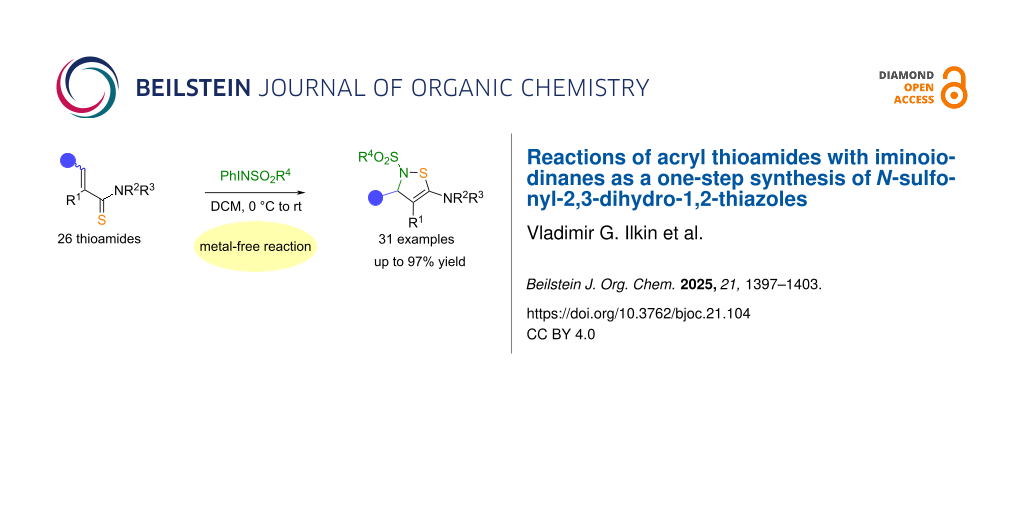
Abstract
A one-step method has been developed for the preparation of 2,3-dihydro-2-sulfonyl-3,4,5-substituted 1,2-thiazoles by the reaction of acryl thioamides and iminoiodinanes. A library of 31 examples of tetrasubstituted 1,2-thiazoles was thus synthesized in high yields. The effectiveness of the synthesis method for these poorly studied 1,2-thiazoles was confirmed by scaling the reaction using gram amounts of the starting thioamide.

Graphical Abstract
Introduction
1,2-Thiazoles (isothiazoles) exhibit a wide range of biological activity (Figure 1): antipoliovirus [1], anticancer [2-7], against Parkinson's disease [8], and diabetes [9-11], and are also used as microbiocides [12,13]. These data [1-5] inspired researchers to expand the chemistry of these compounds and to search for new synthetic methods and chemical transformations [14,15].
![[1860-5397-21-104-1]](/bjoc/content/figures/1860-5397-21-104-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Representatives of biologically active 1,2-thiazoles.
Figure 1: Representatives of biologically active 1,2-thiazoles.
Unlike the aromatic congeners, the chemistry of non-aromatic dihydro-1,2-thiazoles is less represented in the literature [16-18]. 2,5-Dihydro-1,2-thiazoles were synthesized by oxidative cyclization of N-arylamides of 3-(alkylamino)prop-2-enethiocarboxylic acids with iodine (Scheme 1А) [16].
![[1860-5397-21-104-i1]](/bjoc/content/inline/1860-5397-21-104-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of 2,5-dihydro-1,2-thiazoles.
Scheme 1: Synthesis of 2,5-dihydro-1,2-thiazoles.
2,3-Dihydro-1,2-thiazoles were first synthesized in 1997 by the reaction of 2,2-dimethyl-N-alkylsulfonyl-N-benzylaminoacetonitrile with strong bases in acetonitrile (Scheme 1B) [17]. Very recently, our group reported the synthesis of spirocyclic 2,3-dihydro-1,2-triazoles by the reaction of thioamides containing a cycloalkylidene fragment with N-tosyl and N-mesyliminoiodinanes (Scheme 1C) [18]. In the present work, an effective method for the synthesis of 2,3-dihydro-N-sulfonyl-1,2-thiazoles was developed based on the optimized reaction of acrylic acid thioamides containing aryl- and hetarylidene fragments with N-tosyl and N-mesyliminoiodinanes (Scheme 1D).
Results and Discussion
The reaction of thioamide 1a with PhINTs (2a) was chosen as a model for searching the optimal synthesis conditions (Table 1).
Table 1: Optimization of the reaction of thioamide 1a with iodonium salts [I].
![[Graphic 1]](/bjoc/content/inline/1860-5397-21-104-i3.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Cat (mol %) | [I] (equiv) | Time | Solvent (Т, °С) | Yield, % |
| 1 | Rh2(Piv)4 (0.5) | PhINTs (1.5) | 17 h | CHCl3 (50) | 48 |
| 2 | [Cu(MeCN)4]OTf (5) | PhINTs (1.5) | 17 h | CHCl3 (50) | 72 |
| 3 | [Cu(MeCN)4]PF6 (5) | PhINTs (1.5) | 10 min | DCM (0→24) | 82 |
| 4 | Cu(OAc)2 (5) | PhINTs (1.5) | 10 min | DCM (0→24) | 78 |
| 5 | ‒ | PhINTs (1.1) | 20 min | DCM (0→24) | 56 |
| 6 | ‒ | PhINTs (2.0) | 10 min | DCM (0→24) | 67 |
| 7 | ‒ | PhINTs (1.5) | 10 min | DCM (0→24) | 78 |
| 8 | ‒ | PhINTs (1.5) | 10 min | EtOH (0→24) | ‒a |
| 9 | ‒ | PhINTs (1.5) | 10 min | DMSO (0→24) | ‒a |
| 10 | ‒ | DMPb (1.5) | 10 min | DCM (0→24) | 35c |
| 11 | ‒ | HTIBd (1.5) | 10 min | DCM (0→24) | ‒a |
| 12 | ‒ | TsNH2 + PIDAe (1.5) | 10 min | DCM (0→24) | 70 |
Conditions: thioamide 1a (0.19 mmol), iodonium salt [I] (1.1–2.0 equiv), solvent (2.5 mL). aThe reaction does not occur. bDMP = Dess–Martin periodinane. cYield of amide. dHTIB = hydroxy(tosyloxy)iodobenzene. ePIDA = PhI(OAc)2.
We found that when processing thioamide 1a with PhINTs (2a, 1.5 equiv) in chloroform at 50 °C in the presence of Rh2(Piv)4 (0.5 equiv), 2,3-dihydro-N-sulfonyl-1,2-thiazole 3aa is formed with a 48% yield (Table 1, entry 1). When using [Cu(MeCN)4]OTf instead of Rh2(Piv)4, the target product 3aa was obtained in higher yield (72%, Table 1, entry 2) and with [Cu(MeCN)4]PF6, the yield of 1,2-thiazole 3aa increased to 82%, while the reaction time decreased significantly (Table 1, entry 3). Using Cu(OAc)2 as a catalyst led to a slight decrease in the yield of the target product to 78% over the same time (Table 1, entry 4). When the reaction was carried out in the absence of metal catalysts (Table 1, entries 5 and 6), the yields of the target product 3aa were generally lower (56–67%), and in some cases (Table 1, entries 8–11) the reaction did not occur. In the presence of metal catalyst PhINTs form a nitrenoid specie, containing electrophilic nitrogen. In metal-free conditions PhINTs participates in reactions as ylide with a nucleophilic nitrogen. We expected different reactivity of the two different forms of PhINTs. However, our expectations were not fulfilled.
The exception is the data from entry 7 (Table 1), where the yield of compound 3aa was 78%. Thus, the conditions described in entry 7 (absence of a catalyst, use of 1.5 equiv of PhINTs 2a and dichloromethane (DCM) as a solvent at room temperature for 10 min) are optimal and were used for the synthesis of a number of 2,3-dihydro-N-sulfonyl-1,2-thiazoles 3 (Scheme 2, method A). The conditions described in entry 12 (Table 1; Scheme 2, method B) are also noteworthy, since they do not require the preliminary synthesis of iodonium salts 2, which saves time and effort, however, the yields of 1,2-thiazoles 3aa–ae are slightly lower (70 vs 78%) than by method A. We also investigated the effect of chiral catalysts or ligands in the reaction of thioamide 1a with 2a; however, we were unable to achieve high enantiomeric purity of product 3aa (see Supporting Information File 1, Table S1).
![[1860-5397-21-104-i2]](/bjoc/content/inline/1860-5397-21-104-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 2,3-dihydro-N-sulfonyl-1,2-thiazoles 3. Conditions: aMethod A: thioamide 1 (1.0 equiv), PhINMs or PhINTs (2a, 1.5–2.0 equiv), 0→24 °C, 6‒60 min. bMethod B: thioamide 1 (1 equiv), aryl sulfonamide (1.2 equiv), PhI(OAc)2 (1.5 equiv), 0→24 °C, 10–30 min.
Scheme 2: Synthesis of 2,3-dihydro-N-sulfonyl-1,2-thiazoles 3. Conditions: aMethod A: thioamide 1 (1.0 equiv)...
Using the optimal reaction conditions (method A), twenty-seven 2,3-dihydro-N-sulfonyl-1,2-thiazoles 3 were synthesized in 69–96% yields (Scheme 2, method A). According to method B (Scheme 2), five compounds 3aa–ae were obtained in moderate yields (40–70%). The formation of the final compounds starts by analogy with an aza-Michael reaction first with the addition of iminoiodinane accompanied with reorganization of double bonds including attack of the negatively charged sulfur atom on nitrogen with subsequent elimination of iodobenzene as good leaving group.
The diversity of the structure of the target compounds is provided by the variation of substituents in positions 2–5 of the 1,2-thiazole ring. Thus, arylsulfonyl groups containing a methyl, fluorine, chlorine, or nitro group at the aryl moiety, or a mesyl substituent were introduced into position 2 of the thiazole ring and various aryls, quinolinyl, pyridinyl, furyl, isopropyl, and cyclohexyl substituents were introduced into position 3; cyano and various carbonyl groups were added to position 4 and cyclic amine residues were added to position 5 which increase the solubility of the synthesized compounds 3 (Scheme 2). An analysis of the yields of compounds 3aa–af allowed us to conclude that these decreased when electron-accepting groups (R4) were present in the iminoiodinane reactant. Thus, the replacement of the methyl group in 3af with a tolyl group in 3aa led to a 15% decrease of the yield of the target product. This effect can be explained by an increase of the electron density on the nitrogen atom of the iodonium salt 2 when the tosyl group is replaced by a less electron-deficient mesyl group. Using optimal reaction conditions (method A), twenty seven 2,3-dihydro-N-sulfonyl-1,2-thiazoles were synthesized in 69‒96% yields. We scaled the reaction of thioamide 1q with iodonium salt 2a and found that a 20-fold increase of thioamide loading only slightly (by 6%) reduced the yield of the target product 3qa (see Supporting Information File 1). These data indicate the possibility of using the developed method for the synthesis of compounds 3 on a larger or even commercial scale.
The structures of compounds 3 were confirmed by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry. The 1H NMR spectra are characterized by signals from protons of the aromatic rings in the range of 8.08–7.32 ppm, singlets for the proton at the C-3 position of the 2,3-dihydro-1,2-thiazole ring in the range of 6.44–6.06 ppm, and multiplets corresponding to the morpholine fragment in the range of 3.80–2.89 ppm or protons of residues of other amino groups in the range of 3.51–1.31 ppm. In the 13C NMR spectra, characteristic signals of the C-4 atom of the 2,3-dihydro-1,2-thiazole ring are in the range of 75.3–72.4 ppm and signals of the carbon atom of the cyano group in the range of 117.7–116.6 ppm. The structure of 2,3-dihydro-1,2-thiazole 3aa was additionally confirmed by single crystal X-ray diffraction (Figure 2). According to the X-ray diffraction data, compound 3aa crystallizes in the centrosymmetric spatial group of the monoclinic system.
![[1860-5397-21-104-2]](/bjoc/content/figures/1860-5397-21-104-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Compound 3aa in thermal ellipsoids 50% probability.
Figure 2: Compound 3aa in thermal ellipsoids 50% probability.
The molecule has a tweezer-like conformation due to π–π interactions between the electron-donating tolyl and the electron-acceptor fragment NC‒C=C‒S. The heterocyclic fragment in the molecule is non-planar, the atom N(2) deviates from the RMS plane S(1)C(3)C(4)C(5) by 0.461 Å. The atom of the N(1) morpholine fragment has a planar configuration with significant asymmetry in the lengths of C–N bonds. Due to the electron-acceptor effects of substituents, the C(3)‒H bond exhibits significant polarity and is involved in the formation of a weak intermolecular hydrogen bond of the C‒H···N≡C type with distances H(3)···N(3) 2.53(3) Å, C(3)···N(3) 3.436(4) Å and angle C(3)H(3)N(3) 157(2)° (symmetry transformation [1−x, 1−y, 1−z]).
Conclusion
Thus, we found that thioamides of acrylic acid easily interact with iodonium salts. The search for optimal conditions for the process has been carried out. The optimized reaction found to proceed in the absence of metal catalysts, using 1.5 equiv of the iodonium salt at room temperature in DCM. Using the optimized procedure, a library of 31 novel 2,3-dihydro-1,2-thiazoles was synthesized.
Experimental
Thioamides 1a,f,g,l,r [19], 1b,c,d,e,j,u,x [20] and iodonium salts 2a [21] and 2f [22] were synthesized according to the previously described methods. The structures of all thioamides used in this study are provided in Supporting Information File 1.
Preparation of thioamides 1h,i,k,n,o,s,t,v,w,y,z (general procedure). A mixture of the corresponding thioacetamide (1.0 equiv), aldehyde (1.1–4.0 equiv) and DBU (0.1 equiv or 1.0 equiv for 1h,o) in ethanol was stirred for 2–23 h at room temperature. For thioamide 1i, the reaction time was 96 h at 80 °C. The formed precipitate was filtered off and washed with cold ethanol and diethyl ether.
Preparation of 2-sulfonyl-2,3-dihydro-1,2-thiazoles 3 (general procedure). Method A. The corresponding thioamide 1 (1.0 equiv) and DCM (1 mL) was added to an oven-dried standard microwave vial with a volume of 10 mL. The resulting solution was stirred for 10 min in an ice bath, then iodonium salt 2a or 2f (1.5–2.0 equiv) was added in one portion. The reaction vessel was removed from the ice bath and the reaction mass was stirred for 6–60 min, then transferred to a silica gel column and the corresponding 2-sulfonyl-2,3-dihydro-1,2-thiazole 3 was isolated.
Method B. The corresponding aryl sulfonamide (1.2 equiv), PhI(OAc)2 (1.5 equiv) and DCM (0.5 mL) was added to an oven-dried standard microwave vial with a volume of 10 mL. The resulting suspension was stirred for 10 min in an ice bath, then thioamide 1 (1.0 equiv) dissolved in DCM (1.5 mL) was added dropwise. The reaction vessel was removed from the ice bath and the reaction mass was stirred for 10–30 min, then transferred to a silica gel column and the corresponding 2-sulfonyl-1,3-thiazole 3 was isolated.
X-ray structure determination of 3aa. Crystal data for C21H21N3O3S2 (M = 427.53 g/mol): monoclinic, space group P21/n, a = 14.8770(14) Å, b = 8.7007(6) Å, c = 17.5854(15) Å, β = 110.474(11), V = 2132.5(3) Å3, Z = 4, T = 295(2) K, μ(Mo Kα) = 0.277 mm−1, Dcalc = 1.332 g/cm3, 14068 reflections measured (7.492° ≤ 2Θ ≤ 62.136°), 5660 unique (Rint = 0.0465, Rsigma = 0.0585). The final R1 = 0.0584, wR2 = 0.1508 (I > 2σ(I)) and R1 = 0.1081, wR2 = 0.2069 (all data). GooF = 1.027. Largest diff. peak/hole 0.31/−0.40 eÅ−3.
The experiment was performed on an automatic four-circle X-ray diffractometer "Xcalibur 3" with a CCD detector according to the standard procedure (Mo Kα irradiation, graphite monochromator, ω-scanning in 1° increments at T = 295(2) K). An empirical correction for absorption has been introduced. Using the Olex2 [23] software shell, the structure was solved using the SHELXT program and refined using the SHELXL [24,25] program with a full-matrix F2 MNC for non-hydrogen atoms. The H-atoms in C–H bonds are placed in the calculated positions and refined in the "rider" model in the isotropic approximation.
CCDC 2401684 (3aa) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via https://www.ccdc.cam.ac.uk (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information of this article.
References
-
Garozzo, A.; Stivala, A.; Tempera, G.; Castro, A. Antiviral Res. 2010, 88, 325–328. doi:10.1016/j.antiviral.2010.10.003
Return to citation in text: [1] [2] -
Lippa, B.; Morris, J.; Corbett, M.; Kwan, T. A.; Noe, M. C.; Snow, S. L.; Gant, T. G.; Mangiaracina, M.; Coffey, H. A.; Foster, B.; Knauth, E. A.; Wessel, M. D. Bioorg. Med. Chem. Lett. 2006, 16, 3444–3448. doi:10.1016/j.bmcl.2006.04.003
Return to citation in text: [1] [2] -
Carlessi, L.; Buscemi, G.; Larson, G.; Hong, Z.; Wu, J. Z.; Delia, D. Mol. Cancer Ther. 2007, 6, 935–944. doi:10.1158/1535-7163.mct-06-0567
Return to citation in text: [1] [2] -
Coffey, K.; Blackburn, T. J.; Cook, S.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Hewitt, L.; Huberman, K.; McNeill, H. V.; Newell, D. R.; Roche, C.; Ryan-Munden, C. A.; Watson, A.; Robson, C. N. PLoS One 2012, 7, e45539. doi:10.1371/journal.pone.0045539
Return to citation in text: [1] [2] -
Baerfacker, L.; Prechtl, S.; Siemeister, G.; Wengner, A. M.; Ackerstaff, J.; Nowak-Reppel, K.; Bader, B.; Lienau, P.; Stoeckigt, D. Amino-substituted isothiazoles. Int. Pat. Appl. WO 2014/118186 A1, Aug 7, 2014.
Return to citation in text: [1] [2] -
Yu, G. Isothiazole derivative PIM kinase inhibitor and its preparation method and application in pharmaceutical manufacturing. Chin. Pat. Appl. CN105254624A, Jan 20, 2016.
Return to citation in text: [1] -
Ambati, S. R.; Gudala, S.; Sharma, A.; Penta, S.; Reddy, V. L.; Bomma, Y.; Janapala, V. R.; Pola, S. J. Heterocycl. Chem. 2017, 54, 2333–2341. doi:10.1002/jhet.2822
Return to citation in text: [1] -
Banerjee, A.; Yadav, P. S.; Bajpai, M.; Sangana, R. R.; Gullapalli, S.; Gudi, G. S.; Gharat, L. A. Bioorg. Med. Chem. Lett. 2012, 22, 3223–3228. doi:10.1016/j.bmcl.2012.03.025
Return to citation in text: [1] -
Shintani, Y.; Niwa, M.; Monma, S.; Ishiyama, S.; Shigeta, Y.; Kamiyama, T.; Shimazawa, Y.; Okada, K. Substituted azole compound and therapeutic agent for diabetes. Int. Pat. Appl. WO 2015/170775 A1, Nov 12, 2015.
Return to citation in text: [1] -
Sui, Z.; Cai, C.; Zhang, X. GPR120 Agonists for the Treatment of Type II Diabetes. U.S. Pat. Appl. US 2014/0275179 A1, Sept 18, 2014.
Return to citation in text: [1] -
Zhang, X.; Cai, C.; Sui, Z.; Macielag, M.; Wang, Y.; Yan, W.; Suckow, A.; Hua, H.; Bell, A.; Haug, P.; Clapper, W.; Jenkinson, C.; Gunnet, J.; Leonard, J.; Murray, W. V. ACS Med. Chem. Lett. 2017, 8, 947–952. doi:10.1021/acsmedchemlett.7b00233
Return to citation in text: [1] -
Bobbio, C.; Corsi, C.; Jeanmart, S. A. M.; Wendeborn, S. V. Isoxazole, isothiazole, furane and thiophene compounds as microbicides. Int. Pat. Appl. WO 2012/010567 A1, Jan 26, 2012.
Return to citation in text: [1] -
Byrappa, S.; Harsha Raj, M.; Kungyal, T.; Kudva N, N. U.; Salimath, B. P.; Lokanatha Rai, K. M. Eur. J. Med. Chem. 2017, 126, 218–224. doi:10.1016/j.ejmech.2016.09.094
Return to citation in text: [1] -
Kletskov, A. V.; Bumagin, N. A.; Zubkov, F. I.; Grudinin, D. G.; Potkin, V. I. Synthesis 2020, 52, 159–188. doi:10.1055/s-0039-1690688
Return to citation in text: [1] -
De Oliveira Silva, A.; McQuade, J.; Szostak, M. Adv. Synth. Catal. 2019, 361, 3050–3067. doi:10.1002/adsc.201900072
Return to citation in text: [1] -
Skrastiņa, I.; Baran, A.; Muceniece, D.; Popelis, J. Chem. Heterocycl. Compd. 2014, 50, 87–102. doi:10.1007/s10593-014-1451-1
Return to citation in text: [1] [2] -
Ingate, S. T.; Marco, J. L.; Witvrouw, M.; Pannecouque, C.; De Clercq, E. Tetrahedron 1997, 53, 17795–17814. doi:10.1016/s0040-4020(97)10244-7
Return to citation in text: [1] [2] -
Ilkin, V. G.; Gridnev, I. D.; Novikov, M. S.; Silaichev, P. S.; Beryozkina, T. V.; Slepukhin, P. A.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2025, 90, 2245–2257. doi:10.1021/acs.joc.4c02623
Return to citation in text: [1] [2] -
Ilkin, V. G.; Filimonov, V. O.; Utepova, I. A.; Beryozkina, T. V.; Slepukhin, P. A.; Tumashov, A. A.; Dehaen, W.; Bakulev, V. A. Org. Chem. Front. 2024, 11, 3537–3545. doi:10.1039/d3qo02025h
Return to citation in text: [1] -
Ilkin, V. G.; Silaichev, P. S.; Filimonov, V. O.; Beryozkina, T. V.; Krasilnikov, V. A.; Belskaya, N. P.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2025, 90, 3590–3602. doi:10.1021/acs.joc.4c02812
Return to citation in text: [1] -
Evans, D. A.; Faul, M. M.; Bilodeau, M. T. J. Org. Chem. 1991, 56, 6744–6746. doi:10.1021/jo00024a008
Return to citation in text: [1] -
Taylor, S.; Gullick, J.; McMorn, P.; Bethell, D.; Bulman Page, P. C.; Hancock, F. E.; King, F.; Hutchings, G. J. J. Chem. Soc., Perkin Trans. 2 2001, 1714–1723. doi:10.1039/b104522a
Return to citation in text: [1] -
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1]
| 24. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
| 25. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 1. | Garozzo, A.; Stivala, A.; Tempera, G.; Castro, A. Antiviral Res. 2010, 88, 325–328. doi:10.1016/j.antiviral.2010.10.003 |
| 12. | Bobbio, C.; Corsi, C.; Jeanmart, S. A. M.; Wendeborn, S. V. Isoxazole, isothiazole, furane and thiophene compounds as microbicides. Int. Pat. Appl. WO 2012/010567 A1, Jan 26, 2012. |
| 13. | Byrappa, S.; Harsha Raj, M.; Kungyal, T.; Kudva N, N. U.; Salimath, B. P.; Lokanatha Rai, K. M. Eur. J. Med. Chem. 2017, 126, 218–224. doi:10.1016/j.ejmech.2016.09.094 |
| 22. | Taylor, S.; Gullick, J.; McMorn, P.; Bethell, D.; Bulman Page, P. C.; Hancock, F. E.; King, F.; Hutchings, G. J. J. Chem. Soc., Perkin Trans. 2 2001, 1714–1723. doi:10.1039/b104522a |
| 9. | Shintani, Y.; Niwa, M.; Monma, S.; Ishiyama, S.; Shigeta, Y.; Kamiyama, T.; Shimazawa, Y.; Okada, K. Substituted azole compound and therapeutic agent for diabetes. Int. Pat. Appl. WO 2015/170775 A1, Nov 12, 2015. |
| 10. | Sui, Z.; Cai, C.; Zhang, X. GPR120 Agonists for the Treatment of Type II Diabetes. U.S. Pat. Appl. US 2014/0275179 A1, Sept 18, 2014. |
| 11. | Zhang, X.; Cai, C.; Sui, Z.; Macielag, M.; Wang, Y.; Yan, W.; Suckow, A.; Hua, H.; Bell, A.; Haug, P.; Clapper, W.; Jenkinson, C.; Gunnet, J.; Leonard, J.; Murray, W. V. ACS Med. Chem. Lett. 2017, 8, 947–952. doi:10.1021/acsmedchemlett.7b00233 |
| 23. | Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. J. Appl. Crystallogr. 2009, 42, 339–341. doi:10.1107/s0021889808042726 |
| 8. | Banerjee, A.; Yadav, P. S.; Bajpai, M.; Sangana, R. R.; Gullapalli, S.; Gudi, G. S.; Gharat, L. A. Bioorg. Med. Chem. Lett. 2012, 22, 3223–3228. doi:10.1016/j.bmcl.2012.03.025 |
| 20. | Ilkin, V. G.; Silaichev, P. S.; Filimonov, V. O.; Beryozkina, T. V.; Krasilnikov, V. A.; Belskaya, N. P.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2025, 90, 3590–3602. doi:10.1021/acs.joc.4c02812 |
| 2. | Lippa, B.; Morris, J.; Corbett, M.; Kwan, T. A.; Noe, M. C.; Snow, S. L.; Gant, T. G.; Mangiaracina, M.; Coffey, H. A.; Foster, B.; Knauth, E. A.; Wessel, M. D. Bioorg. Med. Chem. Lett. 2006, 16, 3444–3448. doi:10.1016/j.bmcl.2006.04.003 |
| 3. | Carlessi, L.; Buscemi, G.; Larson, G.; Hong, Z.; Wu, J. Z.; Delia, D. Mol. Cancer Ther. 2007, 6, 935–944. doi:10.1158/1535-7163.mct-06-0567 |
| 4. | Coffey, K.; Blackburn, T. J.; Cook, S.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Hewitt, L.; Huberman, K.; McNeill, H. V.; Newell, D. R.; Roche, C.; Ryan-Munden, C. A.; Watson, A.; Robson, C. N. PLoS One 2012, 7, e45539. doi:10.1371/journal.pone.0045539 |
| 5. | Baerfacker, L.; Prechtl, S.; Siemeister, G.; Wengner, A. M.; Ackerstaff, J.; Nowak-Reppel, K.; Bader, B.; Lienau, P.; Stoeckigt, D. Amino-substituted isothiazoles. Int. Pat. Appl. WO 2014/118186 A1, Aug 7, 2014. |
| 6. | Yu, G. Isothiazole derivative PIM kinase inhibitor and its preparation method and application in pharmaceutical manufacturing. Chin. Pat. Appl. CN105254624A, Jan 20, 2016. |
| 7. | Ambati, S. R.; Gudala, S.; Sharma, A.; Penta, S.; Reddy, V. L.; Bomma, Y.; Janapala, V. R.; Pola, S. J. Heterocycl. Chem. 2017, 54, 2333–2341. doi:10.1002/jhet.2822 |
| 21. | Evans, D. A.; Faul, M. M.; Bilodeau, M. T. J. Org. Chem. 1991, 56, 6744–6746. doi:10.1021/jo00024a008 |
| 16. | Skrastiņa, I.; Baran, A.; Muceniece, D.; Popelis, J. Chem. Heterocycl. Compd. 2014, 50, 87–102. doi:10.1007/s10593-014-1451-1 |
| 18. | Ilkin, V. G.; Gridnev, I. D.; Novikov, M. S.; Silaichev, P. S.; Beryozkina, T. V.; Slepukhin, P. A.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2025, 90, 2245–2257. doi:10.1021/acs.joc.4c02623 |
| 16. | Skrastiņa, I.; Baran, A.; Muceniece, D.; Popelis, J. Chem. Heterocycl. Compd. 2014, 50, 87–102. doi:10.1007/s10593-014-1451-1 |
| 17. | Ingate, S. T.; Marco, J. L.; Witvrouw, M.; Pannecouque, C.; De Clercq, E. Tetrahedron 1997, 53, 17795–17814. doi:10.1016/s0040-4020(97)10244-7 |
| 18. | Ilkin, V. G.; Gridnev, I. D.; Novikov, M. S.; Silaichev, P. S.; Beryozkina, T. V.; Slepukhin, P. A.; Dehaen, W.; Bakulev, V. A. J. Org. Chem. 2025, 90, 2245–2257. doi:10.1021/acs.joc.4c02623 |
| 19. | Ilkin, V. G.; Filimonov, V. O.; Utepova, I. A.; Beryozkina, T. V.; Slepukhin, P. A.; Tumashov, A. A.; Dehaen, W.; Bakulev, V. A. Org. Chem. Front. 2024, 11, 3537–3545. doi:10.1039/d3qo02025h |
| 14. | Kletskov, A. V.; Bumagin, N. A.; Zubkov, F. I.; Grudinin, D. G.; Potkin, V. I. Synthesis 2020, 52, 159–188. doi:10.1055/s-0039-1690688 |
| 15. | De Oliveira Silva, A.; McQuade, J.; Szostak, M. Adv. Synth. Catal. 2019, 361, 3050–3067. doi:10.1002/adsc.201900072 |
| 1. | Garozzo, A.; Stivala, A.; Tempera, G.; Castro, A. Antiviral Res. 2010, 88, 325–328. doi:10.1016/j.antiviral.2010.10.003 |
| 2. | Lippa, B.; Morris, J.; Corbett, M.; Kwan, T. A.; Noe, M. C.; Snow, S. L.; Gant, T. G.; Mangiaracina, M.; Coffey, H. A.; Foster, B.; Knauth, E. A.; Wessel, M. D. Bioorg. Med. Chem. Lett. 2006, 16, 3444–3448. doi:10.1016/j.bmcl.2006.04.003 |
| 3. | Carlessi, L.; Buscemi, G.; Larson, G.; Hong, Z.; Wu, J. Z.; Delia, D. Mol. Cancer Ther. 2007, 6, 935–944. doi:10.1158/1535-7163.mct-06-0567 |
| 4. | Coffey, K.; Blackburn, T. J.; Cook, S.; Golding, B. T.; Griffin, R. J.; Hardcastle, I. R.; Hewitt, L.; Huberman, K.; McNeill, H. V.; Newell, D. R.; Roche, C.; Ryan-Munden, C. A.; Watson, A.; Robson, C. N. PLoS One 2012, 7, e45539. doi:10.1371/journal.pone.0045539 |
| 5. | Baerfacker, L.; Prechtl, S.; Siemeister, G.; Wengner, A. M.; Ackerstaff, J.; Nowak-Reppel, K.; Bader, B.; Lienau, P.; Stoeckigt, D. Amino-substituted isothiazoles. Int. Pat. Appl. WO 2014/118186 A1, Aug 7, 2014. |
| 17. | Ingate, S. T.; Marco, J. L.; Witvrouw, M.; Pannecouque, C.; De Clercq, E. Tetrahedron 1997, 53, 17795–17814. doi:10.1016/s0040-4020(97)10244-7 |
© 2025 Ilkin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.