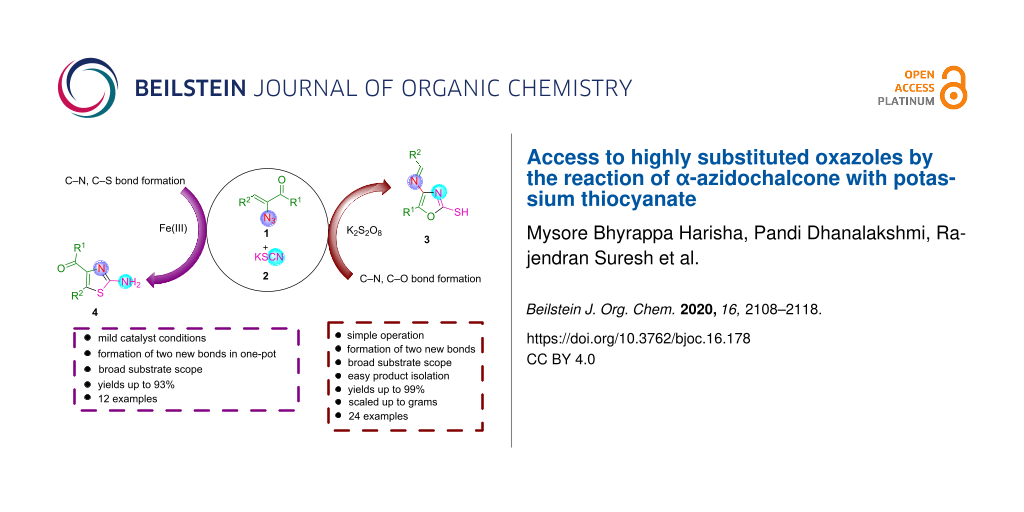
Abstract
The reactivity of α-azidochalcones has been explored for the preparation of highly substituted oxazoles via a 2H-azirine intermediate. The azidochalcones, when treated with potassium thiocyanate in the presence of potassium persulfate, lead to 2,4,5-trisubstituted oxazoles in good yields. Incidentally, 2-aminothiazoles are the products when ferric nitrate is employed instead of persulfate in the above reaction.

Graphical Abstract
Introduction
Vinyl azide is one of the most versatile and potent building blocks explored in the synthesis of several heterocycles [1-5]. It can undergo photolysis or thermolysis to afford highly strained three-membered 2H-azirine, which can act as the precursor for nitrogen heterocycles. As a part of our synthetic design towards the construction of five-membered heterocycles, we have previously reported the synthesis of highly substituted imidazoles [6], indoles [7] and pyrroles [8] starting from different azidochalcones. In continuation, employing α-azidochalcones as the precursor [9], we herein report the preparation of highly substituted oxazoles and thiazoles.
Oxazoles are ubiquitously found in various natural products [10-14], pharmaceuticals [15-18], functional materials [19,20] as well as in several organic building blocks [21-26]. Some oxazoles play a significant role in biological properties such as TRPV1 antagonistical activity, antifungal, analgesic, anti-inflammatory, antiproliferative, antileukemia, anticancer [27-32] and enzyme inhibitory activities [33-42]. 2,4,5-Trisubstituted oxazoles are embedded in some natural products and pharmaceuticals with a broad range of biological activities prompting the development of efficient synthetic strategies for this useful heterocycle [43,44] (Figure 1).
![[1860-5397-16-178-1]](/bjoc/content/figures/1860-5397-16-178-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of biologically active oxazole and aminothiazole scaffolds.
Figure 1: Examples of biologically active oxazole and aminothiazole scaffolds.
In the recent past, the readily accessible 2H-azirine, an efficient source of nitrogen, was employed as starting material for the synthesis of oxazole with various coupling partners such as aldehyde, trifluoroacetic acid, etc. [8,45-50] (Scheme 1).
![[1860-5397-16-178-i1]](/bjoc/content/inline/1860-5397-16-178-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Strategies for the synthesis of 2,4,5-trisubstituted oxazole from azirine. a) I2, PPh3; b) NaH, 1H-pyrazole; c) 2-bromoacetyl bromide, NaN3; d) heating; e) t-BuOK; f) Ph-CHO, visible light; g) KSCN, K2S2O8.
Scheme 1: Strategies for the synthesis of 2,4,5-trisubstituted oxazole from azirine. a) I2, PPh3; b) NaH, 1H-...
Thiazole is a common structural motif that is found in a wide variety of naturally existing alkaloids and a number of pharmaceutically active compounds [51-53]. 2-Aminothiazole has a thiourea-like character with a tendency to modulate promiscuously multiple biological targets. Thiazole derivatives also exhibit a broad spectrum of biological activities including antiviral, antiprion, anti-inflammatory, antimicrobial, anitubercular, psychotropic and anticancer [54-60]. The marketed cancer drug dasatinib [61] continues to prove its worth.
In this work, it is shown that highly substituted oxazoles and aminothiazoles could be accessed directly from the reaction of substituted α-azidochalcones with potassium thiocyanate. Thiocyanate is a known ambident reagent with two potential sites of attack, enabling the selective and efficient construction of C–C and C–N bonds towards biologically important heterocyclic skeletons [62-64].
Results and Discussion
Previously, we have reported the TMSOTf-catalyzed synthesis of highly substituted imidazoles from α-azidochalcones under mild conditions [65]. As a sequel, the synthesis of oxazoles with an arylimino substituent has been accomplished in this work. The biologically important arylimino group [66-69] integrated with a highly substituted oxazole skeleton with a thiol group is expected to have potential synergetic bioactivity [70].
During the exploration of the reactivity of azidochalcones with thiocyanate in the presence of the oxidizing agent, 1i was chosen as the model α-azidochalcone to react with potassium thiocyanate 2 in the presence of several oxidants and metal salts (Table 1). The initial attempts employing iodine, CAN and ZnCl2 upon refluxing in acetonitrile for 6 hours did not yield any product (Table 1, entries 1, 2 and 4).
Table 1: Optimisation studies.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-178-i9.svg?max-width=637&scale=1.0)
|
||||
| Entry | Catalyst | Solvent | Conditions | Yieldb |
| 1 | I2 | CH3CN | KSCN, reflux, 6 h | nrc |
| 2 | CAN | CH3CN | KSCN, reflux, 6 h | nr |
| 3 | FeCl3 | CH3CN | KSCN, reflux, 6 h | –d |
| 4 | ZnCl2 | CH3CN | KSCN, reflux, 6 h | nr |
| 5 | K2S2O8 (0.5 equiv) | CH3CN | KSCN, reflux, 6 h | 97e |
| 6 | K2S2O8 (1 equiv) | CH3CN | KSCN, reflux, 6h | 96 |
| 7 | K2S2O8 (0.1 equiv) | CH3CN | KSCN, reflux, 6 h | 85 |
| 8 | K2S2O8 | CH3CN | KSCN, rt, 24 h | nr |
| 9 | – | CH3CN | KSCN, reflux, 6 h | nr |
| 10 | K2S2O8 | ethanol | KSCN, reflux, 6 h | nr |
| 11 | K2S2O8 | MeOH | KSCN, reflux, 6 h | nr |
| 12 | K2S2O8 | water | KSCN, reflux, 6 h | nr |
| 13 | K2S2O8 | 1,4-dioxan | KSCN, reflux, 6 h | nr |
| 14 | K2S2O8 | THF | KSCN, reflux, 6 h | nr |
| 15 | K2S2O8 | toluene | KSCN, reflux, 6 h | nr |
| 16 | K2S2O8 | DMF | KSCN, 120 °C, 6 h | nr |
| 17 | K2S2O8 | DCE | KSCN, reflux, 6 h | nr |
| 18e | K2S2O8 | CH3CN | NH4SCN, reflux, 6 h | 65 |
| 19e | K2S2O8 | CH3CN | NH4SCN, rt, 18 h | nr |
| 20e | K2S2O8 | DCE | NH4SCN, 90 °C, 6 h | nr |
aReaction conditions: α-azidochalcone 1 (1 equiv), potassium thiocyanate 2 (3 equiv), oxidant/metal salt (0.5 equiv) in various solvents (2 mL); bisolated yield after recrystallization; cno reaction; disolated product was identified as 2-aminothiazole; ereaction conditions: α-azidochalcone 1i (1 equiv), ammonium thiocyanate 2a (2 equiv), potassium persulfate (0.5 equiv) solvent (2 mL).
When potassium persulfate (K2S2O8) is employed, highly substituted oxazole 3i has been obtained. With the observation that potassium persulfate can efficiently catalyze the reaction to furnish highly substituted oxazole 3i, we carried out the reaction of 1i and 2 in acetonitrile in the presence of various equivalents of potassium persulfate, in an attempt to evaluate the catalytic efficiency of persulfate. The reaction was found occurring most efficiently with 1 equiv of 1i, 3 equiv of potassium thiocyanate 2, and 0.5 equiv of potassium persulfate with a yield of 97% of 3i. The yellow solid obtained after filtration of the reaction mixture afforded pure product 3i without the requirement of any further work-up or purification protocol. The analytically pure sample was obtained by recrystallization from cold diethyl ether. However, in the absence of potassium persulfate (Table 1, entry 9), no product was observed indicating that potassium persulfate is essential to facilitate the reaction. After determining the optimal amount of persulfate, we have examined various solvents (Table 1, entries 10–17) to study the outcome of the reaction. These solvent screening studies indicated that acetonitrile is a suitable solvent for this reaction. Having established conditions for the high-yielding synthesis of oxazole 3, the scope of this transformation with various α-azidochalcones was explored (Scheme 2).
![[1860-5397-16-178-i2]](/bjoc/content/inline/1860-5397-16-178-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the α-azidochalcones. The reactions were carried out at reflux temperature, using 1 (1 mmol), 2 (3 mmol), potassium persulfate (0.5 mmol) in acetonitrile (2 mL) for 6 h. Yields refer to pure products after recrystallization.
Scheme 2: Scope of the α-azidochalcones. The reactions were carried out at reflux temperature, using 1 (1 mmo...
As shown in Scheme 2, an efficient conversion of α-azidochalcones 1a–u to highly substituted functionalized oxazoles 3a–u has been achieved with both electron-poor and electron-rich aryl substituents. Both nitro- and bromo-substituted systems can be further functionalized to get a new set of products. The tolerance of the reaction for a variety of aryl substituents illustrates the generality of this method for the preparation of a range of highly substituted oxazoles. Further, the scalability of the reaction using an optimized protocol was investigated by conducting the reaction on a multigram scale (Scheme 3). It was found that the reaction between 1i and 2 on a multigram scale proceeded to afford 3i in 97% yield.
![[1860-5397-16-178-i3]](/bjoc/content/inline/1860-5397-16-178-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
In Figure 2, photograph a shows the reaction mixture just at the start of the reaction and b is the photograph after the completion of the reaction.
![[1860-5397-16-178-2]](/bjoc/content/figures/1860-5397-16-178-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Large-scale synthesis of 3i. a) At the start of the reaction, b) after the reaction.
Figure 2: Large-scale synthesis of 3i. a) At the start of the reaction, b) after the reaction.
Further, the utility of the thiol group in 3 for the generation of a library of compounds was demonstrated by the simple acetylation and alkylation (Scheme 4 and Scheme 5). The acetylation of the thiol group in 3d proceeded smoothly with acetyl chloride in the presence of sodium hydride to afford 5 in good yield.
![[1860-5397-16-178-i4]](/bjoc/content/inline/1860-5397-16-178-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The structure of the product and the site of acetylation was confirmed by X-ray crystallography of a single crystal of 5 [71] (Figure 3).
![[1860-5397-16-178-3]](/bjoc/content/figures/1860-5397-16-178-3.png?scale=2.0&max-width=1024&background=FFFFFF)
The methylated and benzylated derivatives 6 and 7 were also obtained from 3m. S-Methylation of 3m was achieved in 91% yield with methyl iodide in the presence of NaH/THF and the S-benzylation has been carried out by a similar procedure (Scheme 5).
![[1860-5397-16-178-i5]](/bjoc/content/inline/1860-5397-16-178-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of S-methyl/benzylated products 6 and 7.
Scheme 5: Synthesis of S-methyl/benzylated products 6 and 7.
To derive the mechanism of the reaction, a few control experiments have been executed (Scheme 6). Initially, the reaction of 1m with potassium thiocyanate 2 under the optimal conditions in the presence of TEMPO furnished azirine and TEMPO, while the same reaction in the presence of BHT afforded the BHT-coupled thiocyanate product (SO-(2,6-di-tert-butyl-4-methylphenyl) (thioperoxocyanate)). These observations unambiguously indicate that the reaction proceeds through a radical pathway. Potassium persulfate helps to generate a thiocyanate radical and in the absence of potassium persulfate the reaction did not proceed. This experiment supports the role of potassium persulfate as an oxidant.
![[1860-5397-16-178-i6]](/bjoc/content/inline/1860-5397-16-178-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Based on these experiments, the following plausible mechanism for the formation of 2,4,5-trisubstituted oxazoles can be proposed (Scheme 7). It is known that the thiocyanate radical is generated from potassium thiocyanate by the reaction with potassium persulfate [72]. The N-end of thiocyanate radical reacts with the C=N bond to give the intermediate A which undergoes homolytic cleavage yielding B. Subsequent cyclisation results in the oxazole ring.
![[1860-5397-16-178-i7]](/bjoc/content/inline/1860-5397-16-178-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Plausible mechanism proposed for the formation of 2,4,5-trisubstituted oxazoles 3.
Scheme 7: Plausible mechanism proposed for the formation of 2,4,5-trisubstituted oxazoles 3.
During these optimization trials, it was interesting to note the formation of 2-aminothiazole, when ferric chloride was employed along with thiocyanate (Table 1, entry 3). There is a report pertaining to this transformation with Fe(II) salts [73]. We further wanted to capitalize on this result and optimize the methodology to access a series of 2-aminothiazoles as the reported methods [74-79] to access 2,4,5-trisubstituted aminothiazoles, especially 4-aroyl-2-amiothiazoles, suffered from low yields, harsh reaction conditions, expensive and detrimental metal precursors as well as the pollution concerning α-halocarbonyl compounds.
Initially, we started the reaction with (Z)-2-azido-1,3-bis(4-chlorophenyl)prop-2-en-1-one (1d) and commercially available potassium thiocyanate (2) as a representative model system. To optimize the best reaction condition, we began this study by performing the reaction with ferric chloride in acetonitrile at 80 °C for 6 h (Table 2, entry 1).
Table 2: Screening of iron salts and solventsa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-178-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | Catalyst | Temp | Time [h] | Yieldb [%] |
| 1 | CH3CN | FeCl3 | reflux | 6 | 85 |
| 2 | DCE | FeCl3 | reflux | 5 | nr |
| 3 | toluene | FeCl3 | reflux | 5 | nr |
| 4 | CH3CN | FeCl3 | rt | 18 | nr |
| 5 | CH3CN | Fe2O3 | reflux | 6 | 10 |
| 6 | CH3CN | Fe(NO3)3 | reflux | 3 | 60 |
| 7 | CH3CN | Fe(NO3)3 | reflux | 6 | 93 |
| 8 | DCE | Fe(NO3)3 | reflux | 6 | nr |
| 9 | THF | Fe(NO3)3 | reflux | 6 | nr |
| 10 | CH3CN | K3(Fe)(CN)6 | reflux | 6 | nr |
aReaction conditions: azidochalcone 1 (1 equiv), potassium thiocyanate 2 (3 equiv), Fe(III) (0.5 equiv), solvent (2 mL). bIsolated yield after column chromatography.
This reaction has led to the exclusive formation of 4,5-disubstituted 2-aminothiazole 4d. The catalytic activities of different Fe(III) salts and the solvents were screened in the reaction. When ferric chloride was employed in DCE or toluene, the expected product was not obtained (Table 2, entries 2 and 3). The reaction failed to proceed in acetonitrile at room temperature also (Table 2, entry 4). As a further variation, we examined Fe2O3 as catalyst in acetonitrile which resulted only in 10% conversion (Table 2, entry 5). Further screening was performed with ferric nitrate and the 2-aminothiazole was obtained in 60% yield (Table 2, entry 6). The product was obtained in excellent yield when the reaction mixture was heated for 6 h (Table 2, entry 7). Potassium ferricyanide has also been proved ineffective (Table 2, entry 10).
Using the optimized experimental conditions, the Fe(III)-mediated formation of 4,5-disubstituted 2-aminothiazoles 4 was examined for the substrate scope (Scheme 8).
![[1860-5397-16-178-i8]](/bjoc/content/inline/1860-5397-16-178-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Reaction of vinyl azide 1 and 3 with ferric nitrate. Reactions were carried out at reflux temperature, using 1 (1 mmol), 2 (3 mmol), ferric nitrate (0.5 mmol) in acetonitrile (2 mL) for 6 h. Yields refer to the pure products after column chromatography.
Scheme 8: Reaction of vinyl azide 1 and 3 with ferric nitrate. Reactions were carried out at reflux temperatu...
All the synthesized compounds 4a–l were confirmed by 1D and 2D NMR, IR spectroscopy and HRMS techniques. Additional evidence of the structures of these compounds was obtained based on the single-crystal X-ray analysis of 4h [71] (Figure 4).
![[1860-5397-16-178-4]](/bjoc/content/figures/1860-5397-16-178-4.png?scale=2.0&max-width=1024&background=FFFFFF)
The nitrogen end of the thiocyanate attacks the azirine to form the oxazole ring and the sulfur end of the thiocyanate involves in the reaction resulting in the 2-aminothiazole ring. The mechanism for the formation of 4 may be similar to that suggested earlier [73].
Conclusion
In conclusion, we have demonstrated selective routes for the synthesis of highly substituted oxazoles and 2-aminothiazoles from α-azidochalcones and potassium thiocyanate employing potassium persulfate and ferric nitrate, respectively. This new route gains a streamlined workup and the elimination of air-sensitive techniques to afford the product in good yield in a greener medium over a short time frame. The current method involves a broad substrate scope, excellent functional group tolerance and leaves the active site for further synthetic transformation. The overall strategy allows the generation of new C–N and C–O bonds in one-pot.
Experimental
General considerations: The melting points reported in the work are uncorrected. Unless stated otherwise, solvents and chemicals were obtained from commercial sources and used without further purification. Infrared spectra were recorded on a Perkin Elmer instrument with neat sample and only major peaks are reported in cm−1. The 1H and 13C NMR spectra of the new compounds were measured at 400 MHz and 100 MHz, respectively, using Bruker and JEOL NMR instruments in DMSO-d6. Chemical shifts are reported in parts per million (δ), coupling constants (J values) are reported in Hertz (Hz) relative to tetramethylsilane. Spin multiplicities are indicated by the following symbols: s (singlet), d (doublet), t (triplet), m (multiplet), dd (doublet of doublets), td (triplet of doublets), ddd (doublet of doublets of doublets), bs (broad singlet). Mass spectra were measured with Micromass Q-Time of flight (HRESIMS).
General procedure for the preparation of 3: To a solution of azidochalcone 1 (1 mmol) in dry acetonitrile (2 mL) were added potassium thiocyanate 2 (3 mmol) and potassium persulfate (0.5 mmol). The mixture was stirred magnetically at reflux temperature for 6 hours under nitrogen atmosphere. After completion of the reaction (monitored by TLC), the solid that separated was filtered, washed with water and acetonitrile and recrystallized with cold diethyl ether to obtain pure yellow product 3.
General procedure for the preparation of 4: To a solution of azidochalcone 1 (1 mmol) in dry acetonitrile (2 mL) were added potassium thiocyanate 2 (3 mmol) and ferric nitrate (0.5 mmol). The reaction mixture was stirred magnetically at reflux for 6 h. After completion of the reaction (monitored by TLC), the product was diluted with water, extracted with ethyl acetate (15 mL) and purified by column chromatography (100–200 mesh silica gel) using ethyl acetate/petroleum ether mixture to afford product 4.
Supporting Information
| Supporting Information File 1: Full experimental details, compound characterisation, and copies of NMR spectra. | ||
| Format: PDF | Size: 7.1 MB | Download |
Funding
M. B. H. is grateful to Eurofins-Advinus Limited, Bangalore for support. P. D. thanks the Science and Engineering Research Board (SERB), India, for a National Post-Doctoral Fellowship (FILE NO.PDF/2018/000099). S. M. thanks CSIR, New Delhi for the award of Emeritus Scientist Scheme (21(1030)/16/EMR-II dated 18-11-2016).
References
-
L'abbé, G. Angew. Chem., Int. Ed. Engl. 1975, 14, 775–782. doi:10.1002/anie.197507751
Angew. Chemie. 1975, 87, 831–838. doi:10.1002/ange.19750872304
Return to citation in text: [1] -
Chiba, S. Chimia 2012, 66, 377–381. doi:10.2533/chimia.2012.377
Return to citation in text: [1] -
Jung, N.; Bräse, S. Angew. Chem., Int. Ed. 2012, 51, 12169–12171. doi:10.1002/anie.201206409
Angew. Chem. 2012, 124, 12335–12337. doi:10.1002/ange.201206409
Return to citation in text: [1] -
Hayashi, H.; Kaga, A.; Chiba, S. J. Org. Chem. 2017, 82, 11981–11989. doi:10.1021/acs.joc.7b02455
Return to citation in text: [1] -
Hu, B.; DiMagno, S. G. Org. Biomol. Chem. 2015, 13, 3844–3855. doi:10.1039/c5ob00099h
Return to citation in text: [1] -
Rajaguru, K.; Suresh, R.; Mariappan, A.; Muthusubramanian, S.; Bhuvanesh, N. Org. Lett. 2014, 16, 744–747. doi:10.1021/ol403456b
Return to citation in text: [1] -
Rajaguru, K.; Mariappan, A.; Muthusubramanian, S.; Bhuvanesh, N. Org. Chem. Front. 2017, 4, 124–129. doi:10.1039/c6qo00541a
Return to citation in text: [1] -
Suresh, R.; Muthusubramanian, S.; Nagaraj, M.; Manickam, G. Tetrahedron Lett. 2013, 54, 1779–1784. doi:10.1016/j.tetlet.2012.11.065
Return to citation in text: [1] [2] -
Rajaguru, K.; Mariappan, A.; Suresh, R.; Manivannan, P.; Muthusubramanian, S. Beilstein J. Org. Chem. 2015, 11, 2021–2028. doi:10.3762/bjoc.11.219
Return to citation in text: [1] -
Tilvi, S.; Singh, K. S. Curr. Org. Chem. 2016, 20, 898–929. doi:10.2174/1385272819666150804000046
Return to citation in text: [1] -
Wipf, P. Chem. Rev. 1995, 95, 2115–2134. doi:10.1021/cr00038a013
Return to citation in text: [1] -
Shin-ya, K.; Wierzba, K.; Matsuo, K.-i.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. J. Am. Chem. Soc. 2001, 123, 1262–1263. doi:10.1021/ja005780q
Return to citation in text: [1] -
Dalisay, D. S.; Rogers, E. W.; Edison, A. S.; Molinski, T. F. J. Nat. Prod. 2009, 72, 732–738. doi:10.1021/np8007649
Return to citation in text: [1] -
Wilson, Z. E.; Fenner, S.; Ley, S. V. Angew. Chem., Int. Ed. 2015, 54, 1284–1288. doi:10.1002/anie.201410063
Angew. Chem. 2015, 127, 1300–1304. doi:10.1002/ange.201410063
Return to citation in text: [1] -
Joshi, S.; Bisht, A. S.; Juyal, D. Pharma Innovation 2017, 6, 109–117.
Return to citation in text: [1] -
Fernandes, E.; Costa, D.; Toste, S. A.; Lima, J. L. F. C.; Reis, S. Free Radical Biol. Med. 2004, 37, 1895–1905. doi:10.1016/j.freeradbiomed.2004.09.001
Return to citation in text: [1] -
Wang, W.-L.; Yao, D.-Y.; Gu, M.; Fan, M.-Z.; Li, J.-Y.; Xing, Y.-C.; Nan, F.-J. Bioorg. Med. Chem. Lett. 2005, 15, 5284–5287. doi:10.1016/j.bmcl.2005.08.046
Return to citation in text: [1] -
Reddy, B. A.; Hymavathi, R. V.; Swamy, G. N. J. Chem. Sci. 2013, 125, 495–509. doi:10.1007/s12039-013-0417-7
Return to citation in text: [1] -
Baranov, M. S.; Lukyanov, K. A.; Ivashkin, P. E.; Yampolsky, I. V. Synth. Commun. 2013, 43, 2337–2342. doi:10.1080/00397911.2012.706350
Return to citation in text: [1] -
Huong, V. T. T.; Tai, T. B.; Nguyen, M. T. J. Phys. Chem. A 2014, 118, 3335–3343. doi:10.1021/jp500899k
Return to citation in text: [1] -
Boyd, G. V. Oxazoles and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon: Oxford, UK, 1984; Vol. 6, pp 177–233. doi:10.1016/b978-008096519-2.00086-2
Return to citation in text: [1] -
Hartner, F. W., Jr. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: Oxford, UK, 1996; Vol. 3, pp 261–318.
Return to citation in text: [1] -
Knight, D. W. Oxazole and its Derivatives. In Heterocycles in natural product synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 403–458. doi:10.1002/9783527634880.ch12
Return to citation in text: [1] -
Revuelta, J.; Machetti, F.; Cicchi, S. Five‐Membered Heterocycles: 1,3‐Azoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J.; Vaquero, J. J.; Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; Vol. 2, pp 809–923. doi:10.1002/9783527637737.ch10
Return to citation in text: [1] -
Jin, Z. Nat. Prod. Rep. 2011, 28, 1143–1191. doi:10.1039/c0np00074d
Return to citation in text: [1] -
Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084–3213. doi:10.1021/cr300333u
Return to citation in text: [1] -
Perner, R. J.; Koenig, J. R.; DiDomenico, S.; Gomtsyan, A.; Schmidt, R. G.; Lee, C.-H.; Hsu, M. C.; McDonald, H. A.; Gauvin, D. M.; Joshi, S.; Turner, T. M.; Reilly, R. M.; Kym, P. R.; Kort, M. E. Bioorg. Med. Chem. 2010, 18, 4821–4829. doi:10.1016/j.bmc.2010.04.099
Return to citation in text: [1] -
Ryu, C.-K.; Lee, R.-Y.; Kim, N. Y.; Kim, Y. H.; Song, A. L. Bioorg. Med. Chem. Lett. 2009, 19, 5924–5926. doi:10.1016/j.bmcl.2009.08.062
Return to citation in text: [1] -
Argade, N. D.; Kalrale, B. K.; Gill, C. H. E-J. Chem. 2008, 5, 120–129. doi:10.1155/2008/265131
Return to citation in text: [1] -
Merla, B.; Oberboersch, S.; Sundermann, B.; Englberger, W.; Hennies, H. H.; Graubaum, H. Substituted oxazole compounds with analgesic activity. U.S. Patent US7608619B2, Oct 27, 2009.
Return to citation in text: [1] -
Kean, W. F. Curr. Med. Res. Opin. 2004, 20, 1275–1277. doi:10.1185/030079904125004420
Return to citation in text: [1] -
Liu, X.-H.; Lv, P.-C.; Xue, J.-Y.; Song, B.-A.; Zhu, H.-L. Eur. J. Med. Chem. 2009, 44, 3930–3935. doi:10.1016/j.ejmech.2009.04.019
Return to citation in text: [1] -
Desroy, N.; Moreau, F.; Briet, S.; Fralliec, G. L.; Floquet, S.; Durant, L.; Vongsouthi, V.; Gerusz, V.; Denis, A.; Escaich, S. Bioorg. Med. Chem. 2009, 17, 1276–1289. doi:10.1016/j.bmc.2008.12.021
Return to citation in text: [1] -
Heng, S.; Gryncel, K. R.; Kantrowitz, E. R. Bioorg. Med. Chem. 2009, 17, 3916–3922. doi:10.1016/j.bmc.2009.04.030
Return to citation in text: [1] -
Copp, B. R. Nat. Prod. Rep. 2003, 20, 535–557. doi:10.1039/b212154a
Return to citation in text: [1] -
Lack, N. A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F. Q.; Chan, K. H.; Feau, C.; LeBlanc, E.; Guns, E. T.; Guy, R. K.; Rennie, P. S.; Cherkasov, A. J. Med. Chem. 2011, 54, 8563–8573. doi:10.1021/jm201098n
Return to citation in text: [1] -
Hashimoto, H.; Imamura, K.; Haruta, J.-i.; Wakitani, K. J. Med. Chem. 2002, 45, 1511–1517. doi:10.1021/jm010484p
Return to citation in text: [1] -
Momose, Y.; Maekawa, T.; Yamano, T.; Kawada, M.; Odaka, H.; Ikeda, H.; Sohda, T. J. Med. Chem. 2002, 45, 1518–1534. doi:10.1021/jm010490l
Return to citation in text: [1] -
Brown, P.; Davies, D. T.; O'Hanlo, P. J.; Wilson, J. M. J. Med. Chem. 1996, 39, 446–457. doi:10.1021/jm9503862
Return to citation in text: [1] -
Yang, W. S.; Shimada, K.; Delva, D.; Patel, M.; Ode, E.; Skouta, R.; Stockwell, B. R. ACS Med. Chem. Lett. 2012, 3, 35–38. doi:10.1021/ml200195s
Return to citation in text: [1] -
Shaw, A. Y.; Henderson, M. C.; Flynn, G.; Samulitis, B.; Han, H.; Stratton, S. P.; Chow, H.-H. S.; Hurley, L. H.; Dorr, R. T. J. Pharmacol. Exp. Ther. 2009, 331, 636–647. doi:10.1124/jpet.109.156406
Return to citation in text: [1] -
Giddens, A. C.; Boshoff, H. I. M.; Franzblau, S. G.; Barry, C. E., III; Copp, B. R. Tetrahedron Lett. 2005, 46, 7355–7357. doi:10.1016/j.tetlet.2005.08.119
Return to citation in text: [1] -
Yamada, K.; Kamimura, N.; Kunishima, M. Beilstein J. Org. Chem. 2017, 13, 1478–1485. doi:10.3762/bjoc.13.146
Return to citation in text: [1] -
Ibrar, A.; Khan, I.; Abbas, N.; Farooq, U.; Khan, A. RSC Adv. 2016, 6, 93016–93047. doi:10.1039/c6ra19324b
And references cited therein.
Return to citation in text: [1] -
Ning, Y.; Otani, Y.; Ohwada, T. J. Org. Chem. 2017, 82, 6313–6326. doi:10.1021/acs.joc.7b00904
Return to citation in text: [1] -
Lopes, S.; Nunes, C. M.; Fausto, R.; Pinho e Melo, T. M. V. D. J. Mol. Struct. 2009, 919, 47–53. doi:10.1016/j.molstruc.2008.08.014
Return to citation in text: [1] -
Duan, X.; Yang, K.; Lu, J.; Kong, X.; Liu, N.; Ma, J. Org. Lett. 2017, 19, 3370–3373. doi:10.1021/acs.orglett.7b01305
Return to citation in text: [1] -
Zeng, T.-T.; Xuan, J.; Ding, W.; Wang, K.; Lu, L.-Q.; Xiao, W.-J. Org. Lett. 2015, 17, 4070–4073. doi:10.1021/acs.orglett.5b01994
Return to citation in text: [1] -
Xie, H.; Yuan, D.; Ding, M.-W. J. Org. Chem. 2012, 77, 2954–2958. doi:10.1021/jo202588j
Return to citation in text: [1] -
Rossa, T. A.; Suveges, N. S.; Sá, M. M.; Cantillo, D.; Kappe, C. O. Beilstein J. Org. Chem. 2018, 14, 506–514. doi:10.3762/bjoc.14.36
Return to citation in text: [1] -
Metzger, J. V. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon: New York, NY, USA, 1984; Vol. 6, pp 235–332.
Return to citation in text: [1] -
Lewis, J. R. Nat. Prod. Rep. 1999, 16, 389–416. doi:10.1039/a802500b
Return to citation in text: [1] -
Fink, B. E.; Mortensen, D. S.; Stauffer, S. R.; Aron, Z. D.; Katzenellenbogen, J. A. Chem. Biol. 1999, 6, 205–219. doi:10.1016/s1074-5521(99)80037-4
Return to citation in text: [1] -
Liu, R.; Huang, Z.; Murray, M. G.; Guo, X.; Liu, G. J. Med. Chem. 2011, 54, 5747–5768. doi:10.1021/jm200394x
Return to citation in text: [1] -
Gallardo-Godoy, A.; Gever, J.; Fife, K. L.; Silber, B. M.; Prusiner, S. B.; Renslo, A. R. J. Med. Chem. 2011, 54, 1010–1021. doi:10.1021/jm101250y
Return to citation in text: [1] -
Suh, J.; Yum, E. K.; Cheon, H. G.; Cho, Y. S. Chem. Biol. Drug Des. 2012, 80, 89–98. doi:10.1111/j.1747-0285.2012.01371.x
Return to citation in text: [1] -
Annadurai, S.; Martinez, R.; Canney, D. J.; Eidem, T.; Dunman, P. M.; Abou-Gharbia, M. Bioorg. Med. Chem. Lett. 2012, 22, 7719–7725. doi:10.1016/j.bmcl.2012.09.095
Return to citation in text: [1] -
Roy, K. K.; Singh, S.; Sharma, S. K.; Srivastava, R.; Chaturvedi, V.; Saxena, A. K. Bioorg. Med. Chem. Lett. 2011, 21, 5589–5593. doi:10.1016/j.bmcl.2011.06.076
Return to citation in text: [1] -
Zablotskaya, A.; Segal, I.; Germane, S.; Shestakova, I.; Domracheva, I.; Nesterova, A.; Geronikaki, A.; Lukevics, E. Chem. Heterocycl. Compd. 2002, 38, 859–866. doi:10.1023/a:1020698107686
Return to citation in text: [1] -
Romagnoli, R.; Baraldi, P. G.; Salvador, M. K.; Camacho, M. E.; Preti, D.; Tabrizi, M. A.; Bassetto, M.; Brancale, A.; Hamel, E.; Bortolozzi, R.; Basso, G.; Viola, G. Bioorg. Med. Chem. 2012, 20, 7083–7094. doi:10.1016/j.bmc.2012.10.001
Return to citation in text: [1] -
Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R. V.; Shen, Z.; Cook, L. S.; Doweyko, A. M.; Pitt, S.; Pang, S.; Shen, D. R.; Fang, Q.; de Fex, H. F.; McIntyre, K. W.; Shuster, D. J.; Gillooly, K. M.; Behnia, K.; Schieven, G. L.; Wityak, J.; Barrish, J. C. J. Med. Chem. 2006, 49, 6819–6832. doi:10.1021/jm060727j
Return to citation in text: [1] -
Schranck, J.; Tlili, A.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 7642–7644. doi:10.1002/anie.201303015
Return to citation in text: [1] -
Vincent, G. Cycloadditions with Stereoselective C–N Bond Formation in Total Syntheses. In tereoselective Synthesis of Drugs and Natural Products; Andrushko, V.; Andrushko, N., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; Vol. 2, pp 1251 ff. doi:10.1002/9781118596784.ssd041
Return to citation in text: [1] -
Lamberth, C.; Dinges, J. The Significance of Heterocycles for Pharmaceuticals and Agrochemicals. In Bioactive Heterocyclic Compound Classes: Pharmaceuticals; Lamberth, C.; Dinges, J., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp 1–20. doi:10.1002/9783527664412.ch1
Return to citation in text: [1] -
Harisha, M. B.; Dhanalakshmi, P.; Suresh, R.; Kumar, R. R.; Muthusubramanian, S.; Bhuvanesh, N. ChemistrySelect 2019, 4, 2954–2958. doi:10.1002/slct.201801543
Return to citation in text: [1] -
Qin, W.; Long, S.; Panunzio, M.; Biondi, S. Molecules 2013, 18, 12264–12289. doi:10.3390/molecules181012264
Return to citation in text: [1] -
Dhar, D. N.; Taploo, C. L. J. Sci. Ind. Res. 1982, 41, 501–506.
Return to citation in text: [1] -
Przybylski, P.; Huczynski, A.; Pyta, K.; Brzezinski, B.; Bartl, F. Curr. Org. Chem. 2009, 13, 124–148. doi:10.2174/138527209787193774
Return to citation in text: [1] -
da Silva, C. M.; da Silva, D. L.; Modolo, L. V.; Alves, R. B.; de Resende, M. A.; Martins, C. V. B.; de Fátima, Â. J. Adv. Res. 2011, 2, 1–8. doi:10.1016/j.jare.2010.05.004
Return to citation in text: [1] -
Velik, J.; Baliharova, V.; Skalova, L.; Szotakova, B.; Wsol, V.; Lamka, J. J. Vet. Pharmacol. Ther. 2003, 26, 297–302. doi:10.1046/j.1365-2885.2003.00484.x
Return to citation in text: [1] -
CCDC deposits 1959141 (4h) and 1959142 (5) contain the supplementary crystallographic data for this article, which can be obtained free of charge from the Cambridge Crystallographic Data Centre.
Return to citation in text: [1] [2] -
Yang, D.; Yan, K.; Wei, W.; Li, G.; Lu, S.; Zhao, C.; Tian, L.; Wang, H. J. Org. Chem. 2015, 80, 11073–11079. doi:10.1021/acs.joc.5b01637
Return to citation in text: [1] -
Zhang, G.; Chen, B.; Guo, X.; Guo, S.; Yu, Y. Adv. Synth. Catal. 2015, 357, 1065–1069. doi:10.1002/adsc.201400856
Return to citation in text: [1] [2] -
Xu, Z.; Ba, M.; Zhou, H.; Cao, Y.; Tang, C.; Yang, Y.; He, R.; Liang, Y.; Zhang, X.; Li, Z.; Zhu, L.; Guo, Y.; Guo, C. Eur. J. Med. Chem. 2014, 85, 27–42. doi:10.1016/j.ejmech.2014.07.072
Return to citation in text: [1] -
Zhu, D.; Chen, J.; Xiao, H.; Liu, M.; Ding, J.; Wu, H. Synth. Commun. 2009, 39, 2895–2906. doi:10.1080/00397910802691874
Return to citation in text: [1] -
Jalani, H. B.; Pandya, A. N.; Pandya, D. H.; Sharma, J. A.; Sudarsanam, V.; Vasu, K. K. Tetrahedron Lett. 2013, 54, 5403–5406. doi:10.1016/j.tetlet.2013.07.122
Return to citation in text: [1] -
Zhu, Y.-P.; Yuan, J.-J.; Zhao, Q.; Lian, M.; Gao, Q.-H.; Liu, M.-C.; Yang, Y.; Wu, A.-X. Tetrahedron 2012, 68, 173–178. doi:10.1016/j.tet.2011.10.074
Return to citation in text: [1] -
Sinha, S.; Doble, M.; Manju, S. L. Eur. J. Med. Chem. 2018, 158, 34–50. doi:10.1016/j.ejmech.2018.08.098
Return to citation in text: [1] -
Darji, N. D.; Pasha, T. Y.; Bhandari, A.; Molvi, K. I.; Desai, S. A.; Makwana, M. V. Pharma Chem. 2012, 4, 808–812.
Return to citation in text: [1]
| 1. |
L'abbé, G. Angew. Chem., Int. Ed. Engl. 1975, 14, 775–782. doi:10.1002/anie.197507751
Angew. Chemie. 1975, 87, 831–838. doi:10.1002/ange.19750872304 |
| 2. | Chiba, S. Chimia 2012, 66, 377–381. doi:10.2533/chimia.2012.377 |
| 3. |
Jung, N.; Bräse, S. Angew. Chem., Int. Ed. 2012, 51, 12169–12171. doi:10.1002/anie.201206409
Angew. Chem. 2012, 124, 12335–12337. doi:10.1002/ange.201206409 |
| 4. | Hayashi, H.; Kaga, A.; Chiba, S. J. Org. Chem. 2017, 82, 11981–11989. doi:10.1021/acs.joc.7b02455 |
| 5. | Hu, B.; DiMagno, S. G. Org. Biomol. Chem. 2015, 13, 3844–3855. doi:10.1039/c5ob00099h |
| 9. | Rajaguru, K.; Mariappan, A.; Suresh, R.; Manivannan, P.; Muthusubramanian, S. Beilstein J. Org. Chem. 2015, 11, 2021–2028. doi:10.3762/bjoc.11.219 |
| 54. | Liu, R.; Huang, Z.; Murray, M. G.; Guo, X.; Liu, G. J. Med. Chem. 2011, 54, 5747–5768. doi:10.1021/jm200394x |
| 55. | Gallardo-Godoy, A.; Gever, J.; Fife, K. L.; Silber, B. M.; Prusiner, S. B.; Renslo, A. R. J. Med. Chem. 2011, 54, 1010–1021. doi:10.1021/jm101250y |
| 56. | Suh, J.; Yum, E. K.; Cheon, H. G.; Cho, Y. S. Chem. Biol. Drug Des. 2012, 80, 89–98. doi:10.1111/j.1747-0285.2012.01371.x |
| 57. | Annadurai, S.; Martinez, R.; Canney, D. J.; Eidem, T.; Dunman, P. M.; Abou-Gharbia, M. Bioorg. Med. Chem. Lett. 2012, 22, 7719–7725. doi:10.1016/j.bmcl.2012.09.095 |
| 58. | Roy, K. K.; Singh, S.; Sharma, S. K.; Srivastava, R.; Chaturvedi, V.; Saxena, A. K. Bioorg. Med. Chem. Lett. 2011, 21, 5589–5593. doi:10.1016/j.bmcl.2011.06.076 |
| 59. | Zablotskaya, A.; Segal, I.; Germane, S.; Shestakova, I.; Domracheva, I.; Nesterova, A.; Geronikaki, A.; Lukevics, E. Chem. Heterocycl. Compd. 2002, 38, 859–866. doi:10.1023/a:1020698107686 |
| 60. | Romagnoli, R.; Baraldi, P. G.; Salvador, M. K.; Camacho, M. E.; Preti, D.; Tabrizi, M. A.; Bassetto, M.; Brancale, A.; Hamel, E.; Bortolozzi, R.; Basso, G.; Viola, G. Bioorg. Med. Chem. 2012, 20, 7083–7094. doi:10.1016/j.bmc.2012.10.001 |
| 8. | Suresh, R.; Muthusubramanian, S.; Nagaraj, M.; Manickam, G. Tetrahedron Lett. 2013, 54, 1779–1784. doi:10.1016/j.tetlet.2012.11.065 |
| 61. | Das, J.; Chen, P.; Norris, D.; Padmanabha, R.; Lin, J.; Moquin, R. V.; Shen, Z.; Cook, L. S.; Doweyko, A. M.; Pitt, S.; Pang, S.; Shen, D. R.; Fang, Q.; de Fex, H. F.; McIntyre, K. W.; Shuster, D. J.; Gillooly, K. M.; Behnia, K.; Schieven, G. L.; Wityak, J.; Barrish, J. C. J. Med. Chem. 2006, 49, 6819–6832. doi:10.1021/jm060727j |
| 7. | Rajaguru, K.; Mariappan, A.; Muthusubramanian, S.; Bhuvanesh, N. Org. Chem. Front. 2017, 4, 124–129. doi:10.1039/c6qo00541a |
| 8. | Suresh, R.; Muthusubramanian, S.; Nagaraj, M.; Manickam, G. Tetrahedron Lett. 2013, 54, 1779–1784. doi:10.1016/j.tetlet.2012.11.065 |
| 45. | Ning, Y.; Otani, Y.; Ohwada, T. J. Org. Chem. 2017, 82, 6313–6326. doi:10.1021/acs.joc.7b00904 |
| 46. | Lopes, S.; Nunes, C. M.; Fausto, R.; Pinho e Melo, T. M. V. D. J. Mol. Struct. 2009, 919, 47–53. doi:10.1016/j.molstruc.2008.08.014 |
| 47. | Duan, X.; Yang, K.; Lu, J.; Kong, X.; Liu, N.; Ma, J. Org. Lett. 2017, 19, 3370–3373. doi:10.1021/acs.orglett.7b01305 |
| 48. | Zeng, T.-T.; Xuan, J.; Ding, W.; Wang, K.; Lu, L.-Q.; Xiao, W.-J. Org. Lett. 2015, 17, 4070–4073. doi:10.1021/acs.orglett.5b01994 |
| 49. | Xie, H.; Yuan, D.; Ding, M.-W. J. Org. Chem. 2012, 77, 2954–2958. doi:10.1021/jo202588j |
| 50. | Rossa, T. A.; Suveges, N. S.; Sá, M. M.; Cantillo, D.; Kappe, C. O. Beilstein J. Org. Chem. 2018, 14, 506–514. doi:10.3762/bjoc.14.36 |
| 6. | Rajaguru, K.; Suresh, R.; Mariappan, A.; Muthusubramanian, S.; Bhuvanesh, N. Org. Lett. 2014, 16, 744–747. doi:10.1021/ol403456b |
| 51. | Metzger, J. V. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon: New York, NY, USA, 1984; Vol. 6, pp 235–332. |
| 52. | Lewis, J. R. Nat. Prod. Rep. 1999, 16, 389–416. doi:10.1039/a802500b |
| 53. | Fink, B. E.; Mortensen, D. S.; Stauffer, S. R.; Aron, Z. D.; Katzenellenbogen, J. A. Chem. Biol. 1999, 6, 205–219. doi:10.1016/s1074-5521(99)80037-4 |
| 21. | Boyd, G. V. Oxazoles and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W., Eds.; Pergamon: Oxford, UK, 1984; Vol. 6, pp 177–233. doi:10.1016/b978-008096519-2.00086-2 |
| 22. | Hartner, F. W., Jr. In Comprehensive Heterocyclic Chemistry; Katritzky, A. R.; Rees, C. W.; Scriven, E. F. V., Eds.; Elsevier: Oxford, UK, 1996; Vol. 3, pp 261–318. |
| 23. | Knight, D. W. Oxazole and its Derivatives. In Heterocycles in natural product synthesis; Majumdar, K. C.; Chattopadhyay, S. K., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp 403–458. doi:10.1002/9783527634880.ch12 |
| 24. | Revuelta, J.; Machetti, F.; Cicchi, S. Five‐Membered Heterocycles: 1,3‐Azoles. In Modern Heterocyclic Chemistry; Alvarez-Builla, J.; Vaquero, J. J.; Barluenga, J., Eds.; Wiley-VCH: Weinheim, Germany, 2011; Vol. 2, pp 809–923. doi:10.1002/9783527637737.ch10 |
| 25. | Jin, Z. Nat. Prod. Rep. 2011, 28, 1143–1191. doi:10.1039/c0np00074d |
| 26. | Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084–3213. doi:10.1021/cr300333u |
| 33. | Desroy, N.; Moreau, F.; Briet, S.; Fralliec, G. L.; Floquet, S.; Durant, L.; Vongsouthi, V.; Gerusz, V.; Denis, A.; Escaich, S. Bioorg. Med. Chem. 2009, 17, 1276–1289. doi:10.1016/j.bmc.2008.12.021 |
| 34. | Heng, S.; Gryncel, K. R.; Kantrowitz, E. R. Bioorg. Med. Chem. 2009, 17, 3916–3922. doi:10.1016/j.bmc.2009.04.030 |
| 35. | Copp, B. R. Nat. Prod. Rep. 2003, 20, 535–557. doi:10.1039/b212154a |
| 36. | Lack, N. A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F. Q.; Chan, K. H.; Feau, C.; LeBlanc, E.; Guns, E. T.; Guy, R. K.; Rennie, P. S.; Cherkasov, A. J. Med. Chem. 2011, 54, 8563–8573. doi:10.1021/jm201098n |
| 37. | Hashimoto, H.; Imamura, K.; Haruta, J.-i.; Wakitani, K. J. Med. Chem. 2002, 45, 1511–1517. doi:10.1021/jm010484p |
| 38. | Momose, Y.; Maekawa, T.; Yamano, T.; Kawada, M.; Odaka, H.; Ikeda, H.; Sohda, T. J. Med. Chem. 2002, 45, 1518–1534. doi:10.1021/jm010490l |
| 39. | Brown, P.; Davies, D. T.; O'Hanlo, P. J.; Wilson, J. M. J. Med. Chem. 1996, 39, 446–457. doi:10.1021/jm9503862 |
| 40. | Yang, W. S.; Shimada, K.; Delva, D.; Patel, M.; Ode, E.; Skouta, R.; Stockwell, B. R. ACS Med. Chem. Lett. 2012, 3, 35–38. doi:10.1021/ml200195s |
| 41. | Shaw, A. Y.; Henderson, M. C.; Flynn, G.; Samulitis, B.; Han, H.; Stratton, S. P.; Chow, H.-H. S.; Hurley, L. H.; Dorr, R. T. J. Pharmacol. Exp. Ther. 2009, 331, 636–647. doi:10.1124/jpet.109.156406 |
| 42. | Giddens, A. C.; Boshoff, H. I. M.; Franzblau, S. G.; Barry, C. E., III; Copp, B. R. Tetrahedron Lett. 2005, 46, 7355–7357. doi:10.1016/j.tetlet.2005.08.119 |
| 19. | Baranov, M. S.; Lukyanov, K. A.; Ivashkin, P. E.; Yampolsky, I. V. Synth. Commun. 2013, 43, 2337–2342. doi:10.1080/00397911.2012.706350 |
| 20. | Huong, V. T. T.; Tai, T. B.; Nguyen, M. T. J. Phys. Chem. A 2014, 118, 3335–3343. doi:10.1021/jp500899k |
| 43. | Yamada, K.; Kamimura, N.; Kunishima, M. Beilstein J. Org. Chem. 2017, 13, 1478–1485. doi:10.3762/bjoc.13.146 |
| 44. |
Ibrar, A.; Khan, I.; Abbas, N.; Farooq, U.; Khan, A. RSC Adv. 2016, 6, 93016–93047. doi:10.1039/c6ra19324b
And references cited therein. |
| 15. | Joshi, S.; Bisht, A. S.; Juyal, D. Pharma Innovation 2017, 6, 109–117. |
| 16. | Fernandes, E.; Costa, D.; Toste, S. A.; Lima, J. L. F. C.; Reis, S. Free Radical Biol. Med. 2004, 37, 1895–1905. doi:10.1016/j.freeradbiomed.2004.09.001 |
| 17. | Wang, W.-L.; Yao, D.-Y.; Gu, M.; Fan, M.-Z.; Li, J.-Y.; Xing, Y.-C.; Nan, F.-J. Bioorg. Med. Chem. Lett. 2005, 15, 5284–5287. doi:10.1016/j.bmcl.2005.08.046 |
| 18. | Reddy, B. A.; Hymavathi, R. V.; Swamy, G. N. J. Chem. Sci. 2013, 125, 495–509. doi:10.1007/s12039-013-0417-7 |
| 10. | Tilvi, S.; Singh, K. S. Curr. Org. Chem. 2016, 20, 898–929. doi:10.2174/1385272819666150804000046 |
| 11. | Wipf, P. Chem. Rev. 1995, 95, 2115–2134. doi:10.1021/cr00038a013 |
| 12. | Shin-ya, K.; Wierzba, K.; Matsuo, K.-i.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. J. Am. Chem. Soc. 2001, 123, 1262–1263. doi:10.1021/ja005780q |
| 13. | Dalisay, D. S.; Rogers, E. W.; Edison, A. S.; Molinski, T. F. J. Nat. Prod. 2009, 72, 732–738. doi:10.1021/np8007649 |
| 14. |
Wilson, Z. E.; Fenner, S.; Ley, S. V. Angew. Chem., Int. Ed. 2015, 54, 1284–1288. doi:10.1002/anie.201410063
Angew. Chem. 2015, 127, 1300–1304. doi:10.1002/ange.201410063 |
| 27. | Perner, R. J.; Koenig, J. R.; DiDomenico, S.; Gomtsyan, A.; Schmidt, R. G.; Lee, C.-H.; Hsu, M. C.; McDonald, H. A.; Gauvin, D. M.; Joshi, S.; Turner, T. M.; Reilly, R. M.; Kym, P. R.; Kort, M. E. Bioorg. Med. Chem. 2010, 18, 4821–4829. doi:10.1016/j.bmc.2010.04.099 |
| 28. | Ryu, C.-K.; Lee, R.-Y.; Kim, N. Y.; Kim, Y. H.; Song, A. L. Bioorg. Med. Chem. Lett. 2009, 19, 5924–5926. doi:10.1016/j.bmcl.2009.08.062 |
| 29. | Argade, N. D.; Kalrale, B. K.; Gill, C. H. E-J. Chem. 2008, 5, 120–129. doi:10.1155/2008/265131 |
| 30. | Merla, B.; Oberboersch, S.; Sundermann, B.; Englberger, W.; Hennies, H. H.; Graubaum, H. Substituted oxazole compounds with analgesic activity. U.S. Patent US7608619B2, Oct 27, 2009. |
| 31. | Kean, W. F. Curr. Med. Res. Opin. 2004, 20, 1275–1277. doi:10.1185/030079904125004420 |
| 32. | Liu, X.-H.; Lv, P.-C.; Xue, J.-Y.; Song, B.-A.; Zhu, H.-L. Eur. J. Med. Chem. 2009, 44, 3930–3935. doi:10.1016/j.ejmech.2009.04.019 |
| 66. | Qin, W.; Long, S.; Panunzio, M.; Biondi, S. Molecules 2013, 18, 12264–12289. doi:10.3390/molecules181012264 |
| 67. | Dhar, D. N.; Taploo, C. L. J. Sci. Ind. Res. 1982, 41, 501–506. |
| 68. | Przybylski, P.; Huczynski, A.; Pyta, K.; Brzezinski, B.; Bartl, F. Curr. Org. Chem. 2009, 13, 124–148. doi:10.2174/138527209787193774 |
| 69. | da Silva, C. M.; da Silva, D. L.; Modolo, L. V.; Alves, R. B.; de Resende, M. A.; Martins, C. V. B.; de Fátima, Â. J. Adv. Res. 2011, 2, 1–8. doi:10.1016/j.jare.2010.05.004 |
| 62. | Schranck, J.; Tlili, A.; Beller, M. Angew. Chem., Int. Ed. 2013, 52, 7642–7644. doi:10.1002/anie.201303015 |
| 63. | Vincent, G. Cycloadditions with Stereoselective C–N Bond Formation in Total Syntheses. In tereoselective Synthesis of Drugs and Natural Products; Andrushko, V.; Andrushko, N., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; Vol. 2, pp 1251 ff. doi:10.1002/9781118596784.ssd041 |
| 64. | Lamberth, C.; Dinges, J. The Significance of Heterocycles for Pharmaceuticals and Agrochemicals. In Bioactive Heterocyclic Compound Classes: Pharmaceuticals; Lamberth, C.; Dinges, J., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp 1–20. doi:10.1002/9783527664412.ch1 |
| 65. | Harisha, M. B.; Dhanalakshmi, P.; Suresh, R.; Kumar, R. R.; Muthusubramanian, S.; Bhuvanesh, N. ChemistrySelect 2019, 4, 2954–2958. doi:10.1002/slct.201801543 |
| 73. | Zhang, G.; Chen, B.; Guo, X.; Guo, S.; Yu, Y. Adv. Synth. Catal. 2015, 357, 1065–1069. doi:10.1002/adsc.201400856 |
| 74. | Xu, Z.; Ba, M.; Zhou, H.; Cao, Y.; Tang, C.; Yang, Y.; He, R.; Liang, Y.; Zhang, X.; Li, Z.; Zhu, L.; Guo, Y.; Guo, C. Eur. J. Med. Chem. 2014, 85, 27–42. doi:10.1016/j.ejmech.2014.07.072 |
| 75. | Zhu, D.; Chen, J.; Xiao, H.; Liu, M.; Ding, J.; Wu, H. Synth. Commun. 2009, 39, 2895–2906. doi:10.1080/00397910802691874 |
| 76. | Jalani, H. B.; Pandya, A. N.; Pandya, D. H.; Sharma, J. A.; Sudarsanam, V.; Vasu, K. K. Tetrahedron Lett. 2013, 54, 5403–5406. doi:10.1016/j.tetlet.2013.07.122 |
| 77. | Zhu, Y.-P.; Yuan, J.-J.; Zhao, Q.; Lian, M.; Gao, Q.-H.; Liu, M.-C.; Yang, Y.; Wu, A.-X. Tetrahedron 2012, 68, 173–178. doi:10.1016/j.tet.2011.10.074 |
| 78. | Sinha, S.; Doble, M.; Manju, S. L. Eur. J. Med. Chem. 2018, 158, 34–50. doi:10.1016/j.ejmech.2018.08.098 |
| 79. | Darji, N. D.; Pasha, T. Y.; Bhandari, A.; Molvi, K. I.; Desai, S. A.; Makwana, M. V. Pharma Chem. 2012, 4, 808–812. |
| 71. | CCDC deposits 1959141 (4h) and 1959142 (5) contain the supplementary crystallographic data for this article, which can be obtained free of charge from the Cambridge Crystallographic Data Centre. |
| 72. | Yang, D.; Yan, K.; Wei, W.; Li, G.; Lu, S.; Zhao, C.; Tian, L.; Wang, H. J. Org. Chem. 2015, 80, 11073–11079. doi:10.1021/acs.joc.5b01637 |
| 73. | Zhang, G.; Chen, B.; Guo, X.; Guo, S.; Yu, Y. Adv. Synth. Catal. 2015, 357, 1065–1069. doi:10.1002/adsc.201400856 |
| 70. | Velik, J.; Baliharova, V.; Skalova, L.; Szotakova, B.; Wsol, V.; Lamka, J. J. Vet. Pharmacol. Ther. 2003, 26, 297–302. doi:10.1046/j.1365-2885.2003.00484.x |
| 71. | CCDC deposits 1959141 (4h) and 1959142 (5) contain the supplementary crystallographic data for this article, which can be obtained free of charge from the Cambridge Crystallographic Data Centre. |
© 2020 Harisha et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)