1School of Medicine, Southern University of Science and Technology, Shenzhen, 518055, People’s Republic of China
2Academy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen, 518055, People’s Republic of China
Corresponding author email
Associate Editor: D. Y.-K. Chen Beilstein J. Org. Chem.2020,16, 3015–3031.https://doi.org/10.3762/bjoc.16.251 Received 25 Aug 2020,
Accepted 21 Nov 2020,
Published 09 Dec 2020
Many natural products possess interesting medicinal properties that arise from their intriguing chemical structures. The highly-substituted carbocycle is one of the most common structural features in many structurally complicated natural products. However, the construction of highly-substituted, stereo-congested, five-membered carbocycles containing all-carbon quaternary center(s) is, at present, a distinct challenge in modern synthetic chemistry, which can be accessed through the all-carbon [3 + 2] cycloaddition. More importantly, the all-carbon [3 + 2] cycloaddition can forge vicinal all-carbon quaternary centers in a single step and has been demonstrated in the synthesis of complex natural products. In this review, we present the development of all-carbon [3 + 2] cycloadditions and illustrate their application in natural product synthesis reported in the last decade covering 2011–2020 (inclusive).
The highly-substituted, stereo-congested, five-membered carbocycle containing contiguous stereocenters is one of the most common structural features in many structurally complicated, biologically important natural products [1-7] (Figure 1). Meanwhile, the construction of quaternary carbon stereocenter(s) is, at present, a distinct challenge in modern synthetic chemistry [8-11]. Therefore, the synthesis of highly-substituted five-membered carbocycles bearing congested arrays of stereocenters within the polycyclic framework of complex natural products usually require a sophisticated synthetic planning. This issue is not trivial because only a few strategies are available for the efficient synthesis of such an intriguing molecular architecture. More importantly, the all-carbon [3 + 2] cycloaddition can forge vicinal all-carbon quaternary centers [12] in a single-step operation and provides a direct access to various substituted five-membered carbocycles. These characteristics make the all-carbon [3 + 2] cycloaddition an appealing method and/or strategy in the synthesis of complex natural products (Figure 2).
Figure 1:
Highly-substituted five-membered carbocycle in biologically significant natural products.
Figure 1:
Highly-substituted five-membered carbocycle in biologically significant natural products.
Figure 2:
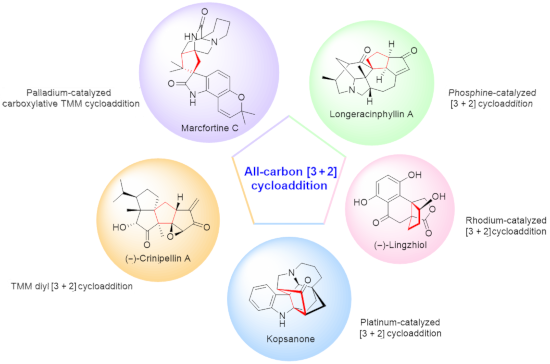
Natural product synthesis featuring the all-carbon [3 + 2] cycloaddition. (Quaternary carbon center(s) created by all-carbon [3 + 2] cyclization are highlighted in cyan; quaternary carbon center(s) created that are removed by subsequent transformations are highlighted in lilac; cyclopentane structures forged by the all-carbon [3 + 2] cyclization are labeled in red). (A) The intermolecular all-carbon [3 + 2] cyclization features as the key reaction. (B) The intramolecular all-carbon [3 + 2] cycloaddition features as the key reaction.
The 1,3-dipolar cycloaddition has been well-documented and widely used for the construction of five-membered heterocycles since the 1960s [13]. However, the development of the all-carbon [3 + 2] cycloaddition, for instance, Berson’s and Little’s [3 + 2] cycloaddition through diyl trapping with an olefin [14,15] and Trost’s palladium-catalyzed trimethylenemethane cycloaddition [16], which allows the preparation of five-membered carbocycles, have been emerged since the 1970s. Thereafter, many novel and important all-carbon [3 + 2] cycloaddition reactions, such as the phosphine-catalyzed [3 + 2] cycloaddition [17], platinum-catalyzed [3 + 2] cycloaddition [18], and Rhodium-catalyzed [3 + 2] cycloaddition [12], were invented and have been extensively used in natural product synthesis in the last decade. Many reviews focusing the method development of the all-carbon [3 + 2] cycloaddition have been published [19-21]. However, there is no review effort, to the best of our knowledge, has been paid attention to the development of the all-carbon [3 + 2] cycloaddition with an emphasis on the natural product synthesis. Therefore, we are motivated to provide a timely and focused review of all-carbon [3 + 2] cycloadditions in natural product synthesis.
In this review, we present the development of the all-carbon [3 + 2] cycloaddition and discuss its application in natural product synthesis reported from 2011–2020. We begin with describing the brief history of the all-carbon [3 + 2] cycloaddition with selected natural product syntheses reported before 2011 [22-26]. Next, we discuss the synthetic methods including the proposed mechanism and/or catalytic cycle and focus on illustrative examples of natural product syntheses. Moreover, several natural product syntheses featuring all-carbon [3 + 2] annulation are elaborated. Lastly, we discuss future directions and opportunities for the all-carbon [3 + 2] cycloaddition.
Review
In 1981, Little and co-workers utilized a trimethylenemethane (TMM) cycloaddition as the key reaction to synthesize the tricyclic compound 25, which led to the synthesis of (±)-hirsutene (14) [22] (Scheme 1A). Refluxing azo compound 22 in acetonitrile generated the proposed biradical intermediate 23 through nitrogen extrusion. This intermediate underwent isomerization to 24 and intramolecular diyl trapping through a [3 + 2] cycloaddition to give fused tricycle 25 in 85% yield. The synthesis of the hydroxykempenone 3β-hydroxykemp-7(8)-en-6-one (7) features Trost’s palladium-catalyzed trimethylenemethane [3 + 2] cycloaddition [27] and was reported by Paquette and co-workers in 1992 [23] (Scheme 1B). Catalytic TMM [3 + 2] cycloaddition of activated octalone 26 and the trimethylenemethane precursor 27 selectively produced adduct 28 in 98% yield, which is a synthetic precursor of 3β-hydroxykemp-7(8)-en-6-one (7).
Scheme 1:
Representative natural product syntheses that feature the all-carbon [3 + 2] cyclization as the key reaction, reported before 2011. (A) TMM cycloaddition of diyl 24 resulted from dinitrogen extrusion/isomerization is used to prepare tricycle 25, which is a synthetic precursor of (±)-hirsutene (14) [22]. (B) Synthesis of 3β-hydroxykemp-7(8)-en-6-one (7) features a palladium-catalyzed intermolecular [3 + 2] cycloaddition to generate tricycle 28[23]. (C) A stereospecific phosphine-catalyzed [3 + 2] cycloaddition completes the synthesis of (±)-hirsutene (14) [25]. (D) Linear alkylidene carbenes involved TMM [3 + 2] cycloaddition produces tricycle 36 in the preparation of (±)-hirsutene (14) [24].
Scheme 1:
Representative natural product syntheses that feature the all-carbon [3 + 2] cyclization as the key...
Another two syntheses of (±)-hirsutene (14), after Little’s pioneering work [22], were accomplished by Krische [25] and Lee [24] independently in 2003. (Scheme 1C and Scheme 1D) In Krische’s synthesis, a stereospecific intramolecular phosphine-catalyzed [3 + 2] cycloaddition of 2-butynoate with electron-deficient alkene 29 afforded cycloadduct 31 in 88% yield as a single diastereomer [25] (Scheme 1C). Later, Lee`s synthesis of (±)-hirsutene (14) used an alkylidene carbene as source of TMM diyl in the intramolecular [3 + 2] cycloaddition [24] (Scheme 1D). Heating of epoxyaziridinyl imine 32 produced tricyclic compound 36 in 57% yield as a single product. The authors proposed that heating of epoxyaziridinyl imine 32 generates alkylidene carbene 33. Transformation of 33 to TMM diyl 35 enables an intramolecular [3 + 2] cycloaddition to give the desired tricyclic product 36.
Trimethylenemethane (TMM) cycloaddition
An intramolecular trimethylenemethane diyl [3 + 2] cycloaddition was reported by Berson [28] and Little [14] independently in the late 1970s, which was used to prepare (±)-hirsutene (14) in 1981 [22] (Scheme 1A). In 2003, Lee and co-workers disclosed an intramolecular trimethylenemethane diyl [3 + 2] cycloaddition with a linear alkylidene carbene as diyl source and was applied in the synthesis of linearly fused triquinane (±)-hirsutene (14) [24] (Scheme 1D). In 2011, the same research group used allenyl diazo compound 38, which was generated from the reaction between aldehyde 37 and p-toluenesulfonehydrazide in the presence of sodium hydride upon heating, to produce diyl 40[29] (Scheme 2A). The intramolecular trimethylenemethane diyl [3 + 2] cycloaddition of 40 led to the formation of angular fused triquinane 41 in 98% yield. The authors suggested that an intramolecular cycloaddition of the diazo group and allene 38 produces tetrahydrocyclopentapyrazole 39. Extrusion of nitrogen from the newly formed 39 produces diyl 40, which undergoes [3 + 2] cycloaddition to produce the angular fused triquinane 41.
Scheme 2:
(A) An intramolecular trimethylenemethane diyl [3 + 2] cycloaddition with allenyl diazo compound 38 as a key intermediate to give angular-fused triquinane 41[29]. (B) Synthesis of (−)-crinipellin A (15) [30]. (C) Synthesis of waihoensene (16) [31].
Scheme 2:
(A) An intramolecular trimethylenemethane diyl [3 + 2] cycloaddition with allenyl diazo compound 38...
With the successful preparation of angular fused triquinane 41 by trimethylenemethane diyl [3 + 2] cycloaddition [29], enabled the synthesis of (−)-crinipellin A (15) [30] and waihoensene (16) [31] by Lee and co-workers in 2014 and 2017, respectively (Scheme 2B and Scheme 2C). The synthesis of (−)-crinipellin A (15) began with the treatment of hydrazone 42 with sodium hydride under reflux to produce the tetraquinane 46 in 87% yield [30] (Scheme 2B). The authors suggested that the diazo compound 43 formed undergoes an intramolecular cycloaddition to give 44. Freshly prepared 44 was converted to diyl 45 followed by another cycloaddition to give the tetraquinane 46. A four-step synthesis from the tetraquinane 46 gave diketone 47. Treatment of sulfoximine 48 with n-butyllithium generated the corresponding anion, which selectively attacked the C-8 ketone moiety of 47 to give alcohol 49 in β-configuration in 80% yield [32]. Chemoselective and stereoselective reduction of the C-9 ketone of 49 was accomplished by treatment with NaBH(OAc)3[33] and produced 50 after a two-step synthesis. Removal of the sulfoximine group in 50 upon refluxing in toluene and subsequent epoxidation afforded 51[32], which was converted to (−)-crinipelline A (15) in two steps.
The synthesis of waihoensene (16) commenced with the conversion of aldehyde 52a to the corresponding hydrazone 52b, which was treated with sodium hydride under reflux to give 56 in 83% yield over two steps [31] (Scheme 2C). This transformation was rationalized as follows: freshly prepared 52b was converted to diazo 53, which was subjected to [3 + 2] cycloaddition to give adduct 54. Formation of diyl 55 from 54 and subsequent [3 + 2] cycloaddition produced the tetracyclic compound 56. Dihydroxylation of freshly prepared 56 with OsO4 and then selective tosylation afforded 57 in 39% yield over two steps. Exposure of 57 to DBU upon heating gave the elimination product 58, which was subjected to an oxidative rearrangement with PDC to give enone 59 in 68% yield. Copper-mediated conjugated addition of methyllithium to enone 59 in the presence of boron trifluoride ether [34,35] produced desired ketone 60 in 75% yield. The resultant ketone 60 was converted to waihoensene (16) in two steps.
In 1986, Trost and co-workers disclosed the palladium-catalyzed intermolecular carboxylative TMM [3 + 2] cycloaddition [36] (Scheme 3). Exposure of coumarin 61 to the silyl-substituted TMM precursor 62 in the presence of a catalytic amount of Pd(PPh3)4 afforded adduct 63 in 81% yield as a single diastereomer (Scheme 3A). Trost and co-workers proposed that the catalytic mechanism involves an oxidative addition of palladium(0) into 62 affording the η3-Pd TMM complex A[37] (Scheme 3B). Methyl trimethylsilyl carbonate (64) is formed as side product, which is in equilibrium with carbon dioxide and methyl trimethylsilyl ether. The electron-rich end of complex A attacks the carbon dioxide to give carboxylate B. Migration of the TMS group on carboxylate B generates the 1,3-dipole on C in the form of TMS carboxylate. An intermolecular [3 + 2] cycloaddition of C and alkene D (see Scheme 3B, inset) gives the cycloaddition adduct E, which is converted to the corresponding carboxylic acid (not shown) upon reaction work-up. This elegant reaction was applied in the synthesis of marcfortine B (8), reported by Trost and co-workers in 2007 [38] and 2013 [39].
The synthesis of marcfortine B (8) began with palladium-catalyzed intermolecular carboxylatve TMM [3 + 2] cycloaddition [36] of enone 65 and TMM donor 62 to forge the highly-substituted spirocyclic cyclopentane 66a[38] (Scheme 4A). Methylation of the resultant cyclopentane 66a gave methyl ester 66b in 93% yield over two steps. A six-step synthesis from ester 66b gave α,β-unsaturated amide 67, which was treated with KHMDS to facilitate an intramolecular Michael addition to give lactam 68 in quantitative yield. The conversion of freshly prepared lactam 68 to xanthante ester 69 was achieved in three steps. Exposure of xanthante ester 69 to AIBN and a catalytic amount of tributylstannane [40] led to a radical cyclization, in which the resultant alkyl radical formed was trapped by AIBN to give a proposed nitrogen-centered radical 70. An 1,4-hydrogen abstraction of the nitrogen-centered radical on 70 produced carbon-centered radical 71, which underwent fragmentation to afford alkene 72 in 61% yield. Marcfortine B (8) was synthesized from alkene 72 in seven steps.
Scheme 4:
Natural product syntheses that make use of palladium-catalyzed intermolecular [3 + 2] cycloadditions of TMM. (A) Synthesis of marcfortine B (8) uses a palladium-catalyzed carboxylatve TMM [3 + 2] cycloaddition [38,39]. (B) Enantioselective synthesis of marcfortine C (9) features a palladium-catalyzed asymmetric cyano-substituted TMM [3 + 2] cycloaddition [39].
Scheme 4:
Natural product syntheses that make use of palladium-catalyzed intermolecular [3 + 2] cycloaddition...
The enantioselective synthesis of marcfortine C (9) commenced with a catalytic asymmetric cyano-substituted TMM cycloaddition of oxindole 73 and TMM donor 75 with Pd(dba)2/74 as catalyst to give a cycloaddition adduct (not shown) [39] (Scheme 4B). Subsequent treatement with t-BuOLi resulted in the isomerization of the exo-olefin followed by exposure to n-butyllithium and Davis‘ oxaziridine 76 to give 77 in 60% yield with 89% ee. A three-step synthesis from 77 gave α,β-unsaturated amide 78, which underwent successive intramolecular Michael addition and hydrolytic nitrile reduction to give 79 in 46% yield in two steps. Extensive studies of the nitrile reduction eventually identified that Et3Al and DIBAL-H could effectively reduce the nitrile group to the corresponding aldehyde and treatment with NaBH4 afforded alcohol 79. Alcohol 79 was converted into the corresponding xanthate ester 80. This ester 80 was exposed to an excessive amount of AIBN and N,O-bis(trimethylsilyl)acetamide in the presence of a catalytic amount of tributylstannane producing bicyclo[2.2.2]diazaoctane 81 in 54% yield. The authors mentioned that the employment of the previously reported conditions for the radical cyclization in the synthesis of marcfortine B (8) led to the decomposition of the starting material. It was suggested that the MOM group of 80 may contribute to undesired side reactions. Synthesis of marcfortine C (9) was accomplished from 81 in two steps.
Phosphine-catalyzed [3 + 2] cycloaddition
In 1995, Lu and co-workers reported a phosphine-catalyzed [3 + 2] cycloaddition, employing electron-deficient olefins and either 2,3-butadienoates or 2-butynoates to give a cyclopentene as product [17] (Scheme 5A). The reaction between ethyl 2,3-butadienoate (82) and diethyl fumarate (83) in the presence of 10 mol % of triphenylphosphine afforded trans-84 in 67% yield. Under the same conditions, the use of diethyl maleate in place of diethyl fumarate (83) will give cis-84 in 46% yield (not shown). Lu and co-workers proposed that the catalytic mechanism involves a reaction between phosphine catalyst A and allene 82 to give B and/or C (Scheme 5B). Catalytic [3 + 2] cycloaddition of B and/or C and alkene D gives the cyclic intermediates E and F in an equilibrium state through a 1,2-proton transfer. The loss of phosphine catalyst from E or F affords the cycloaddition product G and the catalyst is regenerated. It is noteworthy that ethyl 2-butynoate (85) can be used as substrate in place of ethyl 2,3-butadienoate (82) in the phosphine-catalyzed [3 + 2] cycloaddition. Ethyl 2-butynoate (85) enters the catalytic cycle by reacting with phosphine catalyst A to give H and C.
Some total syntheses of hexacyclic Daphniphyllum alkaloids were reported by Li’s group (longeracinphyllin A (10) [41] and daphenylline (11) [42]) and Zhai’s group (daphenylline (11) [43]), applying Lu’s [3 + 2] cycloaddition (Scheme 6). The synthesis of longeracinphyllin A (10), which was reported by Li and co-workers in 2017, used a 1,1’-bis(diphenylphosphino)ferrocene-promoted [3 + 2] cycloaddition [44] of enedione 86 and allenoate 87 to give adduct 88 in 45% yield. This adduct 88 was treated with an excess of LiCH2PO(OMe)2 to afford β-ketophosphonate 89 in 86% yield (Scheme 6A) [41]. Hydrogenation of 89 followed by an intramolecular Horner–Wadsworth–Emmons olefination produced hexacyclic enone 90 in 91% yield over two steps. The conversion of enone 90 to longeracinphyllin A (10) was achieved in three steps.
Scheme 6:
Lu’s [3 + 2] cycloaddition in natural product synthesis. (A) Synthesis of longeracinphyllin A (10) [41]. (B) Synthesis of daphenylline (11) [42]. (C) Synthesis of daphenylline (11) [43].
Scheme 6:
Lu’s [3 + 2] cycloaddition in natural product synthesis. (A) Synthesis of longeracinphyllin A (10) [41]...
The syntheses of daphenylline (11) were reported by Li’s group [42] and Zhai’s group [43] independently in 2017 (Scheme 6B and Scheme 6C). In Li’s synthesis, the common intermediate dienone 86 was subjected to a 1,1’-bis(diphenylphosphino)ferrocene-promoted [3 + 2] cycloaddition [41] with allenyl ketone 91 to give adduct 92a in 52% yield (Scheme 6B). This adduct 92a underwent decarboxylation to afford 92b in 72% yield [42]. Exposure of freshly prepared 92b to triazabicyclodecene [45] led to a ring-expansion/aromatization/aldol cascade producing 93, which was reduced with Et3SiH/TFA smoothly to give indane 94 in 68% yield over two steps. The freshly prepared indane 94 was converted to daphenylline (11) in two steps. The preparation of daphnipaxianine A and himalenine D (not shown) were also disclosed in the same work but are not described here.
Zhai’s synthesis of daphenylline (11) used Lu’s phosphine-catalyzed [3 + 2] cycloaddition [17] of enone 95 and tert-butyl 2-butynoate (96) with PBu3 and K2CO3/MeOH as additive to give the cycloaddition adduct 97 in 83% yield [43] (Scheme 6C). A seven-step synthesis from 97 gave pentacyclic ketone 98. Pentacyclic ketone 98 was exposed to PTSA under reflux to give the Wagner–Meerwein rearrangement product 99 in 85% yield. The synthesis of daphenylline (11) was completed by a seven-step synthesis from benzofuran 99.
In 2019, Lu and co-workers disclosed a novel chiral-phosphine-catalyzed enantioselective [3 + 2] annulation of allenes and isoindigos to give an enantioenriched annulation adduct bearing vicinal quaternary stereocenters [46] (Scheme 7A). Both symmetric and unsymmetric isoindigos can undergo enantioselective [3 + 2] annulation with an allene and produced a chiral adduct with high yield and high ee value. When unsymmetric isoindigo 100 was used as substrate, enantioselective [3 + 2] annulation with allene 101 in the presence of amino acid-derived bifunctional phosphine 102 produced adduct 103 in 90% yield with 92% ee and 4:1 regioisomeric ratio (rr). The authors suggested that the observed regioselectivity could be rationalized by the proposed catalytic mechanism (Scheme 7B). The phosphine (i.e., PR3, A) attacks the allene 101 to generate zwitterion intermediate B, which is subjected to a less hindered attack by the isoindigo 100. The oxindole bearing a chlorine atom on isoindigo 100 makes C-3 more electron deficient than C-3’, which results in the regioselective formation of intermediate C. Cyclization of intermediate C gives D and subsequent proton transfer produces isomer E. It undergoes elimination to afford the annulation product 103 and the phosphine catalyst A is regenerated.
Scheme 7:
(A) Phosphine-catalyzed [3 + 2] annulation of unsymmetric isoindigo 100 with allene in the preparation of spiro adduct 103[46]. (B) The proposed catalytic cycle. (C) Application of phosphine-catalyzed asymmetric [3 + 2] annulation to prepare the chiral adduct 105 with symmetric isoindigo 104 in the formal synthesis of (−)-ditryptophenaline (12).
Scheme 7:
(A) Phosphine-catalyzed [3 + 2] annulation of unsymmetric isoindigo 100 with allene in the preparat...
In the same work, Lu and co-workers applied the enantioselective [3 + 2] annulation to complete the formal synthesis of (−)-ditryptophenaline (12) [46] (Scheme 7C). The synthesis began with the catalytic asymmetric [3 + 2] annulation of symmetric isoindigo 104 and allene 101 with chiral phosphine catalyst 102 to give spirocyclic adduct 105 in 93% yield with 99% ee. The freshly prepared enantioenriched adduct 105 was subjected to ozonolysis [47] followed by decarboxylation to give bisoxindole 106 in 68% yield over two steps. Conversion of 106 to the corresponding acetal and subsequent allylation afforded 108 in 86% yield over two steps. A two-step synthesis from 108 produced 109, which was converted to (−)-ditryptophenaline (12) by using Overman’s protocol [48].
Rhodium-catalyzed [3 + 2] cycloaddition
In 2014, Yang and co-workers reported an efficient rhodium-catalyzed intramolecular [3 + 2] cycloaddition of 110 to give [3.3.0] and [3.4.0] bicyclic systems bearing two quaternary atoms at the bridgehead position [49]. For instance, enynol 110 was treated with 5 mol % of [RhCl(CO)2]2 and carbon monoxide to afford a [3.3.0] bicycle 111 in 87% yield (Scheme 8A). The proposed catalytic cycle of this elegant rhodium-catalyzed intramolecular [3 + 2] cycloaddition begins with the reaction between the rhodium catalyst Rh(I)LCl and alcohol 110 to give complex A through alcoholysis [50,51] (Scheme 8B). Rh(I)-mediated retro-propargylation of the homopropargyl alcohol A afforded complex B. It undergoes an intramolecular Michael addition [52,53] with the allenyl rhodium to the enal and gives the allenyl rhodium species C. A Conia-ene-type reaction [54] between the Rhoda-enolate species and the allene of complex C produces the desired [3.3.0] bicycle D. Protonolysis [55-57] of complex D with the alcohol 110 gives bicyclic product 111 and regenerates the rhodium complex A. This elegant method has been successfully applied by the same research group in their synthesis of lingzhiol (17) [49], lycojaponicumin C (18) [58] and sinensilactam A (20) [59] (Scheme 9).
Scheme 8:
(A) Rhodium-catalyzed intracmolecular [3 + 2] cycloaddition [49]. (B) The proposed catalytic cycle of the reaction.
Scheme 8:
(A) Rhodium-catalyzed intracmolecular [3 + 2] cycloaddition [49]. (B) The proposed catalytic cycle of t...
Scheme 9:
Total synthesis of natural products reported by Yang and co-workers applying rhodium-catalyzed intramolecular [3 + 2] cycloaddition. (A) Synthesis of (−)-lingzhiol (17) [49]. (B) Synthesis of lycojaponicumin C (18) [58]. (C) Synthesis of sinensilactam A (20) [59].
Scheme 9:
Total synthesis of natural products reported by Yang and co-workers applying rhodium-catalyzed intr...
The synthesis of (−)-lingzhiol (17) was reported by Yang and co-workers in 2014 [49] (Scheme 9A). The synthesis began with the conversion of ketone 112 into alcohol 113 in four steps, which involved a hypervalent iodine-mediated ring expansion [60]. A two-step synthesis from 113 gave epoxide 114. Epoxide 114 was converted to the corresponding β-ketoester and subsequent treatment with Waser’s reagent 116[61] afforded alkyne 117 in 62% yield over two steps. Enyne 118, which was prepared in two steps from 117, was subjected to rhodium-catalyzed intramolecular [3 + 2] cycloaddition in the presence of carbon monoxide to give tricycle 119 bearing the desired vicinal quaternary carbon stereocenters in 86% yield. Reduction of aldehyde 119 and subsequent transesterification produced a lactone (not shown). It was exposed to SeO2 to install the allylic hydroxy group to give 120 in 65% yield. Upon catalytic hydrogenation of 120, alcohol 121 was formed. This alcohol 120 was subjected to a bromination [62]/oxidation sequence followed by demethylation to produce (−)-lingzhiol (17).
After the elegant synthesis of (−)-lingzhiol (17) was reported by Yang’s group [49], the same research group disclosed the synthesis of lycojaponicumin C (18) [58] and sinensilactam A (20) [59] in 2017 and 2018, respectively, featuring the rhodium-catalyzed intramolecular [3 + 2] cycloaddition as the key reaction (Scheme 9B and Scheme 9C). Enyne 123, which was prepared from enone 122 in four steps, was subjected to the rhodium-catalyzed intramolecular [3 + 2] cycloaddition under carbon monooxide to give the desired bicyclic [3.3.0] aldehyde 124 in 88% yield. A seven-step synthesis from aldehyde 124 gave azide 125. It was converted to alcohol 126 in seven steps. Alcohol 126 was treated with LDA and vinylMgBr to facilitate a γ-OH directed 1,4-addition [63] to give C-7-vinylated tricycle 127 in 60% yield (74% yield, brsm). A two-step synthesis from 127 produced diene 128, which was subjected to ring-closing metathesis and subsequent Dess–Martin oxidation to give 129 in 63% yield over two steps. Tetracycle 130, which was prepared from 129 in one step, was converted to lycojaponicumin C (18) via Tu’s protocol [64].
The synthesis of sinensilactam A (20) commenced with a three-step synthesis from ketoeseter 131 to give enone 132[59] (Scheme 9C). Selective reduction of the ketone moiety of 132 was accomplished under Luche’s conditions [65] in the presence of calcium chloride [63] to produce the desired alcohol 133 in 75% yield as a single diastereomer. Allylic oxidation of freshly prepared 133 with SeO2 followed by silylation with TBSOTf/Et3N afforded enyne 134. Enyne 134 was subjected to rhodium-catalyzed intramolecular [3 + 2] cycloaddition with a catalytic amount of [Rh(cod)OH]2 to produce 135 and 136 in 85% yield in the ratio of 1:1.16. A six-step synthesis from the major product 136 gave lactone 137. This compond was subjected to successive desilylation, OsO4-mediated dihydroxylation and subsequent oxidative cleavage of the C=C double bond with Pb(OAc)4 to give ketoaldehyde 138 in 64% yield over three steps. The conversion of 138 to sinensilactam A (20) was achieved in two steps.
Platinum-catalyzed [3 + 2] cycloaddition
The platinum-catalyzed intermolecular [3 + 2] cycloaddition of propargyl ether derivatives and vinyl ether producing polycyclic indoles was disclosed by Iwasawa and co-workers in 2011 [18,66] (Scheme 10A). Treatment of Boc-protected aniline 139 and n-butyl vinyl ether (140) with a platinum(II) catalyst afforded tricyclic indole 141 in 83% yield. The authors suggested that this catalytic [3 + 2] cycloaddition reaction may involve an α,β-unsaturated carbene complex intermediate and a mechanism was proposed (Scheme 10B). An nucleophilic attack of the amine nitrogen onto the alkyne 139 under the effect of activated Pt(II) A produces zwitterionic intermediate B. Elimination of the methoxy group from zwitterion B generates the α,β-unsaturated carbene complex intermediate C. C is subjected to the nucleophilic attack of n-butyl vinyl ether (140) and generates alkenyl metallic intermediate D. Intramolecular nucleophilic attack onto the oxonium carbon of D affords the [3 + 2] cycloaddition product 141 with regeneration of the catalyst A.
Scheme 10:
(A) Platinum(II)-catalyzed intermolecular [3 + 2] cycloaddition of propargyl ether 139 and n-butyl vinyl ether (140) gives tricyclic indole 141[18,66]. (B) The proposed mechanism.
Scheme 10:
(A) Platinum(II)-catalyzed intermolecular [3 + 2] cycloaddition of propargyl ether 139 and n-butyl ...
In 2020, Ye and co-workers used a platinum-catalyzed intramolecular [3 + 2] cycloaddition of a propargylic ketal derivative to complete the total synthesis of Kopsia indole alkaloids [67] (Scheme 11). The platinum-catalyzed intramolecular [3 + 2] cycloaddition of propargylic ketal derivative 142 afforded indoline 143 in 58% yield, which possesses three contiguous stereocenters with vicinal all-carbon quaternary centers. (Scheme 11A). According to the proposed mechanism, coordination of the triple bond of 142 to the electrophilic platinum complex A followed by intramolecular nucleophilic attack by the methoxy group gives complex B (Scheme 11B). A facile migration–fragmentation process of complex B eliminates a ketone through fragmentation and produces metal-carbene intermediate C. The freshly prepared metal-carbene C is equilibrated to stabilized 1,3-dipole D. D undergoes a diastereoselective [3 + 2] cycloaddition to give indoline 143 and the active platinum catalyst A is regenerated. After the successful preparation of indoline 143, the synthesis of kopsanone (19) is accomplished (Scheme 11C). Indoline 143 was converted to ketone 144 in three steps, which was subjected to a nucleophilic substitution to give the cyclization product 145 in 76% yield. The hexacyclic compound 145 was converted to kopsanone (19) in three steps.
Scheme 11:
(A) Platinum-catalyzed intramolecular [3 + 2] cycloaddition of propargylic ketal derivative 142 to give indoline 143[67]. (B) The proposed catalytic mechanism. (C) The completion of total synthesis of kopsanone (19).
Scheme 11:
(A) Platinum-catalyzed intramolecular [3 + 2] cycloaddition of propargylic ketal derivative 142 to ...
In 2012, Wang and co-workers reported a Lewis acid-catalyzed intramolecular [3 + 2] cross-cycloaddition (IMCC) of cyclopropane 1,1-diesters with non-activated alkene to generate bridged [n.2.1] carbocyclic skeletons, which is applied to the synthesis of phyllocladanol (21) [68] (Scheme 12A). The IMCC precursor 147 was prepared from aldehyde 146 in nine steps. The IMCC precursor 147 underwent an intramolecular cross-cycloaddition catalyzed by tin tetrachloride to give tetracycle 149 in 81% yield. The authors suggested that the intramolecular [3 + 2] cross-cycloaddition of the less-substituted external carbon atom in the C=C double bond results in the formation of the more stable internal carbenium (i.e., 148) and promotes IMCC to give the bridged [3.2.1] octane 149. The transformation of 149 to phyllocladanol (21) was accomplished in four steps.
Scheme 12:
(A) Synthesis of phyllocladanol (21) features a Lewis acid-catalyzed formal intramolecular [3 + 2] cross-cycloaddition of cyclopropane 1,1-diesters with alkenes [68]. (B) (5,6-Dihydro-1,4-dithiin-2-yl)methanol 151 used as a versatile allyl-cation equivalent in [3 + 2] cycloaddition in the synthesis of (±)-cuparene (13) [69].
Scheme 12:
(A) Synthesis of phyllocladanol (21) features a Lewis acid-catalyzed formal intramolecular [3 + 2] ...
In 2016, Winne and co-workers reported that (5,6-dihydro-1,4-dithiin-2-yl)methanol (151) can be served as a allyl-cation equivalent for the [3 + 2] cycloaddition and was applied in the synthesis of (±)-cuparene (13) [69] (Scheme 12B). An intermolecular [3 + 2] cycloaddition of tetrasubstituted alkene 150 and the dhdt-2-methanol reagent 151 under the effect of trifluoroacetic acid produced adduct 154 in 52% yield. The authors identified that the cyclic nature of the dhdt-2-methanol reagent 151 is essential for the cycloaddition to take place. The use of noncyclic analogues did not give the cycloaddition product. It is suggested that the restricted rotational freedom of 151 and the related enforced conjugation of the sulfur lone pair may block certain undesired cation reactions. Cycloaddition product 154 was subjected to the hydrodesulfurization with Raney nickel as catalyst and subsequent catalytic hydrogenation produced (±)-cuparene (13) in 90% yield.
All-carbon [3 + 2] annulation in natural product synthesis
The all-carbon [3 + 2] cycloaddition demonstrated the ability to assemble intricate polycyclic structures in the synthesis of complex natural products. Besides the all-carbon [3 + 2] cycloaddition reactions and the corresponding applications described above, the all-carbon [3 + 2] annulation, which undergoes other possible mechanistic pathways other than cycloaddition, proved its usefulness in forging highly-substituted five-membered carbocycles. These reactions have been applied successfully in the synthesis of complex natural products. In 2011, Curran and co-workers reported the synthesis of meloscine (158) featuring a tandem radical cyclization of a divinylcyclopropane [70] (Scheme 13A). Slow addition of tributylstannane and AIBN to a refluxing solution of cyclopropane 155 afforded 156 in 38% yield. It was subjected to cleavage of the Boc group followed by N-allylation to give 157 in 73% yield over two steps. A ring-closing metathesis of freshly prepared 157 was effected by the second generation Hoveyda–Grubbs (HG II) catalyst and subsequent base-promoted epimerization produced meloscine (158) in 83% yield.
Scheme 13:
The recent advances of [3 + 2] annulation in natural product synthesis. (A) The preparation of meloscine (158) features a cascade radical annulation of divinylcyclopropane [70]. (B) Thiyl-radical-mediated [3 + 2] annulation reaction realizes the synthesis of (−)-pavidolide B (166) [71,72]. (C) A Danheiser’s [3 + 2] annulation en route to conidiogenone B (171) [73] (inset, the suggested mechanism based on Danheiser’s proposal disclosed in 1981 [74].)
Scheme 13:
The recent advances of [3 + 2] annulation in natural product synthesis. (A) The preparation of melo...
In 2017, Yang and co-workers disclosed the synthesis of (−)-pavidolide B (166) by using a thiyl-radical-mediated [3 + 2] annulation reaction to create four contiguous stereocenters on tricycle 162 in one step [71,72] (Scheme 13B). Exposure of ester 159 to PhSH [75], p-toluidine and a catalytic amount of Ir(dF(CF3)ppy)2(dtbbpy)PF6 under the irradiation of blue LED light [76,77] afforded tricycle 162 in 50% yield. The authors suggested that this process involves an intramolecular 5-exo-conjugated addition of a radical on 160 to the enone and produces 161. The newly formed 161 was subjected to 5-exo radical addition to the allyl sulfane and subsequent loss of a thiyl radical produces 162. A successive hydrolysis/decarboxylation upon heating and cleavage of acetal on 162 afforded aldehyde 163 in 90% yield. Coupling of aldehyde 163 and isoprene (164) with Ni(acac)2 and diethylzinc [78] and then Dess–Martin oxidation gave a diene (not shown, 94% yield over two steps), which was subjected to ring-closing metathesis to give enone 165 in 85% yield. Isomerization of the freshly prepared 165 to more stable α,β-unsaturated enone with RhCl3[79] afforded pavidolide B (166) in 95% yield.
The synthesis of (−)-conidiogenone B (171) featured a Danheiser’s [3 + 2] annulation [74,80] and was reported by Zhai and co-workers in 2020 [73] (Scheme 13C). Treatment of tricycle 167 with allene 168 in the presence of TiCl4 gave the desired 169 carrying two vicinal quaternary carbons. A one-pot desilylation of the newly formed 169 with a trifluoride–acetic acid complex produced the tetraquinane 170a in 89% yield with a 4:1 dr. The conversion of the freshly prepared ketone 170a to 170b was achieved in three steps. Ozonolysis of the C=C double bond of 170b gave a keto aldehyde (not shown), which was subjected to an acid-mediated aldol reaction to give conidiogenone B (171) in 53% yield. The undesired isomer with β,γ-C=C double bond (not shown) was formed in 34% yield and can be isomerized to the more stable α,β-unsaturated enone to afford conidiogenone B (171) in 32% yield upon treatment with RhCl3 in microwave.
The reaction mechanism of Danheiser’s [3 + 2] annulation is shown according to the Danheiser’s proposal [74] (Scheme 13C, inset). Initial complexation of the α,β-unsaturated ketone 167 and titanium tetrachloride produces an alkyoxy allylic carbocation (not shown). This carbocation is subjected to a regiospecific electrophilic substitution of allene 168 to generate a vinyl cation 172, which is stabilized by an adjacent carbon–silicon bond. The 1,2-shift of the silyl group in 172 produces an isomeric vinyl cation, which is intercepted by the titanium enolate and results in the new C–C bond formation to give the five-membered carbocycle 169.
Conclusion
The all-carbon [3 + 2] cycloaddition, together with the [3 + 2] annulation, continue to be an attractive class of reactions for the synthesis of highly-substituted and stereo-congested five-membered carbocycles. Also, one or more quaternary carbons can be created in a single reaction making this class of reactions appealing to complex natural product syntheses. This review outlines the development of the all-carbon [3 + 2] cycloaddition and its application in natural product synthesis reported from 2011–2020 (inclusive). The intermolecular all-carbon [3 + 2] cycloaddition offers a facile approach to install functionalized five-carbon carbocycles, including fused-rings (e.g., longeracinphyllin A (10)) and/or spiro-ring (e.g., marcfortine B (8)), at later stage of the synthesis without the need of pre-installation of necessary functional groups as a reaction precursor, for instance, ring-closing metathesis, intramolecular aldol condensation, and others.
One major issue that still needs to be addressed is the selectivity of the all carbon [3 + 2] cycloadditions, which are usually under substrate-control. Remarkable innovation of the stereoselective palladium-catalyzed trimethylenemethane cycloaddition reported by Trost’s group, which makes use of catalytic amounts of palladium and chiral phosphine ligand 74, was applied successfully in the enantioselective synthesis of marcfortine C (9, Scheme 4B). Another brilliant example is the development of a chiral-phosphine-catalyzed [3 + 2] annulation reported by Lu in 2019, in which the chiral phosphine catalyst confers high stereocontrol on the formation of a spiro adduct bearing two vicinal all-carbon quaternary stereocenters (Scheme 7). We believe that the enantioselective all-carbon [3 + 2] cycloaddition provides a new strategy for the preparation of sp3-carbon-enriched complex scaffolds [81,82] for biological studies and potential new drug development.
The all-carbon [3 + 2] cycloaddition is undoubtedly an efficient synthetic transformation that creates two C–C bonds in a single reaction. However, the prior protection of the reactive functional groups, such as the hydroxy and amino groups, are still necessary for most of the all-carbon [3 + 2] cycloaddition reactions. We predict that further development of the all-carbon [3 + 2] cyclization with the reactive functional groups’ compatibilities and/or without the use of protecting groups [83,84] can improve the synthetic efficiency and make this class of reactions more attractive to the synthetic scientist for applications. Lastly, we anticipate that the all-carbon [3 + 2] cycloaddition will gain further attention from the synthetic community, including scientists from academia and pharmaceutical industry, for methodic innovation and the efficient synthesis of biologically important natural products.
Acknowledgements
The author thanks C. Hui (Max Planck Institute of Molecular Physiology) for helpful discussion during the preparation of this manuscript. The authors would like to thank the anonymous reviewers for their thought-provoking comments and apologize to colleagues whose work was not cited owing to selected coverage.
Funding
Financial support from the Shenzhen Human Resources and Social Security Bureau (50820190066) to Z. Wang is gratefully acknowledged. J. Liu acknowledges the financial support from Shenzhen Science and Technology Innovation Committee (grant nos. JCYJ20190809181011411).
References
Hasler, C. M.; Acs, G.; Blumberg, P. M. Cancer Res.1992,52, 202–208.
Return to citation in text:
[1]
Ogbourne, S. M.; Suhrbier, A.; Jones, B.; Cozzi, S.-J.; Boyle, G. M.; Morris, M.; McAlpine, D.; Johns, J.; Scott, T. M.; Sutherland, K. P.; Gardner, J. M.; Le, T. T. T.; Lenarczyk, A.; Aylward, J. H.; Parsons, P. G. Cancer Res.2004,64, 2833–2839. doi:10.1158/0008-5472.can-03-2837
Return to citation in text:
[1]
Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. J. Am. Chem. Soc.2006,128, 3148–3149. doi:10.1021/ja057404h
Return to citation in text:
[1]
Kiewert, C.; Kumar, V.; Hildmann, O.; Rueda, M.; Hartmann, J.; Naik, R. S.; Klein, J. Brain Res.2007,1128, 70–78. doi:10.1016/j.brainres.2006.10.042
Return to citation in text:
[1]
Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Org. Lett.2009,11, 5190–5193. doi:10.1021/ol9021029
Return to citation in text:
[1]
Ratnayake, R.; Covell, D.; Ransom, T. T.; Gustafson, K. R.; Beutler, J. A. Org. Lett.2009,11, 57–60. doi:10.1021/ol802339w
Return to citation in text:
[1]
Jang, J.-H.; Asami, Y.; Jang, J.-P.; Kim, S.-O.; Moon, D. O.; Shin, K.-S.; Hashizume, D.; Muroi, M.; Saito, T.; Oh, H.; Kim, B. Y.; Osada, H.; Ahn, J. S. J. Am. Chem. Soc.2011,133, 6865–6867. doi:10.1021/ja1110688
Return to citation in text:
[1]
Zeng, X.-P.; Cao, Z.-Y.; Wang, Y.-H.; Zhou, F.; Zhou, J. Chem. Rev.2016,116, 7330–7396. doi:10.1021/acs.chemrev.6b00094
Return to citation in text:
[1]
Li, H.; Lu, Y. Asian J. Org. Chem.2017,6, 1130–1145. doi:10.1002/ajoc.201700220
Return to citation in text:
[1]
Feng, J.; Holmes, M.; Krische, M. J. Chem. Rev.2017,117, 12564–12580. doi:10.1021/acs.chemrev.7b00385
Return to citation in text:
[1]
Li, C.; Ragab, S. S.; Liu, G.; Tang, W. Nat. Prod. Rep.2020,37, 276–292. doi:10.1039/c9np00039a
Return to citation in text:
[1]
Long, R.; Huang, J.; Gong, J.; Yang, Z. Nat. Prod. Rep.2015,32, 1584–1601. doi:10.1039/c5np00046g
Return to citation in text:
[1]
[2]
Huisgen, R. Angew. Chem., Int. Ed. Engl.1963,2, 565–598. doi:10.1002/anie.196305651
Return to citation in text:
[1]
Little, R. D.; Muller, G. W. J. Am. Chem. Soc.1979,101, 7129–7130. doi:10.1021/ja00517a086
Return to citation in text:
[1]
[2]
Little, R. D. Chem. Rev.1986,86, 875–884. doi:10.1021/cr00075a010
Return to citation in text:
[1]
Trost, B. M.; Chan, D. M. T. J. Am. Chem. Soc.1979,101, 6429–6432. doi:10.1021/ja00515a046
Return to citation in text:
[1]
Zhang, C.; Lu, X. J. Org. Chem.1995,60, 2906–2908. doi:10.1021/jo00114a048
Return to citation in text:
[1]
[2]
[3]
[4]
Saito, K.; Sogou, H.; Suga, T.; Kusama, H.; Iwasawa, N. J. Am. Chem. Soc.2011,133, 689–691. doi:10.1021/ja108586d
Return to citation in text:
[1]
[2]
[3]
Little, R. D.; Muller, G. W. J. Am. Chem. Soc.1981,103, 2744–2749. doi:10.1021/ja00400a043
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Paquette, L. A.; Sauer, D. R.; Cleary, D. G.; Kinsella, M. A.; Blackwell, C. M.; Anderson, L. G. J. Am. Chem. Soc.1992,114, 7375–7387. doi:10.1021/ja00045a007
Return to citation in text:
[1]
[2]
[3]
Lee, H.-Y.; Kim, Y. J. Am. Chem. Soc.2003,125, 10156–10157. doi:10.1021/ja036263l
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Wang, J.-C.; Krische, M. J. Angew. Chem., Int. Ed.2003,42, 5855–5857. doi:10.1002/anie.200352218
Return to citation in text:
[1]
[2]
[3]
[4]
Adams, T. E.; El Sous, M.; Hawkins, B. C.; Hirner, S.; Holloway, G.; Khoo, M. L.; Owen, D. J.; Savage, G. P.; Scammells, P. J.; Rizzacasa, M. A. J. Am. Chem. Soc.2009,131, 1607–1616. doi:10.1021/ja808402e
Return to citation in text:
[1]
Trost, B. M.; Chan, D. M. T. J. Am. Chem. Soc.1983,105, 2315–2325. doi:10.1021/ja00346a035
Return to citation in text:
[1]
Berson, J. A.; Duncan, C. D.; Corwin, L. R. J. Am. Chem. Soc.1974,96, 6175–6177. doi:10.1021/ja00826a035
Return to citation in text:
[1]
Kang, T.; Kim, W.-Y.; Yoon, Y.; Kim, B. G.; Lee, H.-Y. J. Am. Chem. Soc.2011,133, 18050–18053. doi:10.1021/ja207591e
Return to citation in text:
[1]
[2]
[3]
Kang, T.; Song, S. B.; Kim, W.-Y.; Kim, B. G.; Lee, H.-Y. J. Am. Chem. Soc.2014,136, 10274–10276. doi:10.1021/ja5054412
Return to citation in text:
[1]
[2]
[3]
Lee, H.; Kang, T.; Lee, H.-Y. Angew. Chem., Int. Ed.2017,56, 8254–8257. doi:10.1002/anie.201704492
Return to citation in text:
[1]
[2]
[3]
Lipshutz, B. H.; Parker, D. A.; Kozlowski, J. A.; Nguyen, S. L. Tetrahedron Lett.1984,25, 5959–5962. doi:10.1016/s0040-4039(01)81732-9
Return to citation in text:
[1]
Van Hijfte, L.; Little, R. D.; Petersen, J. L.; Moeller, K. D. J. Org. Chem.1987,52, 4647–4661. doi:10.1021/jo00230a001
Return to citation in text:
[1]
Trost, B. M.; Mignani, S. M.; Nanninga, T. N. J. Am. Chem. Soc.1986,108, 6051–6053. doi:10.1021/ja00279a070
Return to citation in text:
[1]
[2]
[3]
Greco, G. E.; Gleason, B. L.; Lowery, T. A.; Kier, M. J.; Hollander, L. B.; Gibbs, S. A.; Worthy, A. D. Org. Lett.2007,9, 3817–3820. doi:10.1021/ol7017246
Return to citation in text:
[1]
Trost, B. M.; Cramer, N.; Bernsmann, H. J. Am. Chem. Soc.2007,129, 3086–3087. doi:10.1021/ja070142u
Return to citation in text:
[1]
[2]
[3]
Trost, B. M.; Bringley, D. A.; Zhang, T.; Cramer, N. J. Am. Chem. Soc.2013,135, 16720–16735. doi:10.1021/ja409013m
Return to citation in text:
[1]
[2]
[3]
[4]
Ohno, M.; Ishizaki, K.; Eguchi, S. J. Org. Chem.1988,53, 1285–1288. doi:10.1021/jo00241a030
Return to citation in text:
[1]
Li, J.; Zhang, W.; Zhang, F.; Chen, Y.; Li, A. J. Am. Chem. Soc.2017,139, 14893–14896. doi:10.1021/jacs.7b09186
Return to citation in text:
[1]
[2]
[3]
[4]
Chen, Y.; Zhang, W.; Ren, L.; Li, J.; Li, A. Angew. Chem., Int. Ed.2018,57, 952–956. doi:10.1002/anie.201711482
Return to citation in text:
[1]
[2]
[3]
[4]
Wallace, D. J.; Sidda, R. L.; Reamer, R. A. J. Org. Chem.2007,72, 1051–1054. doi:10.1021/jo062170l
Return to citation in text:
[1]
Hammar, P.; Ghobril, C.; Antheaume, C.; Wagner, A.; Baati, R.; Himo, F. J. Org. Chem.2010,75, 4728–4736. doi:10.1021/jo100488g
Return to citation in text:
[1]
Chan, W.-L.; Tang, X.; Zhang, F.; Quek, G.; Mei, G.-J.; Lu, Y. Angew. Chem., Int. Ed.2019,58, 6260–6264. doi:10.1002/anie.201900758
Return to citation in text:
[1]
[2]
[3]
Li, F.-Z.; Li, S.; Zhang, P.-P.; Huang, Z.-H.; Zhang, W.-B.; Gong, J.; Yang, Z. Chem. Commun.2016,52, 12426–12429. doi:10.1039/c6cc06794h
Return to citation in text:
[1]
Overman, L. E.; Paone, D. V. J. Am. Chem. Soc.2001,123, 9465–9467. doi:10.1021/ja0166141
Return to citation in text:
[1]
Long, R.; Huang, J.; Shao, W.; Liu, S.; Lan, Y.; Gong, J.; Yang, Z. Nat. Commun.2014,5, 5707. doi:10.1038/ncomms6707
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
Zhao, P.; Incarvito, C. D.; Hartwig, J. F. J. Am. Chem. Soc.2006,128, 3124–3125. doi:10.1021/ja058550q
Return to citation in text:
[1]
Zhao, P.; Incarvito, C. D.; Hartwig, J. F. J. Am. Chem. Soc.2006,128, 9642–9643. doi:10.1021/ja063347w
Return to citation in text:
[1]
Ma, S.; Negishi, E.-i. J. Am. Chem. Soc.1995,117, 6345–6357. doi:10.1021/ja00128a025
Return to citation in text:
[1]
Burns, A. R.; McAllister, G. D.; Shanahan, S. E.; Taylor, R. J. K. Angew. Chem., Int. Ed.2010,49, 5574–5577. doi:10.1002/anie.201002416
Return to citation in text:
[1]
Conia, J. M.; Le Perchec, P. Synthesis1975, 1–19. doi:10.1055/s-1975-23652
Return to citation in text:
[1]
Senda, T.; Ogasawara, M.; Hayashi, T. J. Org. Chem.2001,66, 6852–6856. doi:10.1021/jo0103930
Return to citation in text:
[1]
Hayashi, T.; Takahashi, M.; Takaya, Y.; Ogasawara, M. J. Am. Chem. Soc.2002,124, 5052–5058. doi:10.1021/ja012711i
Return to citation in text:
[1]
Sun, Z.-M.; Zhao, P. Angew. Chem., Int. Ed.2009,48, 6726–6730. doi:10.1002/anie.200901097
Return to citation in text:
[1]
Zheng, N.; Zhang, L.; Gong, J.; Yang, Z. Org. Lett.2017,19, 2921–2924. doi:10.1021/acs.orglett.7b01154
Return to citation in text:
[1]
[2]
[3]
Shao, W.; Huang, J.; Guo, K.; Gong, J.; Yang, Z. Org. Lett.2018,20, 1857–1860. doi:10.1021/acs.orglett.8b00380
Return to citation in text:
[1]
[2]
[3]
[4]
Luche, J. L. J. Am. Chem. Soc.1978,100, 2226–2227. doi:10.1021/ja00475a040
Return to citation in text:
[1]
Kusama, H.; Ebisawa, M.; Funami, H.; Iwasawa, N. J. Am. Chem. Soc.2009,131, 16352–16353. doi:10.1021/ja907633b
Return to citation in text:
[1]
[2]
Jia, X.; Lei, H.; Han, F.; Zhang, T.; Chen, Y.; Xu, Z.; Nakliang, P.; Choi, S.; Guo, Y.; Ye, T. Angew. Chem., Int. Ed.2020,59, 12832–12836. doi:10.1002/anie.202005048
Return to citation in text:
[1]
[2]
Zhu, W.; Fang, J.; Liu, Y.; Ren, J.; Wang, Z. Angew. Chem., Int. Ed.2013,52, 2032–2037. doi:10.1002/anie.201206484
Return to citation in text:
[1]
[2]
Hullaert, J.; Winne, J. M. Angew. Chem., Int. Ed.2016,55, 13254–13258. doi:10.1002/anie.201606411
Return to citation in text:
[1]
[2]
Zhang, H.; Curran, D. P. J. Am. Chem. Soc.2011,133, 10376–10378. doi:10.1021/ja2042854
Return to citation in text:
[1]
[2]
Zhang, P.-P.; Yan, Z.-M.; Li, Y.-H.; Gong, J.-X.; Yang, Z. J. Am. Chem. Soc.2017,139, 13989–13992. doi:10.1021/jacs.7b07388
Return to citation in text:
[1]
[2]
Zhang, P.; Li, Y.; Yan, Z.; Gong, J.; Yang, Z. J. Org. Chem.2019,84, 15958–15971. doi:10.1021/acs.joc.9b02230
Return to citation in text:
[1]
[2]
Xu, B.; Xun, W.; Su, S.; Zhai, H. Angew. Chem., Int. Ed.2020,59, 16475–16479. doi:10.1002/anie.202007247
Return to citation in text:
[1]
[2]
Danheiser, R. L.; Carini, D. J.; Basak, A. J. Am. Chem. Soc.1981,103, 1604–1606. doi:10.1021/ja00396a071
Return to citation in text:
[1]
[2]
[3]
Miura, K.; Fugami, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett.1988,29, 5135–5138. doi:10.1016/s0040-4039(00)80701-7
Return to citation in text:
[1]
Bhat, V. T.; Duspara, P. A.; Seo, S.; Abu Bakar, N. S. B.; Greaney, M. F. Chem. Commun.2015,51, 4383–4385. doi:10.1039/c4cc09987g
Return to citation in text:
[1]
Fadeyi, O. O.; Mousseau, J. J.; Feng, Y.; Allais, C.; Nuhant, P.; Chen, M. Z.; Pierce, B.; Robinson, R. Org. Lett.2015,17, 5756–5759. doi:10.1021/acs.orglett.5b03184
Return to citation in text:
[1]
Hasler, C. M.; Acs, G.; Blumberg, P. M. Cancer Res.1992,52, 202–208.
2.
Ogbourne, S. M.; Suhrbier, A.; Jones, B.; Cozzi, S.-J.; Boyle, G. M.; Morris, M.; McAlpine, D.; Johns, J.; Scott, T. M.; Sutherland, K. P.; Gardner, J. M.; Le, T. T. T.; Lenarczyk, A.; Aylward, J. H.; Parsons, P. G. Cancer Res.2004,64, 2833–2839. doi:10.1158/0008-5472.can-03-2837
3.
Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. J. Am. Chem. Soc.2006,128, 3148–3149. doi:10.1021/ja057404h
4.
Kiewert, C.; Kumar, V.; Hildmann, O.; Rueda, M.; Hartmann, J.; Naik, R. S.; Klein, J. Brain Res.2007,1128, 70–78. doi:10.1016/j.brainres.2006.10.042
5.
Kubo, M.; Okada, C.; Huang, J.-M.; Harada, K.; Hioki, H.; Fukuyama, Y. Org. Lett.2009,11, 5190–5193. doi:10.1021/ol9021029
6.
Ratnayake, R.; Covell, D.; Ransom, T. T.; Gustafson, K. R.; Beutler, J. A. Org. Lett.2009,11, 57–60. doi:10.1021/ol802339w
7.
Jang, J.-H.; Asami, Y.; Jang, J.-P.; Kim, S.-O.; Moon, D. O.; Shin, K.-S.; Hashizume, D.; Muroi, M.; Saito, T.; Oh, H.; Kim, B. Y.; Osada, H.; Ahn, J. S. J. Am. Chem. Soc.2011,133, 6865–6867. doi:10.1021/ja1110688
Greco, G. E.; Gleason, B. L.; Lowery, T. A.; Kier, M. J.; Hollander, L. B.; Gibbs, S. A.; Worthy, A. D. Org. Lett.2007,9, 3817–3820. doi:10.1021/ol7017246
Paquette, L. A.; Sauer, D. R.; Cleary, D. G.; Kinsella, M. A.; Blackwell, C. M.; Anderson, L. G. J. Am. Chem. Soc.1992,114, 7375–7387. doi:10.1021/ja00045a007
Paquette, L. A.; Sauer, D. R.; Cleary, D. G.; Kinsella, M. A.; Blackwell, C. M.; Anderson, L. G. J. Am. Chem. Soc.1992,114, 7375–7387. doi:10.1021/ja00045a007
Paquette, L. A.; Sauer, D. R.; Cleary, D. G.; Kinsella, M. A.; Blackwell, C. M.; Anderson, L. G. J. Am. Chem. Soc.1992,114, 7375–7387. doi:10.1021/ja00045a007
Adams, T. E.; El Sous, M.; Hawkins, B. C.; Hirner, S.; Holloway, G.; Khoo, M. L.; Owen, D. J.; Savage, G. P.; Scammells, P. J.; Rizzacasa, M. A. J. Am. Chem. Soc.2009,131, 1607–1616. doi:10.1021/ja808402e


![[1860-5397-16-251-1]](/bjoc/content/figures/1860-5397-16-251-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-2]](/bjoc/content/figures/1860-5397-16-251-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i1]](/bjoc/content/inline/1860-5397-16-251-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i2]](/bjoc/content/inline/1860-5397-16-251-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i3]](/bjoc/content/inline/1860-5397-16-251-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i4]](/bjoc/content/inline/1860-5397-16-251-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i5]](/bjoc/content/inline/1860-5397-16-251-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i6]](/bjoc/content/inline/1860-5397-16-251-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i7]](/bjoc/content/inline/1860-5397-16-251-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i8]](/bjoc/content/inline/1860-5397-16-251-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i9]](/bjoc/content/inline/1860-5397-16-251-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i10]](/bjoc/content/inline/1860-5397-16-251-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i11]](/bjoc/content/inline/1860-5397-16-251-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i12]](/bjoc/content/inline/1860-5397-16-251-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-251-i13]](/bjoc/content/inline/1860-5397-16-251-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)