 3573–3577. doi:10.1002/anie.200800106 |
| 18. | Mugishima, T.; Tsuda, M.; Kasai, Y.; Ishiyama, H.; Fukushi, E.; Kawabata, J.; Watanabe, M.; Akao, K.; Kobayashi, J. J. Org. Chem. 2005, 70, 9430–9435. doi:10.1021/jo051499o |
| 19. | Bond, R. F.; Boeyens, J. C. A.; Holzapfel, C. W.; Steyn, P. S. J. Chem. Soc., Perkin Trans. 1 1979, 1751–1761. doi:10.1039/p19790001751 |
| 34. | Ding, L.-Z.; Zhong, T.-S.; Wu, H.; Wang, Y.-M. Eur. J. Org. Chem. 2014, 5139–5143. doi:10.1002/ejoc.201402687 |
| 35. | Albertshofer, K.; Anderson, K. E.; Barbas, C. F., III. Org. Lett. 2012, 14, 5968–5971. doi:10.1021/ol302876c |
| 36. | Zhang, S. L.; Xie, H.-X.; Zhu, J.; Li, H.; Zhang, X.-S.; Li, J.; Wang, W. Nat. Commun. 2011, 2, 211. doi:10.1038/ncomms1214 |
| 65. | Cai, H.; Zhou, Y.; Zhang, D.; Xu, J.; Liu, H. Chem. Commun. 2014, 50, 14771–14774. doi:10.1039/c4cc06000h |
| 66. | Rong, X.; Yao, H.; Xia, W.; Du, Y.; Zhou, Y.; Liu, H. ACS Comb. Sci. 2016, 18, 220–224. doi:10.1021/acscombsci.5b00197 |
| 67. | Chen, X.; Chen, H.; Ji, X.; Jiang, H.; Yao, Z.-J.; Liu, H. Org. Lett. 2013, 15, 1846–1849. doi:10.1021/ol4004542 |
| 68. | Yao, H.; Zhou, Y.; Chen, X.; Zhang, P.; Xu, J.; Liu, H. J. Org. Chem. 2016, 81, 8888–8899. doi:10.1021/acs.joc.6b01596 |
| 27. | Sun, W.; Zhu, G.; Wu, C.; Hong, L.; Wang, R. Chem. – Eur. J. 2012, 18, 13959–13963. doi:10.1002/chem.201201976 |
| 28. | Li, Y.-M.; Li, X.; Peng, F.-Z.; Li, Z.-Q.; Wu, S.-T.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6200–6203. doi:10.1021/ol202624f |
| 29. | Trost, B. M.; Cramer, N.; Silverman, S. M. J. Am. Chem. Soc. 2007, 129, 12396–12397. doi:10.1021/ja075335w |
| 30. | Corkey, B. K.; Toste, F. D. J. Am. Chem. Soc. 2007, 129, 2764–2765. doi:10.1021/ja068723r |
| 31. | Parthasarathy, K.; Praveen, C.; Jeyaveeran, J. C.; Prince, A. A. M. Bioorg. Med. Chem. Lett. 2016, 26, 4310–4317. doi:10.1016/j.bmcl.2016.07.036 |
| 32. | Arai, T.; Miyazaki, T.; Ogawa, H.; Masu, H. Org. Lett. 2016, 18, 5824–5827. doi:10.1021/acs.orglett.6b02783 |
| 33. | Dhara, K.; Mandal, T.; Das, J.; Dash, J. Angew. Chem., Int. Ed. 2015, 54, 15831–15835. doi:10.1002/anie.201508746 |
| 68. | Yao, H.; Zhou, Y.; Chen, X.; Zhang, P.; Xu, J.; Liu, H. J. Org. Chem. 2016, 81, 8888–8899. doi:10.1021/acs.joc.6b01596 |
| 23. | Zhou, B.; Yang, Y.; Shi, J.; Luo, Z.; Li, Y. J. Org. Chem. 2013, 78, 2897–2907. doi:10.1021/jo302655u |
| 24. | Shi, Y.; Lin, A.; Mao, H.; Mao, Z.; Li, W.; Hu, H.; Zhu, C.; Cheng, Y. Chem. – Eur. J. 2013, 19, 1914–1918. doi:10.1002/chem.201202937 |
| 25. | Albertshofer, K.; Tan, B.; Barbas, C. F., III. Org. Lett. 2012, 14, 1834–1837. doi:10.1021/ol300441z |
| 26. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w |
| 25. | Albertshofer, K.; Tan, B.; Barbas, C. F., III. Org. Lett. 2012, 14, 1834–1837. doi:10.1021/ol300441z |
| 2. | Ball-Jones, N. R.; Badillo, J. J.; Franz, A. K. Org. Biomol. Chem. 2012, 10, 5165–5181. doi:10.1039/C2OB25184A |
| 4. | Zhou, F.; Liu, Y.-L.; Zhou, J. Adv. Synth. Catal. 2010, 352, 1381–1407. doi:10.1002/adsc.201000161 |
| 11. | Galliford, C. V.; Scheidt, K. A. Angew. Chem., Int. Ed. 2007, 46, 8748–8758. doi:10.1002/anie.200701342 |
| 20. | Hong, L.; Wang, R. Adv. Synth. Catal. 2013, 355, 1023–1052. doi:10.1002/adsc.201200808 |
| 21. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 22. | Cheng, D.; Ishihara, Y.; Tan, B.; Barbas, C. F., III. ACS Catal. 2014, 4, 743–762. doi:10.1021/cs401172r |
| 50. | Li, X.; Li, Y.-M.; Peng, F.-Z.; Wu, S.-T.; Li, Z.-Q.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6160–6163. doi:10.1021/ol2024955 |
| 25. | Albertshofer, K.; Tan, B.; Barbas, C. F., III. Org. Lett. 2012, 14, 1834–1837. doi:10.1021/ol300441z |
| 28. | Li, Y.-M.; Li, X.; Peng, F.-Z.; Li, Z.-Q.; Wu, S.-T.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6200–6203. doi:10.1021/ol202624f |
| 49. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. Nat. Chem. 2011, 3, 473–477. doi:10.1038/nchem.1039 |
| 50. | Li, X.; Li, Y.-M.; Peng, F.-Z.; Wu, S.-T.; Li, Z.-Q.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6160–6163. doi:10.1021/ol2024955 |
| 28. | Li, Y.-M.; Li, X.; Peng, F.-Z.; Li, Z.-Q.; Wu, S.-T.; Sun, Z.-W.; Zhang, H.-B.; Shao, Z.-H. Org. Lett. 2011, 13, 6200–6203. doi:10.1021/ol202624f |
| 38. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 39. | Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094 |
| 46. | Jiang, K.; Tiwari, B.; Chi, Y. R. Org. Lett. 2012, 14, 2382–2385. doi:10.1021/ol3008028 |
| 47. | Grossmann, A.; Enders, D. Angew. Chem., Int. Ed. 2012, 51, 314–325. doi:10.1002/anie.201105415 |
| 48. | Menon, R. S.; Biju, A. T.; Nair, V. Chem. Soc. Rev. 2015, 44, 5040–5052. doi:10.1039/c5cs00162e |
| 35. | Albertshofer, K.; Anderson, K. E.; Barbas, C. F., III. Org. Lett. 2012, 14, 5968–5971. doi:10.1021/ol302876c |
| 45. | Peng, J.; Huang, X.; Jiang, L.; Cui, H.-L.; Chen, Y.-C. Org. Lett. 2011, 13, 4584–4587. doi:10.1021/ol201776h |
| 26. | Tan, B.; Candeias, N. R.; Barbas, C. F., III. J. Am. Chem. Soc. 2011, 133, 4672–4675. doi:10.1021/ja110147w |
| 37. | Wang, Y.; Liu, L.; Zhang, T.; Zhong, N.-J.; Wang, D.; Chen, Y.-J. J. Org. Chem. 2012, 77, 4143–4147. doi:10.1021/jo3002535 |
| 38. | Doyle, A. G.; Jacobsen, E. N. Chem. Rev. 2007, 107, 5713–5743. doi:10.1021/cr068373r |
| 39. | Zhong, F.; Han, X.; Wang, Y.; Lu, Y. Angew. Chem., Int. Ed. 2011, 50, 7837–7841. doi:10.1002/anie.201102094 |
| 40. | Voituriez, A.; Pinto, N.; Neel, M.; Retailleau, P.; Marinetti, A. Chem. – Eur. J. 2010, 16, 12541–12544. doi:10.1002/chem.201001791 |
| 41. | Pinto, N.; Neel, M.; Panossian, A.; Retailleau, P.; Frison, G.; Voituriez, A.; Marinetti, A. Chem. – Eur. J. 2010, 16, 1033–1045. doi:10.1002/chem.200901893 |
| 42. | Zhang, J.; Cao, D.; Wang, H.; Zheng, C.; Zhao, G.; Shang, Y. J. Org. Chem. 2016, 81, 10558–10568. doi:10.1021/acs.joc.6b01553 |
| 43. | Zhang, X.-Y.; Shen, Z.; Hu, L.-L.; Wang, L.-J.; Lin, Y.-S.; Xie, J.-W.; Cui, H.-L. Tetrahedron Lett. 2016, 57, 3790–3794. doi:10.1016/j.tetlet.2016.07.035 |
| 44. | Voituriez, A.; Marinetti, A.; Gicquel, M. Synlett 2015, 26, 142–166. doi:10.1055/s-0034-1379251 |
| 51. | Zhu, S.; Yu, S.; Ma, D. Angew. Chem., Int. Ed. 2008, 47, 545–548. doi:10.1002/anie.200704161 |
| 52. | Wiesner, M.; Revell, J. D.; Wennemers, H. Angew. Chem., Int. Ed. 2008, 47, 1871–1874. doi:10.1002/anie.200704972 |
| 53. | Wiesner, M.; Revell, J. D.; Tonazzi, S.; Wennemers, H. J. Am. Chem. Soc. 2008, 130, 5610–5611. doi:10.1021/ja801027s |
| 54. | Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Angew. Chem., Int. Ed. 2008, 47, 4722–4724. doi:10.1002/anie.200801130 |
| 55. | García-García, P.; Ladépêche, A.; Halder, R.; List, B. Angew. Chem., Int. Ed. 2008, 47, 4719–4721. doi:10.1002/anie.200800847 |
| 56. | McCooey, S. H.; Connon, S. J. Org. Lett. 2007, 9, 599–602. doi:10.1021/ol0628006 |
| 57. | Xu, Y.; Zou, W.; Sundén, H.; Ibrahem, S.; Córdova, A. Adv. Synth. Catal. 2006, 348, 418–424. doi:10.1002/adsc.200505373 |
| 58. | Wang, J.; Li, H.; Lou, B.; Zu, L.; Guo, H.; Wang, W. Chem. – Eur. J. 2006, 12, 4321–4332. doi:10.1002/chem.200600115 |
| 59. | Palomo, C.; Vera, S.; Mielgo, A.; Gómez-Bengoa, E. Angew. Chem., Int. Ed. 2006, 45, 5984–5987. doi:10.1002/anie.200602207 |
| 60. | Mossé, S.; Alexakis, A. Org. Lett. 2006, 8, 3577–3580. doi:10.1021/ol0614727 |
| 61. | Luo, S.; Mi, X.; Zhang, L.; Liu, S.; Xu, H.; Cheng, J.-P. Angew. Chem., Int. Ed. 2006, 45, 3093–3097. doi:10.1002/anie.200600048 |
| 62. | Wang, W.; Wang, J.; Li, H. Angew. Chem., Int. Ed. 2005, 44, 1369–1371. doi:10.1002/anie.200461959 |
| 63. | Hayashi, Y.; Gotoh, H.; Hayashi, T.; Shoji, M. Angew. Chem., Int. Ed. 2005, 44, 4212–4215. doi:10.1002/anie.200500599 |
| 64. | Andrey, O.; Alexakis, A.; Tomassini, A.; Bernardinelli, G. Adv. Synth. Catal. 2004, 346, 1147–1168. doi:10.1002/adsc.200404037 |
© 2017 Du et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)