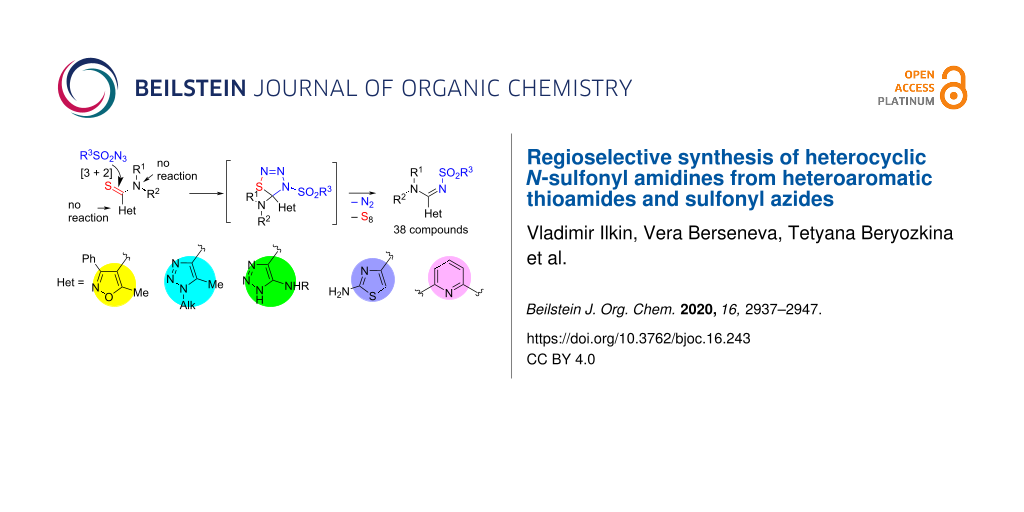
Abstract
N-Sulfonyl amidines bearing 1,2,3-triazole, isoxazole, thiazole and pyridine substituents were successfully prepared for the first time by reactions of primary, secondary and tertiary heterocyclic thioamides with alkyl- and arylsulfonyl azides. For each type of thioamides a reliable procedure to prepare N-sulfonyl amidines in good yields was found. Reactions of 1-aryl-1,2,3-triazole-4-carbothioamides with azides were shown to be accompanied with a Dimroth rearrangement to form 1-unsubstituted 5-arylamino-1,2,3-triazole-4-N-sulfonylcarbimidamides. 2,5-Dithiocarbamoylpyridine reacts with sulfonyl azides to form a pyridine bearing two sulfonyl amidine groups.

Graphical Abstract
Introduction
The biological activity, rich chemistry and technically useful properties of heterocyclic compounds have made them a focal point of science and industry over the years. Heterocyclic compounds including azoles and azines have been found in natural products, and they are included in the structures of nucleic acids, vitamins, antibiotics and in many types of synthetic drugs [1-12]. N-Sulfonyl amidines have received considerable attention because they exhibit various types of pharmaceutical properties and biological activities [13-21] and also have been used as interesting building blocks in organic synthesis [17-20] (Figure 1).
![[1860-5397-16-243-1]](/bjoc/content/figures/1860-5397-16-243-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of biological activity and interesting chemical reactivity of N-sulfonyl amidines.
Figure 1: Examples of biological activity and interesting chemical reactivity of N-sulfonyl amidines.
An N-sulfonyl amidine was recently found to be a key group in acid–base-induced rearrangements of 1,2,3-triazoles and thiadiazoles [22]. A variety of methods have been developed for the synthesis of N-sulfonyl amidines. The most commonly used methods to prepare these compounds include the Cu-catalyzed multicomponent reaction of alkynes, sulfonyl azides and amines [23-31], the reaction of thioacetamide derivatives and cyclic thioamides with sulfonyl azides [22,32,33], the chlorophosphite-mediated Beckmann reaction of oximes with p-toluenesulfonyl azide [34], the sulfonyl ynamide rearrangement by treatment with amines [35], the sodium iodide catalyzed reaction of sulfonamide with formamide [36], and the condensation of sulfonamide derivatives with DMF–DMA [37].
A few representatives of N-sulfonyl amidines of heteroaromatic acids have been prepared and applied [22,32,38-40]. However, no efficient and general method to prepare a series of heterocyclic N-sulfonyl amidines has been elaborated so far. A new approach to N-sulfonyl amidines has been published recently, based on the reaction of thioamides with sulfonyl azides [33,41,42] (Figure 2).
![[1860-5397-16-243-2]](/bjoc/content/figures/1860-5397-16-243-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Data on the synthesis of N′-sulfonylazole-4-carboximidamides.
Figure 2: Data on the synthesis of N′-sulfonylazole-4-carboximidamides.
This method was used successfully for the synthesis of N-sulfonyl amidines of aliphatic acids and benzoic acid, including biologically active compounds. On the other hand, reactions of thioamides with electrophilic reagents have often been used for the synthesis of various types of sulfur containing heterocyclic compounds [43-47]. This gives some promise to the development of a general and efficient method for the synthesis of N-sulfonyl amidines of heteroaromatic acids based on the reaction of heterocyclic thioamides with higly electrophilic sulfonyl azides.
With the purpose of the synthesis of heterocyclic N-sulfonyl amidines bearing various heteroatoms in the ring, namely nitrogen, sulfur and oxygen atoms, we have studied reactions of thioamides of 1,2,3-triazole-, isoxazole-, thiazolecarboxylic acids and 2,5-dithiocarbamoylpyridine with sulfonyl azides. Due to the high dipole moment, the presence of electronegative heteroatoms bearing electron lone pairs, one could propose alternative reactions which might make it difficult to find a general regioselective procedure for the synthesis of the target molecules in good yields. To the best of our knowledge, there are no examples for the synthesis of N-sulfonyl amidines of heteroaromatic acids through this reaction so far.
Results and Discussion
1-Alkyl-1,2,3-triazole-N-sulfonyl amidines
Since 1,2,3-triazole derivatives exhibit valuable biological and technical properties, and take part in various ring transformations and rearrangements [48-51], we decided to study reactions of 1-alkyl-1,2,3-triazole-4-carbothioamides 1a–d with aryl- and alkylsulfonyl azides 2a–f with, with the goal of affecting a “iminosulfonylation” (Scheme 1).
![[1860-5397-16-243-i1]](/bjoc/content/inline/1860-5397-16-243-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 1-alkyl-N-phenyl-N'-(sulfonyl)-1H-1,2,3-triazole-4-carboximidamides 3.
Scheme 1: Synthesis of 1-alkyl-N-phenyl-N'-(sulfonyl)-1H-1,2,3-triazole-4-carboximidamides 3.
The thioamides 1a–d were prepared from the corresponding amides 4a–d by treatment with phosphorus decasulfide (Scheme 1). It is worth noting that amides of 1-alkyl-1,2,3-triazole-4-carboxylic acids are poorly represented in the literature and the methods of their preparation require the addition of alkyl azides to acetylene carboxylic esters and reactions of 2-diazomalonates with aliphatic amines [40,52]. The first approach leads to a mixture of two regioisomers and the second method involves the use of explosive diazo compounds. Therefore, such compounds are better prepared by a recently found method in our laboratory which includes the reaction of 4-acetyl-1,2,3-triazole 5a–d with aniline followed by a Cornforth rearrangement of the 1,2,3-triazole ring [52]. Alkyl- (2a,b) and arylsulfonyl (2c–g) azides were prepared, respectively, from the corresponding sulfonyl chlorides and sodium azides according to published methods (Figure 3) [53].
![[1860-5397-16-243-3]](/bjoc/content/figures/1860-5397-16-243-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
We have found that 1-butyl-1,2,3-triazole-4-carbothioamide (1c) reacts well with benzenesulfonyl azide (2c) in various solvents to form the desired 1-butyl-1,2,3-N-sulfonyl amidine 3n in diverse solvents such as n-butanol, n-propanol, toluene, ethanol, water and even under solvent-free conditions (see Table 1 for the yields and other circumstances).
Table 1: Optimizations of the reaction conditions for the reaction of thioamide 1с with phenylsulfonyl azide 2ca.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-243-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent | T (°C) | 2c (equiv) | t (h) | Yieldb (%) |
| 1 | n-BuOH | 117 | 5 | 21 | 23 |
| 2 | n-BuOH | 100 | 5 | 12.7 | 75 |
| 3 | n-PrOH | 97.4 | 5 | 12.7 | 80 |
| 4 | n-PrOH | 97.4 | 2.5 | 12.7 | 63 |
| 5 | toluene | 105 | 5 | 12.7 | 26 |
| 6 | water | 100 | 5 | 12.7 | 61 |
| 7 | ethanol | 78.4 | 5 | 21 | 74 |
| 8 | neat | 88 | 1.2 | 12.7 | 34 |
| 9 | neat | 88 | 5 | 4 | 86 |
| 10 | neat | 55 | 5 | 24 | 63 |
| 11 | neat | 88 | 2.5 | 5 | 87 |
aReaction conditions: 0.18 mmol of 1c, solvent (1 mL); bisolated yield.
From these data we can conclude that the yield of the final product is optimal for the reaction under solvent-free conditions. 1-Butyl-1,2,3-triazole 1с reacts faster than 1,2,3-triazole-4-carbothioamide 1f while using a lower amount of a sulfonyl azide (Table 1, entry 11 and Table 2, entry 14). Thus solvent-free conditions, a temperature of 88 °C and a thioamide/azide ratio of 1:2.5 are optimal to prepare N-sulfonyl amidine 1c (entry 11, Table 1).
Next, these optimized conditions were used for the synthesis of a small library of 1-alkyl-1,2,3-triazoles 3a–s (Scheme 2).
![[1860-5397-16-243-i2]](/bjoc/content/inline/1860-5397-16-243-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope for the reaction of 1-alkyl-1,2,3-triazole-4-carbothioamides 1a–d with azides 2a–f.
Scheme 2: Scope for the reaction of 1-alkyl-1,2,3-triazole-4-carbothioamides 1a–d with azides 2a–f.
The reaction can be applied without problems to various alkyl substituents in position 1 of the 1,2,3-triazole ring from methyl to decyl and benzyl, goes well with alkylsulfonyl azides and arylsulfonyl azides that were 4-substituted with both electron-withdrawing and electron-donating substituents.
5-Arylamino-1,2,3-triazole-N-sulfonyl amidines
To further expand the scope of the reaction we continued studying the reaction of 1-aryl-1,2,3-triazole-4-carbothioamides 1e–h with aryl- and alkylsulfonyl azides 2a,c,f.
We have found that thioamide 1e did react with benzenesulfonyl azide (2c) neither in water, ethanol nor in the absence of a solvent, conditions that were successfully used in the synthesis of 1-alkyl-1,2,3-triazole-4-N-sulfonylimidamides 3a–s (Scheme 2). On the other hand, we have found the formation of a new product 3t in low yield together with the starting compound 1e and the product of its rearrangement to 5-(4-nitrophenyl)aminotriazole 1j [54], when the reaction was carried out in n-butanol at 105 °C (Table 2). Therefore, we can conclude that compound 3t was the product of a tandem reaction involving first the rearrangement of thioamide 1e to 1j followed by iminosulfonylation of the latter to form amidine 3t (Table 2).
Table 2: Synthesis and optimization of the reaction conditions for the reaction of thioamide 1j with phenylsulfonyl azide (2c)a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-243-i9.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Solvent (additive) | T (°C) | 2c (equiv) | t (h) | Yieldb (%) |
| 1 | n-BuOH | 105 | 1 | 17.5 | 33 |
| 2 | n-BuOH | 105 | 7 | 17.5 | 76 |
| 3 | n-BuOH (Cs2CO3, 10 mol %) | 105 | 5 | 5 | no reaction |
| 4 | n-BuOH (CuI, 10 mol %) | 105 | 5 | 5 | no reaction |
| 5 | n-PrOH | 88 | 5 | 17.5 | 43 |
| 6 | n-PrOH | 97.4 | 5 | 17.5 | 52 |
| 7 | n-PrOH | 97.4 | 7 | 17.5 | 78 |
aReactions conditions: 0.45 mmol of thioamide 1j, solvent (3 mL); bIsolated yield.
To obtain higher yields of sulfonyl amidines we decided to prepare 5-arylamino-1,2,3-triazole-4-carbothioamide 1j by rearrangement of triazole 1e [54] and carried out an optimization with variations of the solvent, temperature and various additives (Table 2). We have shown that optimal conditions include the use of n-propanol, a temperature of 97 °C and a ratio of thioamide 1j and azide 2c of 1:7 which allowed to prepare the desired compound 3t in 78% (Table 2).
With the optimal conditions in hand we prepared a series of N-sulfonyl amidines 3t–aa in good yields (Scheme 3). Thus, a library of N-sulfonyl amidines bearing differently substituted 1,2,3-triazoles was successfully prepared. Among them are compounds bearing an NH-unsubstituted 1,2,3-triazole ring which gives extra possibilities for the modification of the molecules by the reaction with electrophilic reagents to prepare new compounds of this series [55] (Scheme 3).
![[1860-5397-16-243-i3]](/bjoc/content/inline/1860-5397-16-243-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the reaction of 5-arylamino-1,2,3-triazole-4-carbothioamides 1i–l with azides 2a,c–f.
Scheme 3: Scope of the reaction of 5-arylamino-1,2,3-triazole-4-carbothioamides 1i–l with azides 2a,c–f.
To show the practical convenience of the developed method we tried to synthesize these compounds in a one-pot procedure starting from readily available 1-aryl-1,2,3-triazoles 1f,g,t and sulfonyl azides 2c,f (Table 3). Thioamides 1f,g,t were converted to 5-arylamino-1,2,3-triazoles 1i–k by heating at reflux in n-propanol in the presence of DBU and these rearranged thioamides were then treated with sulfonyl azide 2c,f and kept at the same temperature for 17‒31 h. After flash column chromatography, pure N-sulfonyl amidines 3t,u,x were isolated in 41‒65 % yield. The data of Table 3 demonstrates that the yields of sulfonyl amidines 3t,u,x are higher when we used the one-pot protocol in comparison with the two-step method. Furthermore, the one-pot procedure is obviously more simple and less time consuming.
Table 3: Yields of triazoles 3t,u,x following a one-pot procedurea compared to the yields involving the isolation of 5-arylamino-1,2,3-triazoles 1i–k.
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-243-i10.svg?max-width=637&scale=1.0)
|
|||||
| Entry |
Thioamide
1 |
Azide
2 |
Product
3 |
Yield of 3, %
(time) |
|
| one-pot |
with isolation
of 1i–k |
||||
| 1 | 1t | 2f | 3x |
49
(17.5 h) |
41
(via 1i, 27.5 h) |
| 2 | 1g | 2c | 3u |
41
(31 h) |
36
(via 1k, 41 h) |
| 3 | 1f | 2f | 3t |
65
(31 h) |
60
(via 1j, 41 h) |
a1 (0.60‒0.65 mmol), DBU (0.63‒0.65 mmol), 2 (3.56‒4.0 mmol), HOAc (1 mL).
2-Aminothiazole-4-N-sulfonyl amidines
In spite of the presence of a nucleophilic amino group capable to react with sulfonyl azide to form an azide group, the reaction of azides 2 occurred selectively to the thioamide group of compound 1m.
Thus, similar to the reaction of 5-arylamino-1,2,3-triazole-4-carbothioamides 1i–l, the reaction of the primary thioamide of 2-aminothiazole-4-carboxyamide (1m) with sulfonyl azides 2a,c is succesful in n-propanol at reflux temperature, to afford N-sulfonyl amidines 3ab and 3ac bearing a 2-aminothiazole ring in very good yields (Scheme 4).
![[1860-5397-16-243-i4]](/bjoc/content/inline/1860-5397-16-243-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 2-aminothiazole-4-N-sulfonyl amidines.
Scheme 4: Synthesis of 2-aminothiazole-4-N-sulfonyl amidines.
3-Methyl-5-phenyl-isoxazole-4-N-sulfonyl amidines
The primary thioamide 1n containing an isoxazole ring was shown to react with mesyl azide or arylsulfonyl azides in n-propanol at reflux temperature to form the N-sulfonyl amidines 3ad–ag in 49‒76% yields (Scheme 5).
![[1860-5397-16-243-i5]](/bjoc/content/inline/1860-5397-16-243-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of N-sulfonyl amidines of isoxazolylcarboxylic acid.
Scheme 5: Synthesis of N-sulfonyl amidines of isoxazolylcarboxylic acid.
The reaction takes place also in the absence of a solvent, albeit in lower yields. We have found that secondary thioamide 1o does not react with sulfonyl azides 2a,c either in n-propanol or in the absence of a solvent. On the other hand, we have found that the reaction can occur in n-butanol at 118 °C to form compounds 3ah–ai in low yields (38‒45%) accompanied with the formation of tar-like products.
2,5-Bis(N-sulfonylamidino)pyridines
Bis(thioamide) 1p containing a pyridine ring was found to react with sulfonyl azides 2a,c–f either in boiling propanol or in the absence of a solvent to form compounds 3aj–an bearing two N-sulfonyl amidine fragments connected to a pyridine linker. The solvent-free protocol includes the use of a lower amount of azide 2d,c,f (2.5 equiv) in comparison with the reaction in n-propanol (4 equiv of azide) to afford the desired products in the same yield and therefore was selected as the method of choice for the synthesis of 3aj–an (Scheme 6). The synthesis of complexes of bis(sulfonyl amidines) 3aj–an with metals is in progress.
![[1860-5397-16-243-i6]](/bjoc/content/inline/1860-5397-16-243-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of bis(sulfonyl amidines) 3aj–an.
Scheme 6: Synthesis of bis(sulfonyl amidines) 3aj–an.
1H and 13C NMR spectra including 2D HMBC and HSQC experiments of compounds 3a–an, as well as high-resolution mass spectra are consistent with the proposed structures. Carbon signals of the amidine groups of compounds 3 appear at 154.1‒159.7 ppm which is close to 156 ppm which is the value found for N-sulfonyl amidines of 1,2,3-thiadiazole-4-carboxylic acid prepared by another method [22] and was clearly different from the thioamide carbon signal at 185‒187 ppm in the 13C NMR spectra of starting materials 1. A final proof of the structures of the prepared compounds comes from the X-ray data for 3e,t,ag (Schemes 2, 3, and 5). Moreover, the X-ray data reveal the existence of N-sulfonyl amidines 3e,t in the E-isomeric form and N-sulfonyl amidine 3ag in Z-isomeric form. The existence of the latter in the Z-isomeric form can be explained by steric hindrance between the phenyl and the arylsulfonyl groups.
Because of the observed evolution of nitrogen and sulfur in every reaction of heterocyclic thioamides and sulfonyl azides it is logic to propose the formation of a thiatriazole ring via [3 + 2] cycloaddition of the azide group and the C=S moiety of the thioamide group (Scheme 7).
![[1860-5397-16-243-i7]](/bjoc/content/inline/1860-5397-16-243-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Plausible mechanism for the reaction of heterocyclic thioamides with sulfonyl azides.
Scheme 7: Plausible mechanism for the reaction of heterocyclic thioamides with sulfonyl azides.
The formation of nitrene-like products was excluded because of the high selectivity of the process, where only the thioamide group takes part, even with heterocyclic rings that contain other nucleophilic centers, and in one case, an amino group. Thiatriazoles are known to be unstable compounds that readily evolve nitrogen and sulfur upon heating [56].
Conclusion
We have shown that the reaction of sulfonyl azides with thioamides can serve as the basis for a general and efficient method for the regioselective synthesis of N-sulfonyl amidines of azolyl and pyridine carboxylic acids. The most promising aspect for organic synthesis and green chemistry is a solvent-free process which was successfully applied to prepare sulfonyl amidines containing pyridine and isoxazolyl rings and 1-alkyl-1,2,3-triazole-4-N-sulfonylamidino-1,2,3-triazoles. The 1-alkyltriazole thioamides are the most active in the solvent-free method due to their low melting points and good solubility in alkyl- and arylsulfonyl azides. Conversely, thioamides containing 5-arylamino-1,2,3-triazole and 2-aminothiazole rings are not soluble in sulfonyl azides and could be transformed to the corresponding N-sulfonyl amidines by reactions in 1-propanol via two- or one-pot procedures. Pyridine-2,6-dithioamide was shown to react with mesyl and arylsulfonyl azides to form pyridine derivatives bearing two N-sulfonyl amidine moieties in excellent yield. Depending on the structure of the heterocycle the N-sulfonyl amidines exist in either E- or Z-isomeric forms.
Experimental
X-ray diffraction study
X-ray analyses were accomplished on an Xcalibur 3 diffractometer using the standard procedure (graphite-monochromated Mo Kα irradiation, ω-scanning with step 1o, T = 295(2) K (see Supporting Information File 3). Using Olex2 [57], the structures were solved with the Superflip [58] structure solution program using charge flipping and refined with the ShelXL [59] refinement package using least squares minimization. Deposition numbers for compounds 3e (2020829), 3t (2020831) and 3ag (2020830), contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Supporting Information
| Supporting Information File 1: Full experimental details and characterization data of all new compounds, crystal data and structure refinement for 3e, 3t, and 3ag. | ||
| Format: PDF | Size: 948.5 KB | Download |
| Supporting Information File 2: Copies of NMR spectra of all new compounds. | ||
| Format: PDF | Size: 5.6 MB | Download |
| Supporting Information File 3: Crystallographic information files for compounds 3e, 3t and 3ag. | ||
| Format: ZIP | Size: 16.7 KB | Download |
References
-
Wu, S.-M.; Qiu, X.-Y.; Liu, S.-J.; Sun, J. Mini-Rev. Med. Chem. 2020, 20, 908–920. doi:10.2174/1389557520666200302114620
Return to citation in text: [1] -
Wang, Z.; Ma, J.; Li, N.; Li, L. Preparation of heterocyclic compounds as inhibitors of fibroblast growth factor receptor. PCT Int. Appl. WO2020119606 A1, June 18, 2020.
Return to citation in text: [1] -
Lather, A.; Sharma, S.; Khatkar, S.; Khatkar, A. Curr. Pharm. Des. 2020, 26, 1650–1665. doi:10.2174/1381612826666200217115211
Return to citation in text: [1] -
Li, D.; Duan, L.; Zheng, T.; Wang, Y.; Jin, K.; Su, W.; Hong, Y.; Xu, J.; Xia, G.; Ke, Y. Preparation of nitrogen-containing heterocyclic compounds as p38 protein kinase inhibitors. PCT Int. Appl. WO2020108659 A1, June 4, 2020.
Return to citation in text: [1] -
Li, X.; Zhang, S.-Q.; Xu, L.-C.; Hong, X. Angew. Chem., Int. Ed. 2020, 59, 13253–13259. doi:10.1002/anie.202000959
Return to citation in text: [1] -
Evano, G.; Blanchard, N.; Toumi, M. Chem. Rev. 2008, 108, 3054–3131. doi:10.1021/cr8002505
Return to citation in text: [1] -
Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j
Return to citation in text: [1] -
Whitehouse, M. W. Curr. Med. Chem. 2005, 12, 2931–2942. doi:10.2174/092986705774462879
Return to citation in text: [1] -
Zhou, Q.-F.; Ge, F.-F.; Chen, Q.-Q.; Lu, T. RSC Adv. 2016, 6, 1395–1402. doi:10.1039/c5ra17267e
Return to citation in text: [1] -
Katritzky, A. R.; Rees, C. W., Eds. Comprehensive Heterocyclic Chemistry I; Pergamon Press: Oxford, U.K., 1984.
Return to citation in text: [1] -
Katritzky, A. R.; Rees, C. W.; Scriven, E. F., Eds. Comprehensive Heterocyclic Chemistry II; Elsevier : Oxford, U.K., 1996.
Return to citation in text: [1] -
Katritzky, A. R.; Ramsden, C.; Scriven, E. F. V.; Taylor, R., Eds. Comprehensive Heterocyclic Chemistry III; Elsevier : Oxford, U.K., 2008.
Return to citation in text: [1] -
Lee, M. Y.; Kim, M. H.; Kim, J.; Kim, S. H.; Kim, B. T.; Jeong, I. H.; Chang, S.; Kim, S. H.; Chang, S.-Y. Bioorg. Med. Chem. Lett. 2010, 20, 541–545. doi:10.1016/j.bmcl.2009.11.104
Return to citation in text: [1] -
Beryozkina, T.; Bakulev, V.; Dianova, L.; Berseneva, V.; Slepukhin, P.; Leban, J.; Kalaba, P.; Aher, N. Y.; Ilic, M.; Sitte, H. H.; Lubec, G. Synthesis 2016, 1046. doi:10.1055/s-0035-1561350
Return to citation in text: [1] -
Rupakova, N. A.; Bakulev, V. A.; Knippschild, U.; García-Reyes, B.; Eltsov, O. S.; Slesarev, G. P.; Beliaev, N.; Slepukhin, P. A.; Witt, L.; Peifer, C.; Beryozkina, T. V. ARKIVOC 2017, No. iii, 225–240. doi:10.24820/ark.5550190.p010.200
Return to citation in text: [1] -
Suja, T. D.; Divya, K. V. L.; Naik, L. V.; Kumar, A. R.; Kamal, A. Bioorg. Med. Chem. Lett. 2016, 26, 2072–2076. doi:10.1016/j.bmcl.2016.02.071
Return to citation in text: [1] -
Scholz, T. H.; Sondey, J. M.; Randall, W. C.; Schwam, H.; Thompson, W. J.; Mallorga, P. J.; Sugrue, M. F.; Graham, S. L. J. Med. Chem. 1993, 36, 2134–2141. doi:10.1021/jm00067a012
Return to citation in text: [1] [2] -
Deprez, P.; Heckmann, B.; Corbier, A.; Vevert, J.-P.; Fortin, M.; Guillaume, J. Bioorg. Med. Chem. Lett. 1995, 5, 2605–2610. doi:10.1016/0960-894x(95)00478-c
Return to citation in text: [1] [2] -
Heitsch, H.; Becker, R. H. A.; Kleemann, H.-W.; Wagner, A. Bioorg. Med. Chem. 1997, 5, 673–678. doi:10.1016/s0968-0896(97)00012-6
Return to citation in text: [1] [2] -
Bekhit, A. A.; Ashour, H. M. A.; Abdel Ghany, Y. S.; Bekhit, A. E.-D. A.; Baraka, A. Eur. J. Med. Chem. 2008, 43, 456–463. doi:10.1016/j.ejmech.2007.03.030
Return to citation in text: [1] [2] -
Song, Z.-L.; Chen, H.-L.; Wang, Y.-H.; Goto, M.; Gao, W.-J.; Cheng, P.-L.; Morris-Natschke, S. L.; Liu, Y.-Q.; Zhu, G.-X.; Wang, M.-J.; Lee, K.-H. Bioorg. Med. Chem. Lett. 2015, 25, 2690–2693. doi:10.1016/j.bmcl.2015.04.060
Return to citation in text: [1] -
Filimonov, V. O.; Dianova, L. N.; Galata, K. A.; Beryozkina, T. V.; Novikov, M. S.; Berseneva, V. S.; Eltsov, O. S.; Lebedev, A. T.; Slepukhin, P. A.; Bakulev, V. A. J. Org. Chem. 2017, 82, 4056–4071. doi:10.1021/acs.joc.6b02736
Return to citation in text: [1] [2] [3] [4] -
Yavari, I.; Ahmadian, S.; Ghazanfarpur-Darjani, M.; Solgi, Y. Tetrahedron Lett. 2011, 52, 668–670. doi:10.1016/j.tetlet.2010.11.135
Return to citation in text: [1] -
He, X.; Shang, Y.; Hu, J.; Ju, K.; Jiang, W.; Wang, S. Sci. China: Chem. 2012, 55, 214–222. doi:10.1007/s11426-011-4405-9
Return to citation in text: [1] -
Yang, T.; Cui, H.; Zhang, C.; Zhang, L.; Su, C.-Y. Inorg. Chem. 2013, 52, 9053–9059. doi:10.1021/ic4012229
Return to citation in text: [1] -
Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038–2039. doi:10.1021/ja0432968
Return to citation in text: [1] -
Kim, J.; Stahl, S. S. J. Org. Chem. 2015, 80, 2448–2454. doi:10.1021/jo5029198
Return to citation in text: [1] -
Hwang, S. J.; Cho, S. H.; Chang, S. Pure Appl. Chem. 2008, 80, 873–879. doi:10.1351/pac200880050873
Return to citation in text: [1] -
Yoo, E. J.; Ahlquist, M.; Bae, I.; Sharpless, K. B.; Fokin, V. V.; Chang, S. J. Org. Chem. 2008, 73, 5520–5528. doi:10.1021/jo800733p
Return to citation in text: [1] -
Kim, J.; Lee, S. Y.; Lee, J.; Do, Y.; Chang, S. J. Org. Chem. 2008, 73, 9454–9457. doi:10.1021/jo802014g
Return to citation in text: [1] -
Krstulović, L.; Ismaili, H.; Bajić, M.; Višnjevac, A.; Glavaš-Obrovac, L.; Žinić, B. Croat. Chem. Acta 2012, 85, 525–534. doi:10.5562/cca2198
Return to citation in text: [1] -
Filimonov, V. O.; Dianova, L. N.; Beryozkina, T. V.; Mazur, D.; Beliaev, N. A.; Volkova, N. N.; Ilkin, V. G.; Dehaen, W.; Lebedev, A. T.; Bakulev, V. A. J. Org. Chem. 2019, 84, 13430–13446. doi:10.1021/acs.joc.9b01599
Return to citation in text: [1] [2] -
Aswad, M.; Chiba, J.; Tomohiro, T.; Hatanaka, Y. Chem. Commun. 2013, 49, 10242–10244. doi:10.1039/c3cc46055j
Return to citation in text: [1] [2] -
Fleury, L. M.; Wilson, E. E.; Vogt, M.; Fan, T. J.; Oliver, A. G.; Ashfeld, B. L. Angew. Chem., Int. Ed. 2013, 52, 11589–11593. doi:10.1002/anie.201305141
Return to citation in text: [1] -
DeKorver, K. A.; Johnson, W. L.; Zhang, Y.; Hsung, R. P.; Dai, H.; Deng, J.; Lohse, A. G.; Zhang, Y.-S. J. Org. Chem. 2011, 76, 5092–5103. doi:10.1021/jo200780x
Return to citation in text: [1] -
Chen, S.; Xu, Y.; Wan, X. Org. Lett. 2011, 13, 6152–6155. doi:10.1021/ol2024604
Return to citation in text: [1] -
Chandna, N.; Chandak, N.; Kumar, P.; Kapoor, J. K.; Sharma, P. K. Green Chem. 2013, 15, 2294–2301. doi:10.1039/c3gc40797g
Return to citation in text: [1] -
Gobis, K.; Foks, H.; Sławiński, J.; Sikorski, A.; Trzybiński, D.; Augustynowicz-Kopeć, E.; Napiórkowska, A.; Bojanowski, K. Monatsh. Chem. 2013, 144, 647–658. doi:10.1007/s00706-012-0888-0
Return to citation in text: [1] -
Himbert, G.; Schwickerath, W. Liebigs Ann. Chem. 1984, 85–97. doi:10.1002/jlac.198419840110
Return to citation in text: [1] -
van Loevezijn, A.; Venhorst, J.; Iwema Bakker, W. I.; de Korte, C. G.; de Looff, W.; Verhoog, S.; van Wees, J.-W.; van Hoeve, M.; van de Woestijne, R. P.; van der Neut, M. A. W.; Borst, A. J. M.; van Dongen, M. J. P.; de Bruin, N. M. W. J.; Keizer, H. G.; Kruse, C. G. J. Med. Chem. 2011, 54, 7030–7054. doi:10.1021/jm200466r
Return to citation in text: [1] [2] -
Dianova, L.; Berseneva, V.; Beryozkina, T.; Efimov, I.; Kosterina, M.; Eltsov, O.; Dehaen, W.; Bakulev, V. Eur. J. Org. Chem. 2015, 6917–6923. doi:10.1002/ejoc.201500968
Return to citation in text: [1] -
Il’kin, V. G.; Berseneva, V. S.; Slepukhin, P. A.; Bakulev, V. A. Chem. Heterocycl. Compd. 2018, 54, 1153–1160. doi:10.1007/s10593-019-02407-7
Return to citation in text: [1] -
Shafran, Y.; Glukhareva, T.; Dehaen, W.; Bakulev, V. Adv. Heterocycl. Chem. 2018, 126, 109–172. doi:10.1016/bs.aihch.2017.12.001
Return to citation in text: [1] -
Bakulev, V. A.; Dehaen, W. The Chemistry of 1,2,3-Thiadiazoles; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004.
Return to citation in text: [1] -
Berseneva, V. S.; Morzherin, Y. Y.; Dehaen, W.; Luyten, I.; Bakulev, V. A. Tetrahedron 2001, 57, 2179–2184. doi:10.1016/s0040-4020(01)00049-7
Return to citation in text: [1] -
Denisova, A. B.; Sosnovskikh, V. Y.; Dehaen, W.; Toppet, S.; Van Meervelt, L.; Bakulev, V. A. J. Fluorine Chem. 2002, 115, 183–192. doi:10.1016/s0022-1139(02)00060-x
Return to citation in text: [1] -
Bel'skaya, N. P.; Demina, M. A.; Sapognikova, S. G.; Fan, Z.-J.; Zhang, H.-K.; Dehaen, W.; Bakulev, V. A. ARKIVOC 2008, No. xvi, 9–21. doi:10.3998/ark.5550190.0009.g02
Return to citation in text: [1] -
Watkinson, M. Top. Heterocycl. Chem. 2012, 28, 109–136. doi:10.1007/7081_2011_69
Return to citation in text: [1] -
Belskaya, N.; Subbotina, J.; Lesogorova, S. Top. Heterocycl. Chem. 2014, 40, 51–116. doi:10.1007/7081_2014_125
Return to citation in text: [1] -
Bakulev, V.; Dehaen, W.; Beryozkina, T. Top. Heterocycl. Chem. 2015, 40, 1–50. doi:10.1007/7081_2014_131
Return to citation in text: [1] -
Bakulev, V. A.; Tarasov, E. V.; Morzherin, Y. Y.; Luyten, I.; Toppet, S.; Dehaen, W. Tetrahedron 1998, 54, 8501–8514. doi:10.1016/s0040-4020(98)00449-9
Return to citation in text: [1] -
Khazhieva, I. S.; Glukhareva, T. V.; El’tsov, O. S.; Morzherin, Y. Y.; Minin, A. A.; Pozdina, V. A.; Ulitko, M. V. Pharm. Chem. J. 2015, 49, 296–300. doi:10.1007/s11094-015-1273-1
Return to citation in text: [1] [2] -
Kang, T.; Kim, H.; Kim, J. G.; Chang, S. Chem. Commun. 2014, 50, 12073–12075. doi:10.1039/c4cc05655h
Return to citation in text: [1] -
Ilkin, V. G.; Dianova, L. N.; Bakulev, V. A.; Berseneva, V. S.; Saveliev, D. A.; Beryozkina, T. V. Chem. Heterocycl. Compd. 2020, 56, 1335–1340. doi:10.1007/s10593-020-02819-w
Return to citation in text: [1] [2] -
Bakulev, V. A.; Beryozkina, T. A. Chem. Heterocycl. Compd. 2016, 52, 4–6. doi:10.1007/s10593-016-1821-y
Return to citation in text: [1] -
Dehaen, W.; Bakulev, V. A. 1,2,3,4-Thiatriazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, U.K., 2008; Vol. 6, pp 441–484. doi:10.1016/b978-008044992-0.00519-8
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Palatinus, L.; Chapuis, G. J. Appl. Crystallogr. 2007, 40, 786–790. doi:10.1107/s0021889807029238
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1]
| 1. | Wu, S.-M.; Qiu, X.-Y.; Liu, S.-J.; Sun, J. Mini-Rev. Med. Chem. 2020, 20, 908–920. doi:10.2174/1389557520666200302114620 |
| 2. | Wang, Z.; Ma, J.; Li, N.; Li, L. Preparation of heterocyclic compounds as inhibitors of fibroblast growth factor receptor. PCT Int. Appl. WO2020119606 A1, June 18, 2020. |
| 3. | Lather, A.; Sharma, S.; Khatkar, S.; Khatkar, A. Curr. Pharm. Des. 2020, 26, 1650–1665. doi:10.2174/1381612826666200217115211 |
| 4. | Li, D.; Duan, L.; Zheng, T.; Wang, Y.; Jin, K.; Su, W.; Hong, Y.; Xu, J.; Xia, G.; Ke, Y. Preparation of nitrogen-containing heterocyclic compounds as p38 protein kinase inhibitors. PCT Int. Appl. WO2020108659 A1, June 4, 2020. |
| 5. | Li, X.; Zhang, S.-Q.; Xu, L.-C.; Hong, X. Angew. Chem., Int. Ed. 2020, 59, 13253–13259. doi:10.1002/anie.202000959 |
| 6. | Evano, G.; Blanchard, N.; Toumi, M. Chem. Rev. 2008, 108, 3054–3131. doi:10.1021/cr8002505 |
| 7. | Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395–3442. doi:10.1021/cr050041j |
| 8. | Whitehouse, M. W. Curr. Med. Chem. 2005, 12, 2931–2942. doi:10.2174/092986705774462879 |
| 9. | Zhou, Q.-F.; Ge, F.-F.; Chen, Q.-Q.; Lu, T. RSC Adv. 2016, 6, 1395–1402. doi:10.1039/c5ra17267e |
| 10. | Katritzky, A. R.; Rees, C. W., Eds. Comprehensive Heterocyclic Chemistry I; Pergamon Press: Oxford, U.K., 1984. |
| 11. | Katritzky, A. R.; Rees, C. W.; Scriven, E. F., Eds. Comprehensive Heterocyclic Chemistry II; Elsevier : Oxford, U.K., 1996. |
| 12. | Katritzky, A. R.; Ramsden, C.; Scriven, E. F. V.; Taylor, R., Eds. Comprehensive Heterocyclic Chemistry III; Elsevier : Oxford, U.K., 2008. |
| 23. | Yavari, I.; Ahmadian, S.; Ghazanfarpur-Darjani, M.; Solgi, Y. Tetrahedron Lett. 2011, 52, 668–670. doi:10.1016/j.tetlet.2010.11.135 |
| 24. | He, X.; Shang, Y.; Hu, J.; Ju, K.; Jiang, W.; Wang, S. Sci. China: Chem. 2012, 55, 214–222. doi:10.1007/s11426-011-4405-9 |
| 25. | Yang, T.; Cui, H.; Zhang, C.; Zhang, L.; Su, C.-Y. Inorg. Chem. 2013, 52, 9053–9059. doi:10.1021/ic4012229 |
| 26. | Bae, I.; Han, H.; Chang, S. J. Am. Chem. Soc. 2005, 127, 2038–2039. doi:10.1021/ja0432968 |
| 27. | Kim, J.; Stahl, S. S. J. Org. Chem. 2015, 80, 2448–2454. doi:10.1021/jo5029198 |
| 28. | Hwang, S. J.; Cho, S. H.; Chang, S. Pure Appl. Chem. 2008, 80, 873–879. doi:10.1351/pac200880050873 |
| 29. | Yoo, E. J.; Ahlquist, M.; Bae, I.; Sharpless, K. B.; Fokin, V. V.; Chang, S. J. Org. Chem. 2008, 73, 5520–5528. doi:10.1021/jo800733p |
| 30. | Kim, J.; Lee, S. Y.; Lee, J.; Do, Y.; Chang, S. J. Org. Chem. 2008, 73, 9454–9457. doi:10.1021/jo802014g |
| 31. | Krstulović, L.; Ismaili, H.; Bajić, M.; Višnjevac, A.; Glavaš-Obrovac, L.; Žinić, B. Croat. Chem. Acta 2012, 85, 525–534. doi:10.5562/cca2198 |
| 40. | van Loevezijn, A.; Venhorst, J.; Iwema Bakker, W. I.; de Korte, C. G.; de Looff, W.; Verhoog, S.; van Wees, J.-W.; van Hoeve, M.; van de Woestijne, R. P.; van der Neut, M. A. W.; Borst, A. J. M.; van Dongen, M. J. P.; de Bruin, N. M. W. J.; Keizer, H. G.; Kruse, C. G. J. Med. Chem. 2011, 54, 7030–7054. doi:10.1021/jm200466r |
| 52. | Khazhieva, I. S.; Glukhareva, T. V.; El’tsov, O. S.; Morzherin, Y. Y.; Minin, A. A.; Pozdina, V. A.; Ulitko, M. V. Pharm. Chem. J. 2015, 49, 296–300. doi:10.1007/s11094-015-1273-1 |
| 22. | Filimonov, V. O.; Dianova, L. N.; Galata, K. A.; Beryozkina, T. V.; Novikov, M. S.; Berseneva, V. S.; Eltsov, O. S.; Lebedev, A. T.; Slepukhin, P. A.; Bakulev, V. A. J. Org. Chem. 2017, 82, 4056–4071. doi:10.1021/acs.joc.6b02736 |
| 52. | Khazhieva, I. S.; Glukhareva, T. V.; El’tsov, O. S.; Morzherin, Y. Y.; Minin, A. A.; Pozdina, V. A.; Ulitko, M. V. Pharm. Chem. J. 2015, 49, 296–300. doi:10.1007/s11094-015-1273-1 |
| 17. | Scholz, T. H.; Sondey, J. M.; Randall, W. C.; Schwam, H.; Thompson, W. J.; Mallorga, P. J.; Sugrue, M. F.; Graham, S. L. J. Med. Chem. 1993, 36, 2134–2141. doi:10.1021/jm00067a012 |
| 18. | Deprez, P.; Heckmann, B.; Corbier, A.; Vevert, J.-P.; Fortin, M.; Guillaume, J. Bioorg. Med. Chem. Lett. 1995, 5, 2605–2610. doi:10.1016/0960-894x(95)00478-c |
| 19. | Heitsch, H.; Becker, R. H. A.; Kleemann, H.-W.; Wagner, A. Bioorg. Med. Chem. 1997, 5, 673–678. doi:10.1016/s0968-0896(97)00012-6 |
| 20. | Bekhit, A. A.; Ashour, H. M. A.; Abdel Ghany, Y. S.; Bekhit, A. E.-D. A.; Baraka, A. Eur. J. Med. Chem. 2008, 43, 456–463. doi:10.1016/j.ejmech.2007.03.030 |
| 43. | Shafran, Y.; Glukhareva, T.; Dehaen, W.; Bakulev, V. Adv. Heterocycl. Chem. 2018, 126, 109–172. doi:10.1016/bs.aihch.2017.12.001 |
| 44. | Bakulev, V. A.; Dehaen, W. The Chemistry of 1,2,3-Thiadiazoles; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. |
| 45. | Berseneva, V. S.; Morzherin, Y. Y.; Dehaen, W.; Luyten, I.; Bakulev, V. A. Tetrahedron 2001, 57, 2179–2184. doi:10.1016/s0040-4020(01)00049-7 |
| 46. | Denisova, A. B.; Sosnovskikh, V. Y.; Dehaen, W.; Toppet, S.; Van Meervelt, L.; Bakulev, V. A. J. Fluorine Chem. 2002, 115, 183–192. doi:10.1016/s0022-1139(02)00060-x |
| 47. | Bel'skaya, N. P.; Demina, M. A.; Sapognikova, S. G.; Fan, Z.-J.; Zhang, H.-K.; Dehaen, W.; Bakulev, V. A. ARKIVOC 2008, No. xvi, 9–21. doi:10.3998/ark.5550190.0009.g02 |
| 13. | Lee, M. Y.; Kim, M. H.; Kim, J.; Kim, S. H.; Kim, B. T.; Jeong, I. H.; Chang, S.; Kim, S. H.; Chang, S.-Y. Bioorg. Med. Chem. Lett. 2010, 20, 541–545. doi:10.1016/j.bmcl.2009.11.104 |
| 14. | Beryozkina, T.; Bakulev, V.; Dianova, L.; Berseneva, V.; Slepukhin, P.; Leban, J.; Kalaba, P.; Aher, N. Y.; Ilic, M.; Sitte, H. H.; Lubec, G. Synthesis 2016, 1046. doi:10.1055/s-0035-1561350 |
| 15. | Rupakova, N. A.; Bakulev, V. A.; Knippschild, U.; García-Reyes, B.; Eltsov, O. S.; Slesarev, G. P.; Beliaev, N.; Slepukhin, P. A.; Witt, L.; Peifer, C.; Beryozkina, T. V. ARKIVOC 2017, No. iii, 225–240. doi:10.24820/ark.5550190.p010.200 |
| 16. | Suja, T. D.; Divya, K. V. L.; Naik, L. V.; Kumar, A. R.; Kamal, A. Bioorg. Med. Chem. Lett. 2016, 26, 2072–2076. doi:10.1016/j.bmcl.2016.02.071 |
| 17. | Scholz, T. H.; Sondey, J. M.; Randall, W. C.; Schwam, H.; Thompson, W. J.; Mallorga, P. J.; Sugrue, M. F.; Graham, S. L. J. Med. Chem. 1993, 36, 2134–2141. doi:10.1021/jm00067a012 |
| 18. | Deprez, P.; Heckmann, B.; Corbier, A.; Vevert, J.-P.; Fortin, M.; Guillaume, J. Bioorg. Med. Chem. Lett. 1995, 5, 2605–2610. doi:10.1016/0960-894x(95)00478-c |
| 19. | Heitsch, H.; Becker, R. H. A.; Kleemann, H.-W.; Wagner, A. Bioorg. Med. Chem. 1997, 5, 673–678. doi:10.1016/s0968-0896(97)00012-6 |
| 20. | Bekhit, A. A.; Ashour, H. M. A.; Abdel Ghany, Y. S.; Bekhit, A. E.-D. A.; Baraka, A. Eur. J. Med. Chem. 2008, 43, 456–463. doi:10.1016/j.ejmech.2007.03.030 |
| 21. | Song, Z.-L.; Chen, H.-L.; Wang, Y.-H.; Goto, M.; Gao, W.-J.; Cheng, P.-L.; Morris-Natschke, S. L.; Liu, Y.-Q.; Zhu, G.-X.; Wang, M.-J.; Lee, K.-H. Bioorg. Med. Chem. Lett. 2015, 25, 2690–2693. doi:10.1016/j.bmcl.2015.04.060 |
| 48. | Watkinson, M. Top. Heterocycl. Chem. 2012, 28, 109–136. doi:10.1007/7081_2011_69 |
| 49. | Belskaya, N.; Subbotina, J.; Lesogorova, S. Top. Heterocycl. Chem. 2014, 40, 51–116. doi:10.1007/7081_2014_125 |
| 50. | Bakulev, V.; Dehaen, W.; Beryozkina, T. Top. Heterocycl. Chem. 2015, 40, 1–50. doi:10.1007/7081_2014_131 |
| 51. | Bakulev, V. A.; Tarasov, E. V.; Morzherin, Y. Y.; Luyten, I.; Toppet, S.; Dehaen, W. Tetrahedron 1998, 54, 8501–8514. doi:10.1016/s0040-4020(98)00449-9 |
| 36. | Chen, S.; Xu, Y.; Wan, X. Org. Lett. 2011, 13, 6152–6155. doi:10.1021/ol2024604 |
| 22. | Filimonov, V. O.; Dianova, L. N.; Galata, K. A.; Beryozkina, T. V.; Novikov, M. S.; Berseneva, V. S.; Eltsov, O. S.; Lebedev, A. T.; Slepukhin, P. A.; Bakulev, V. A. J. Org. Chem. 2017, 82, 4056–4071. doi:10.1021/acs.joc.6b02736 |
| 32. | Filimonov, V. O.; Dianova, L. N.; Beryozkina, T. V.; Mazur, D.; Beliaev, N. A.; Volkova, N. N.; Ilkin, V. G.; Dehaen, W.; Lebedev, A. T.; Bakulev, V. A. J. Org. Chem. 2019, 84, 13430–13446. doi:10.1021/acs.joc.9b01599 |
| 38. | Gobis, K.; Foks, H.; Sławiński, J.; Sikorski, A.; Trzybiński, D.; Augustynowicz-Kopeć, E.; Napiórkowska, A.; Bojanowski, K. Monatsh. Chem. 2013, 144, 647–658. doi:10.1007/s00706-012-0888-0 |
| 39. | Himbert, G.; Schwickerath, W. Liebigs Ann. Chem. 1984, 85–97. doi:10.1002/jlac.198419840110 |
| 40. | van Loevezijn, A.; Venhorst, J.; Iwema Bakker, W. I.; de Korte, C. G.; de Looff, W.; Verhoog, S.; van Wees, J.-W.; van Hoeve, M.; van de Woestijne, R. P.; van der Neut, M. A. W.; Borst, A. J. M.; van Dongen, M. J. P.; de Bruin, N. M. W. J.; Keizer, H. G.; Kruse, C. G. J. Med. Chem. 2011, 54, 7030–7054. doi:10.1021/jm200466r |
| 35. | DeKorver, K. A.; Johnson, W. L.; Zhang, Y.; Hsung, R. P.; Dai, H.; Deng, J.; Lohse, A. G.; Zhang, Y.-S. J. Org. Chem. 2011, 76, 5092–5103. doi:10.1021/jo200780x |
| 33. | Aswad, M.; Chiba, J.; Tomohiro, T.; Hatanaka, Y. Chem. Commun. 2013, 49, 10242–10244. doi:10.1039/c3cc46055j |
| 41. | Dianova, L.; Berseneva, V.; Beryozkina, T.; Efimov, I.; Kosterina, M.; Eltsov, O.; Dehaen, W.; Bakulev, V. Eur. J. Org. Chem. 2015, 6917–6923. doi:10.1002/ejoc.201500968 |
| 42. | Il’kin, V. G.; Berseneva, V. S.; Slepukhin, P. A.; Bakulev, V. A. Chem. Heterocycl. Compd. 2018, 54, 1153–1160. doi:10.1007/s10593-019-02407-7 |
| 34. | Fleury, L. M.; Wilson, E. E.; Vogt, M.; Fan, T. J.; Oliver, A. G.; Ashfeld, B. L. Angew. Chem., Int. Ed. 2013, 52, 11589–11593. doi:10.1002/anie.201305141 |
| 22. | Filimonov, V. O.; Dianova, L. N.; Galata, K. A.; Beryozkina, T. V.; Novikov, M. S.; Berseneva, V. S.; Eltsov, O. S.; Lebedev, A. T.; Slepukhin, P. A.; Bakulev, V. A. J. Org. Chem. 2017, 82, 4056–4071. doi:10.1021/acs.joc.6b02736 |
| 32. | Filimonov, V. O.; Dianova, L. N.; Beryozkina, T. V.; Mazur, D.; Beliaev, N. A.; Volkova, N. N.; Ilkin, V. G.; Dehaen, W.; Lebedev, A. T.; Bakulev, V. A. J. Org. Chem. 2019, 84, 13430–13446. doi:10.1021/acs.joc.9b01599 |
| 33. | Aswad, M.; Chiba, J.; Tomohiro, T.; Hatanaka, Y. Chem. Commun. 2013, 49, 10242–10244. doi:10.1039/c3cc46055j |
| 37. | Chandna, N.; Chandak, N.; Kumar, P.; Kapoor, J. K.; Sharma, P. K. Green Chem. 2013, 15, 2294–2301. doi:10.1039/c3gc40797g |
| 54. | Ilkin, V. G.; Dianova, L. N.; Bakulev, V. A.; Berseneva, V. S.; Saveliev, D. A.; Beryozkina, T. V. Chem. Heterocycl. Compd. 2020, 56, 1335–1340. doi:10.1007/s10593-020-02819-w |
| 53. | Kang, T.; Kim, H.; Kim, J. G.; Chang, S. Chem. Commun. 2014, 50, 12073–12075. doi:10.1039/c4cc05655h |
| 54. | Ilkin, V. G.; Dianova, L. N.; Bakulev, V. A.; Berseneva, V. S.; Saveliev, D. A.; Beryozkina, T. V. Chem. Heterocycl. Compd. 2020, 56, 1335–1340. doi:10.1007/s10593-020-02819-w |
| 58. | Palatinus, L.; Chapuis, G. J. Appl. Crystallogr. 2007, 40, 786–790. doi:10.1107/s0021889807029238 |
| 59. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 56. | Dehaen, W.; Bakulev, V. A. 1,2,3,4-Thiatriazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A. R.; Ramsden, C. A.; Scriven, E. F. V.; Taylor, R. J. K., Eds.; Elsevier: Oxford, U.K., 2008; Vol. 6, pp 441–484. doi:10.1016/b978-008044992-0.00519-8 |
| 57. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 55. | Bakulev, V. A.; Beryozkina, T. A. Chem. Heterocycl. Compd. 2016, 52, 4–6. doi:10.1007/s10593-016-1821-y |
| 22. | Filimonov, V. O.; Dianova, L. N.; Galata, K. A.; Beryozkina, T. V.; Novikov, M. S.; Berseneva, V. S.; Eltsov, O. S.; Lebedev, A. T.; Slepukhin, P. A.; Bakulev, V. A. J. Org. Chem. 2017, 82, 4056–4071. doi:10.1021/acs.joc.6b02736 |
© 2020 Ilkin et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)