Abstract
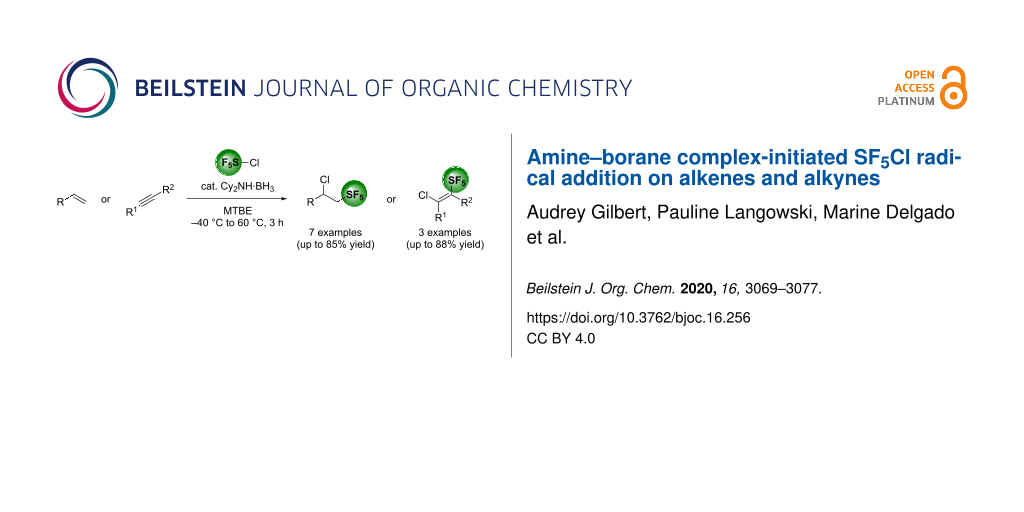
The SF5Cl radical addition on unsaturated compounds was performed using an air-stable amine–borane complex as the radical initiator. This method showed to be complementary to the classic Et3B-mediated SF5Cl addition on alkenes and alkynes. A total of seven alkene and three alkyne derivatives were tested in the reaction, with yields ranging from 3% to 85%.

Graphical Abstract
Introduction
The pentafluorosulfanyl (SF5) substituent has been attracting its share of attention since its discovery in 1950 [1]. Often referred to as a "super CF3", the SF5 shows enhanced properties when compared to its trifluoromethylated (CF3) analog [2]. Indeed, the SF5 moiety is more electronegative, more lipophilic, bulkier, and more thermally and chemically stable than the CF3 substituent [3-8]. Furthermore, it induces a stronger dipole moment, which can dramatically affect the properties of the neighboring functional groups on a molecule [3-8]. Considering the wide range of trifluoromethylated compounds of interest, the synthesis of their SF5-analogs has become a trend to increase the properties of these valuable molecules [9]. Due to the unique properties, the SF5 group has been used in various fields of chemistry, including pharmaceuticals [10-16], agrochemistry [17-20], and materials sciences [21-26]. The applications have, however, been limited by the poor synthetic accessibility of SF5-containing molecules. As such, the development of alternative methods for the introduction of the SF5 group is highly relevant.
Although the number of synthetic routes towards the SF5 substituent remains limited, a few methods have been developed in the past 20 years in order to include a SF5 moiety on various organic substrates [27,28]. The main strategy towards pentafluorosulfanylated aliphatic compounds, reported for the first time by Dolbier and co-workers in 2002, is the Et3B-mediated radical addition of SF5Cl on alkenes and alkynes (Scheme 1) [29,30]. This strategy represented a tremendous step forward in the pentafluorosulfanyl aliphatic chemistry, since it addressed the drawbacks that were previously reported with the use of SF5Cl as a reagent. It allows the SF5Cl addition to occur in the liquid phase (SF5Cl being a gas that boils at −21 °C) in milder reaction conditions and in normal glassware, instead of using special apparatus such as autoclaves and photochemical reactors [31]. Moreover, this method leads to significantly higher yields in shorter reaction times compared to the previous methods. Since this first report, this reaction has been extensively used to obtain a wide range of SF5-containing aliphatic derivatives, and represents the most versatile route towards pentafluorosulfanylated aliphatic compounds [28].
![[1860-5397-16-256-i1]](/bjoc/content/inline/1860-5397-16-256-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Dolbier’s protocol for the SF5Cl radical addition on alkenes.
Scheme 1: Dolbier’s protocol for the SF5Cl radical addition on alkenes.
However, some limitations have emerged from the Dolbier protocol. The SF5Cl addition on unsaturated compounds goes through a free-radical mechanism, and is promoted by the radical activation of SF5Cl by Et3B, which leads to the formation of the propagating species SF5• [32,33]. Trialkylboranes are common low-temperature radical initiators, and Et3B is one of the most used in the literature [34,35]. The use of the reagent allows the radicals to form, even at very low temperature, due to its strong reactivity with oxygen. However, the disadvantage for the use of the reagent comes from the same property: Et3B is an oxygen-sensitive and pyrophoric compound even at low temperatures, which requires the use of air-free techniques in the laboratory. Et3B in the pure form has limited commercial availability and is known to spontaneously react with oxygen to produce a green flame [36]. To avoid the pyrophoric properties of triethylborane, this reagent is mostly sold in low-concentrated solutions (typically 1–2 M in hexane, THF or Et2O). Another concern is the fluctuation in the quality/concentration in commercial solutions, even among the same batch from the same supplier. Finally, in some cases, the reaction yields can be poorly reproducible if the trialkylborane reagent is not freshly prepared [37]. It is therefore of interest to address this challenge in order to widen the scope of this transformation.
Compared to trialkylboranes, amine–borane complexes have shown to be more stable [38]. Indeed, they are usually air-stable, and their preparation from NaBH4, H2SO4 and amines involves traditional mild acidic work-up without degradation. They are therefore much easier to handle on a laboratory scale and can be stored on the shelf for months without noticeable alteration of their properties and purity [39]. Amine–borane complexes have been extensively used in the literature as hydrogen reservoirs [40], as reducing agents in various transformations, including the reduction of aldehydes, amides and ketones, reductive aminations, alkene hydroboration, and carbon bond forming reaction [41,42], as well as various boronate and borinic acid precursors [43-47]. More recently, it has been shown that some of these common amine–borane complexes can also be used as radical initiators for atom transfer radical addition of alkyl halides to alkenes [48]. They were also used in the free-radical polymerization of alkene-containing monomers such as methyl methacrylate or styrene [48-50]. We envisioned that it could be possible to replace the Et3B in Dolbier’s protocol by a stable amine–borane complex that could perform the radical initiation of SF5Cl on its addition on alkenes. This would address the drawbacks associated with the use of Et3B as the radical initiator, and therefore facilitate the access to various pentafluorosulfanylated derivatives.
As shown in Scheme 2, we envisioned that the amine–borane complex-initiated reaction of SF5Cl with alkenes would proceed following a mechanism similar to the amine–borane complex-initiated carbohalogenation of alkenes [48]. The first step would involve the formation of a trialkylborane species via the hydroboration of the alkene, as previously observed by 11B NMR spectroscopy [48,49]. In the presence of oxygen, the trialkylborane would, similarly to Et3B, generate an alkyl radical. The latter would react with SF5Cl to produce a chloroalkane as well as the key SF5• radical. The propagation steps would occur exactly as reported by Dolbier and co-workers for the Et3B-mediated radical addition of SF5Cl on alkenes and alkynes [29,30]. Overall, while similar mechanistically, the use of an amine–borane complex as the initiator would avoid the need to manipulate an oxygen-sensitive and pyrophoric reagent.
![[1860-5397-16-256-i2]](/bjoc/content/inline/1860-5397-16-256-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Proposed mechanism for the amine–borane complex-initiated radical addition of SF5Cl on alkenes.
Scheme 2: Proposed mechanism for the amine–borane complex-initiated radical addition of SF5Cl on alkenes.
Results and Discussion
Table 1 and Table 2 show the selected optimization results for the use of amine–borane complexes in the SF5Cl radical addition on alkenes. We chose allyl benzyl ether (1) as the model substrate for our optimization, since it has been previously shown that this compound undergoes SF5Cl radical addition following Dolbier’s protocol in various solvents with high yields [51]. We started the optimization with 3 equivalents of SF5Cl, 10 mol % of the amine–borane complex, with temperatures going from 30 °C to 60 °C for 3 hours. The addition of all reagents was performed at −40 °C, before the reaction vessel was hermetically sealed and heated to avoid evaporation of SF5Cl, since it is gaseous above −21 °C. The reaction was performed in common organic solvents that remain liquid both at −40 °C and at the tested reaction temperatures. The results in hexane, ethyl acetate (EtOAc), and methyl tert-butyl ether (MTBE) are shown in Table 1 and Table 2, but more solvents were tested and did not lead to higher yields of the desired compound (see Supporting Information File 1 for the complete optimization results). Three commercially available borane complexes were tested in the reaction, i.e., diisopropylamine borane (DIPAB), dicyclohexylamine borane (DICAB), and N,N-diisopropylethylamine borane (DIPEA·BH3) (Figure 1). The use of DIPAB and DICAB led to higher yields, and the results are respectively shown in Table 1 and Table 2 (see Supporting Information File 1 for the results with DIPEA·BH3).
![[1860-5397-16-256-1]](/bjoc/content/figures/1860-5397-16-256-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures and acronyms of the amine-borane complexes investigated.
Figure 1: Structures and acronyms of the amine-borane complexes investigated.
We started the optimization with DIPAB as the amine–borane complex. To our delight, in all tested solvents and temperatures, we observed the formation of the desired product 2a, which is in accordance with our hypothesis that the amine–borane complexes can indeed be used as radical initiators under thermal activation in the SF5Cl addition on alkenes. DIPAB showed to be generally more productive at lower temperatures. Indeed, when performing the reaction in EtOAc, a full conversion was observed at all tested temperatures, but with a decrease in the yield when the reaction temperature was increased (Table 1, entries 5–8). The same effect was observed in MTBE, with the best results obtained at 30 °C and 50 °C, while 60 °C led to a low yield of 26% (Table 1, entries 9–12). Surprisingly, the intermediate temperature of 40 °C led to only 6% of the desired compound, and the reason for this result remains unclear (Table 1, entry 10). Moreover, the use of hexane as the solvent did not show to be compatible with DIPAB as the amine–borane complex, since it led to low yields at all tested temperatures (Table 1, entries 1–4). When using DIPAB as the radical initiator, the best result was obtained in EtOAc at 30 °C, with a yield of 72% of the desired addition product (Table 1, entry 5).
Table 1: Selected optimization results for the SF5Cl addition on allyl benzyl ether (1) using DIPAB as the radical initiator.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-256-i5.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent | x (°C) | conversion (%)a | yield (%)b |
| 1 | hexane | 30 | 28 | 2 |
| 2 | hexane | 40 | 32 | 6 |
| 3 | hexane | 50 | 33 | 7 |
| 4 | hexane | 60 | 55 | 5 |
| 5 | EtOAc | 30 | 100 | 72 |
| 6 | EtOAc | 40 | 100 | 64 |
| 7 | EtOAc | 50 | 100 | 5 |
| 8 | EtOAc | 60 | 100 | 29 |
| 9 | MTBE | 30 | 77 | 40 |
| 10 | MTBE | 40 | 69 | 6 |
| 11 | MTBE | 50 | 79 | 41 |
| 12 | MTBE | 60 | 92 | 26 |
aDisappearance of the starting material, estimated by 1H NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard. bYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard.
We next turned our attention to the amine–borane complex DICAB as the radical initiator for the addition of SF5Cl on allyl benzyl ether (1) (Table 2). In this case, increasing the reaction temperature generally led to higher yields. When the reaction was performed in hexane, the yields went from 1–2% to 72% when heating the reaction at 50 °C, compared to 30 °C and 40 °C, while a low yield of 32% was obtained with the reaction temperature of 60 °C (Table 2, entries 1–4). This tendency was also observed when using EtOAc as the solvent (Table 2, entries 5–8), with the best yield of 62% obtained at 50 °C (Table 2, entry 7). We hypothesized that a higher temperature is necessary to activate the crystalline and more sterically hindered DICAB, compared to the liquid DIPAB, but that a too high reaction temperature such as 60 °C tends to increase the degradation pathways instead of the formation of the desired compound. However, this effect was not observed when performing the reaction in MTBE. A high yield of 86% was obtained at 60 °C (Table 2, entry 12), while performing the reaction at 40 °C led to a higher yield than at 50 °C (Table 2, entries 10 and 11). With the use of DICAB, the best result obtained was with MTBE as the solvent at 60 °C, affording the product with a yield of 86% (Table 2, entry 12).
Table 2: Selected optimization results for the SF5Cl addition on allyl benzyl ether (1) using DICAB as the radical initiator.
![[Graphic 2]](/bjoc/content/inline/1860-5397-16-256-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent | x (°C) | conversion (%)a | yield (%)b |
| 1 | hexane | 30 | 25 | 1 |
| 2 | hexane | 40 | 17 | 2 |
| 3 | hexane | 50 | 82 | 72 |
| 4 | hexane | 60 | 76 | 32 |
| 5 | EtOAc | 30 | 100 | traces |
| 6 | EtOAc | 40 | 100 | 4 |
| 7 | EtOAc | 50 | 100 | 62 |
| 8 | EtOAc | 60 | 100 | 56 |
| 9 | MTBE | 30 | 49 | 3 |
| 10 | MTBE | 40 | 100 | 65 |
| 11 | MTBE | 50 | 94 | 21 |
| 12 | MTBE | 60 | 100 | 86 (77)c |
aDisappearance of the starting material, estimated by 1H NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard. bYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard. cIsolated yield.
At this point, we performed some control reactions in order to get more insight into the reaction (Table 3). We first increased the amount of the amine–borane complex added to the reaction mixture to evaluate if this would promote the desired reaction. Hexane and EtOAc were tested at 50 °C with 20 mol % instead of 10 mol % of DICAB (Table 3, entries 1 and 2). This showed to be slightly beneficial in the case of EtOAc (Table 3, entry 1), while it led to a low yield in the case of hexane (Table 3, entry 2). Moreover, increasing the reaction time from 3 to 6 hours led to higher yields of the desired SF5-containing adduct, with yields of 80% and 89% with EtOAc and hexane, respectively (Table 3, entries 3 and 4). Finally, we performed the reaction in hexane with no amine–borane complex added, and only trace amounts of the final compound 2a were detected (Table 3, entry 5).
Table 3: Effect of time and amount of the amine–borane complex on the SF5Cl addition on allyl benzyl ether (1).
![[Graphic 3]](/bjoc/content/inline/1860-5397-16-256-i7.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | DICAB (equiv) | time (h) | conversion (%)a | yield (%)b |
| 1 | EtOAc | 0.2 | 3 | 100 | 80 |
| 2 | hexane | 0.2 | 3 | 61 | 37 |
| 3 | EtOAc | 0.1 | 6 | 100 | 86 |
| 4 | hexane | 0.1 | 6 | 94 | 89 |
| 5 | hexane | 0 | 6 | 20 | traces |
aDisappearance of the starting material, estimated by 1H NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard. bYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard.
We next wondered if it could be possible to start the reaction at a higher temperature than −40 °C without significantly affecting the reaction yield. We performed the reaction with 10 mol % of DICAB, using our standard conditions, but with the addition of all reagents at 0 °C or at room temperature (20–21 °C) before sealing the reaction vessel and heating the reaction mixture to 50 °C (Table 4). EtOAc, hexane, and MTBE were tested in these conditions, and with the exception of hexane when starting the reaction at 0 °C (Table 4, entry 3), all reactions led to the desired final compound in good to excellent yields (Table 4, entries 1, 2, and 4–6). Indeed, we obtained a 93% yield of the addition product 2a when performing the reaction in EtOAc from 0 °C to 50 °C (Table 4, entry 1). The low yield of 2% that was obtained with hexane (Table 4, entry 3) is, however, rather surprising, since the reaction in that solvent from room temperature to 50 °C led to 81% of the desired compound (Table 4, entry 4). When repeating the reaction, we rapidly realized that these higher initial temperatures conditions were not reproducible, which we believe came from the fact that at 0 °C and room temperature, SF5Cl is gaseous. Therefore, its addition to the reaction mixture might have been inefficient in some case, while getting added properly in some other cases, which would explain the reproducibility problems. Moreover, this effect was not observed when repeating some of the reactions where the initial temperature was −40 °C, which is in accordance with this hypothesis.
Table 4: Effect of the initial temperature on the SF5Cl addition on allyl benzyl ether (1).
![[Graphic 4]](/bjoc/content/inline/1860-5397-16-256-i8.svg?max-width=637&scale=1.0)
|
||||
| entry | solvent | x (°C) | conversion (%)a | yield (%)b |
| 1 | EtOAc | 0 | 100 | 93 |
| 2 | EtOAc | 20 | 100 | 71 |
| 3 | hexane | 0 | 40 | 2 |
| 4 | hexane | 20 | 100 | 81 |
| 5 | MTBE | 0 | 100 | 75 |
| 6 | MTBE | 20 | 100 | 79 |
aDisappearance of the starting material, estimated by 1H NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard. bYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard.
Finally, we hypothesized that reducing the amount of the amine–borane complex in the reaction could increase the yield by avoiding a surge of the radical species early in the reaction. We performed the reactions with 3.3 mol % of the amine–borane complex in hexane, EtOAc, and MTBE and at 50 °C and 60 °C (Table 5). Unfortunately, the reaction yields did not increase, and these reaction conditions proved to be less efficient for the SF5Cl addition on allyl benzyl ether than the ones previously discussed. The only case where the yield was promoted by a decreased amount of the amine–borane complex was when the reaction was performed in hexane at 50 °C and with the use of DICAB as the radical initiator (Table 5, entry 2). Indeed, this led to a product yield of 86%, which is equal to the best yield obtained so far, in MTBE with 10 mol % of DICAB at 60 °C (Table 2, entry 12). However, the latter led to a full conversion, which is not the case with the reaction in hexane, with a conversion of 93%. Considering the similar polarity of the starting material 1 and the final product 2a, we chose the reaction conditions in MTBE as the optimal conditions, in order to facilitate the purification process.
Table 5: Effect of the decreased amount of the amine-borane complex on the SF5Cl addition on allyl benzyl ether (1).
![[Graphic 5]](/bjoc/content/inline/1860-5397-16-256-i9.svg?max-width=637&scale=1.0)
|
|||||
| entry | solvent | amine–borane complex | x (°C) | conversion (%)a | yield (%)b |
| 1 | hexane | DIPAB | 50 | 25 | 2 |
| 2 | hexane | DICAB | 50 | 93 | 86 |
| 3 | hexane | DIPAB | 60 | 65 | 41 |
| 4 | hexane | DICAB | 60 | 30 | 22 |
| 5 | EtOAc | DIPAB | 50 | 54 | 9 |
| 6 | EtOAc | DICAB | 50 | 66 | 2 |
| 7 | EtOAc | DIPAB | 60 | 100 | 6 |
| 8 | EtOAc | DICAB | 60 | 100 | 37 |
| 9 | MTBE | DIPAB | 50 | 67 | 20 |
| 10 | MTBE | DICAB | 50 | 100 | 73 |
| 11 | MTBE | DIPAB | 60 | 67 | 24 |
| 12 | MTBE | DICAB | 60 | 100 | 75 |
aDisappearance of the starting material, estimated by 1H NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard. bYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard.
With the optimal reaction conditions in hand, we evaluated the scope of the reaction. Both our DICAB-promoted and the Dolbier’s protocol using Et3B for the SF5Cl radical addition on unsaturated compounds were performed on every substrate, in order to compare the two methods. First, a series of alkenes was assessed for the SF5Cl radical addition using both protocols (Scheme 3). In most cases, the desired pentafluorosulfanylated compounds were obtained in comparable yields with both methods, with a slightly better yield for the Dolbier protocol. Indeed, compound 2a was obtained with an 88% yield when the SF5Cl addition was performed with Et3B, while 77% of the desired final product was obtained with the DICAB-mediated protocol. Moreover, yields of 90% and 85% of compound 2b were respectively obtained when using 4-phenyl-1-butene as the starting material. When performing the reactions on styrene, the desired addition product 2c was only observed with a low NMR yield of 8% with the Et3B-mediated reaction, while the DICAB protocol led to a 15% isolated yield of the 2:1 addition product 2d, with no sign of the desired compound 2c. Furthermore, as expected, a low yield of 15% of the corresponding addition product 2e was obtained with Dolbier’s protocol when performing the reaction on dec-9-en-1-ol, while protecting the alcohol group with an acetate significantly increased the yield, leading to the corresponding pentafluorosulfanylated derivative 2f with a 92% yield. However, this effect was not observed when the DICAB protocol was performed on these two substrates. In both cases, while the final compounds could not be isolated from the reaction mixture, comparison of the NMR yields of both crude mixtures showed that the alcohol was more tolerated in the reaction with DICAB, albeit the final compound 2e was obtained with the moderate NMR yield of 43%. The SF5Cl addition on the acetate derivative led to only 3% NMR yield of the final compound 2f and the reaction mixture showed the presence of various degradation compounds. Finally, the SF5Cl addition was performed on two ester derivatives. When using vinyl benzoate as the starting material, the desired compound 2g was obtained with a 75% yield with Dolbier’s protocol and an 84% yield with the DICAB protocol, while the addition product 2h was obtained with an 81% yield using Et3B as the radical initiator and 70% when the reaction was performed with DICAB.
![[1860-5397-16-256-i3]](/bjoc/content/inline/1860-5397-16-256-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Scope of the Et3B and the DICAB-initiated SF5Cl additions on alkenes. Unless noted otherwise, isolated yields are reported. aYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard.
Scheme 3: Scope of the Et3B and the DICAB-initiated SF5Cl additions on alkenes. Unless noted otherwise, isola...
Next, the SF5Cl radical additions on alkynes using both protocols were performed (Scheme 4). First, 4-phenyl-1-butyne was evaluated, and the desired pentafluorosulfanylated product 2i was obtained with a 79% yield with the Et3B-mediated reaction, and a higher yield of 88% when using DICAB as the radical initiator. In the case of the SF5Cl addition on phenylacetylene, it has been reported in the initial report from Dolbier that the formation of the side-product 2k, resulting from the 2:1 addition of the starting material on the intermediate radical, occurred with the Et3B-mediated reaction [29]. In our hands, Dolbier’s protocol led to only trace amounts of the compound 2k and 18% of the desired addition product 2j, while the reaction with the DICAB conditions led to 23% of the desired compound 2j, and 5% of the 2:1 addition side product 2k. Finally, the reaction was performed on the internal alkyne 6-dodecyne, and the Dolbier’s protocol led to the moderate yield of 65% of the desired compound 2l, while only a 17% NMR yield was obtained in the reaction using DICAB as the radical initiator.
![[1860-5397-16-256-i4]](/bjoc/content/inline/1860-5397-16-256-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Scope of the Et3B and the DICAB-initiated SF5Cl additions on alkynes. Unless noted otherwise, isolated yields are reported. aYield estimated by 19F NMR analysis of the crude mixture using 2-fluoro-4-nitrotoluene as an internal standard.
Scheme 4: Scope of the Et3B and the DICAB-initiated SF5Cl additions on alkynes. Unless noted otherwise, isola...
Conclusion
In conclusion, we have shown that amine–borane complexes can be used as radical initiators under thermal conditions to perform the SF5Cl radical addition on unsaturated compounds. These air-stable complexes can therefore be used as alternatives to the more unstable and pyrophoric Et3B, in order to incorporate the SF5 substituent on aliphatic derivatives. A total of 7 examples of alkene derivatives and 3 examples of alkyne derivatives were evaluated in the reaction, with yields ranging from 3% to 85%. Overall, this reaction represents a complementary method to the Et3B-mediated SF5Cl addition on unsaturated compounds.
Supporting Information
| Supporting Information File 1: General information, synthetic procedures, additional optimization results, NMR spectra for known compounds (1H, 19F) and full characterization of all new compounds. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Silvey, G. A.; Cady, G. H. J. Am. Chem. Soc. 1950, 72, 3624–3626. doi:10.1021/ja01164a084
Return to citation in text: [1] -
Thayer, A. M. Chem. Eng. News 2006, 84 (23), 27–32. doi:10.1021/cen-v084n023.p027
Return to citation in text: [1] -
Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323
Return to citation in text: [1] [2] -
Bassetto, M.; Ferla, S.; Pertusati, F. Future Med. Chem. 2015, 7, 527–546. doi:10.4155/fmc.15.5
Return to citation in text: [1] [2] -
Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u
Return to citation in text: [1] [2] -
Dalvit, C.; Ko, S. Y.; Vulpetti, A. J. Fluorine Chem. 2013, 152, 129–135. doi:10.1016/j.jfluchem.2013.01.017
Return to citation in text: [1] [2] -
Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006
Return to citation in text: [1] [2] -
Jackson, D. A.; Mabury, S. A. Environ. Toxicol. Chem. 2009, 28, 1866–1873. doi:10.1897/09-037.1
Return to citation in text: [1] [2] -
Sowaileh, M. F.; Hazlitt, R. A.; Colby, D. A. ChemMedChem 2017, 12, 1481–1490. doi:10.1002/cmdc.201700356
Return to citation in text: [1] -
Welch, J. T.; Lim, D. S. Bioorg. Med. Chem. 2007, 15, 6659–6666. doi:10.1016/j.bmc.2007.08.012
Return to citation in text: [1] -
Wipf, P.; Mo, T.; Geib, S. J.; Caridha, D.; Dow, G. S.; Gerena, L.; Roncal, N.; Milner, E. E. Org. Biomol. Chem. 2009, 7, 4163–4165. doi:10.1039/b911483a
Return to citation in text: [1] -
Chia, P. W.; Brennan, S. C.; Slawin, A. M. Z.; Riccardi, D.; O'Hagan, D. Org. Biomol. Chem. 2012, 10, 7922–7927. doi:10.1039/c2ob26402a
Return to citation in text: [1] -
Zhang, Y.; Wang, Y.; He, C.; Liu, X.; Lu, Y.; Chen, T.; Pan, Q.; Xiong, J.; She, M.; Tu, Z.; Qin, X.; Li, M.; Tortorella, M. D.; Talley, J. J. J. Med. Chem. 2017, 60, 4135–4146. doi:10.1021/acs.jmedchem.6b01484
Return to citation in text: [1] -
Moraski, G. C.; Bristol, R.; Seeger, N.; Boshoff, H. I.; Tsang, P. S.-Y.; Miller, M. J. ChemMedChem 2017, 12, 1108–1115. doi:10.1002/cmdc.201700170
Return to citation in text: [1] -
Mori, S.; Tsuemoto, N.; Kasagawa, T.; Nakano, E.; Fujii, S.; Kagechika, H. Chem. Pharm. Bull. 2019, 67, 1278–1283. doi:10.1248/cpb.c19-00522
Return to citation in text: [1] -
Naclerio, G. A.; Abutaleb, N. S.; Onyedibe, K. I.; Seleem, M. N.; Sintim, H. O. RSC Med. Chem. 2020, 11, 102–110. doi:10.1039/c9md00391f
Return to citation in text: [1] -
Banks, B. J. Parasiticidal pyrazoles. Eur. Pat. Appl. EP0933363A1, Aug 4, 1999.
Return to citation in text: [1] -
Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. (Tokyo, Jpn.) 2007, 32, 255–259. doi:10.1584/jpestics.g06-50
Return to citation in text: [1] -
Kay, I. T.; Barton, J. E. D.; Collins, D. J.; Kowalczyk, B.; Mitchell, G.; Shribbs, J. M.; Cox, J. M.; Barnes, N. J.; Smith, S. C. Herbicides. WO Pat. Appl. WO/94/13652, June 23, 1994.
Return to citation in text: [1] -
Alt, G. H.; Pratt, J. K.; Phillips, W. G.; Srouji, G. H. Substituted thiazoles and their use as fungicides. Eur. Pat. Appl. EP0371950A2, June 6, 1990.
Return to citation in text: [1] -
Zhang, G.; Lee, Y.-J.; Gautam, P.; Lin, C.-C.; Liu, C.-L.; Chan, J. M. W. J. Mater. Chem. C 2019, 7, 7865–7871. doi:10.1039/c9tc00756c
Return to citation in text: [1] -
Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(sici)1521-3773(19990712)38:13/14<1989::aid-anie1989>3.0.co;2-k
Return to citation in text: [1] -
Henwood, A. F.; Webster, J.; Cordes, D.; Slawin, A. M. Z.; Jacquemin, D.; Zysman-Colman, E. RSC Adv. 2017, 7, 25566–25574. doi:10.1039/c7ra03190d
Return to citation in text: [1] -
Iida, N.; Tanaka, K.; Tokunaga, E.; Mori, S.; Saito, N.; Shibata, N. ChemistryOpen 2015, 4, 698–702. doi:10.1002/open.201500165
Return to citation in text: [1] -
Chan, J. M. W. J. Mater. Chem. C 2019, 7, 12822–12834. doi:10.1039/c9tc01949a
Return to citation in text: [1] -
Pal, A. K.; Henwood, A. F.; Cordes, D. B.; Slawin, A. M. Z.; Samuel, I. D. W.; Zysman-Colman, E. Inorg. Chem. 2017, 56, 7533–7544. doi:10.1021/acs.inorgchem.7b01075
Return to citation in text: [1] -
Beier, P. Pentafluorosulfanylation of Aromatics and Heteroaromatics. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Ma, J.-A.; Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 551–570. doi:10.1002/9783527824342.ch18
Return to citation in text: [1] -
Haufe, G. Pentafluorosulfanylation of Aliphatic Substrates. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Ma, J.-A.; Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 571–609. doi:10.1002/9783527824342.ch19
Return to citation in text: [1] [2] -
Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o
Return to citation in text: [1] [2] [3] -
Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003
Return to citation in text: [1] [2] -
Verma, R. D.; Kirchmeier, R. L.; Shreeve, J. M. Adv. Inorg. Chem. 1994, 41, 125–169. doi:10.1016/s0898-8838(08)60171-3
Return to citation in text: [1] -
Sidebottom, H. W.; Tedder, J. M.; Walton, J. C. Trans. Faraday Soc. 1969, 65, 2103–2109. doi:10.1039/tf9696502103
Return to citation in text: [1] -
Uematsu, R.; Saka, C.; Sumiya, Y.; Ichino, T.; Taketsugu, T.; Maeda, S. Chem. Commun. 2017, 53, 7302–7305. doi:10.1039/c7cc02541f
Return to citation in text: [1] -
Ollivier, C.; Renaud, P. Chem. Rev. 2001, 101, 3415–3434. doi:10.1021/cr010001p
Return to citation in text: [1] -
Renaud, P.; Beauseigneur, A.; Brecht-Forster, A.; Becattini, B.; Darmency, V.; Kandhasamy, S.; Montermini, F.; Ollivier, C.; Panchaud, P.; Pozzi, D.; Scanlan, E. M.; Schaffner, A.-P.; Weber, V. Pure Appl. Chem. 2007, 79, 223–233. doi:10.1351/pac200779020223
Return to citation in text: [1] -
Frankland, E. J. Chem. Soc. 1862, 15, 363–381. doi:10.1039/js8621500363
Return to citation in text: [1] -
Székely, A.; Klussmann, M. Chem. – Asian J. 2019, 14, 105–115. doi:10.1002/asia.201801636
Return to citation in text: [1] -
Brotherton, R. J.; Weber, C. J.; Guibert, C. R.; Little, J. L. Boron Compounds. Ullmann's Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2000. doi:10.1002/14356007.a04_309
Return to citation in text: [1] -
A 11B NMR spectrum of the amino-borane complex DICAB was taken over one year after the reception of the compound (see Supporting Information File 1). The 11B NMR spectrum shows a clean signal from the DICAB complex, with no sign of oxidation or degradation. This indicates that these complexes can be stored (without the need for argon) for a long time without noticeable degradation.
Return to citation in text: [1] -
Staubitz, A.; Robertson, A. P. M.; Manners, I. Chem. Rev. 2010, 110, 4079–4124. doi:10.1021/cr100088b
Return to citation in text: [1] -
Couturier, M.; Tucker, J. L.; Andresen, B. M.; Dubé, P.; Negri, J. T. Org. Lett. 2001, 3, 465–467. doi:10.1021/ol006969+
Return to citation in text: [1] -
Marciasini, L. D.; Richard, J.; Cacciuttolo, B.; Sartori, G.; Birepinte, M.; Chabaud, L.; Pinet, S.; Pucheault, M. Tetrahedron 2019, 75, 164–171. doi:10.1016/j.tet.2018.11.036
Return to citation in text: [1] -
Guerrand, H. D. S.; Marciasini, L. D.; Gendrineau, T.; Pascu, O.; Marre, S.; Pinet, S.; Vaultier, M.; Aymonier, C.; Pucheault, M. Tetrahedron 2014, 70, 6156–6161. doi:10.1016/j.tet.2014.04.036
Return to citation in text: [1] -
Guerrand, H. D. S.; Vaultier, M.; Pinet, S.; Pucheault, M. Adv. Synth. Catal. 2015, 357, 1167–1174. doi:10.1002/adsc.201401153
Return to citation in text: [1] -
Richard, J.; Birepinte, M.; Charbonnier, J. B.; Liautard, V.; Pinet, S.; Pucheault, M. Synthesis 2017, 49, 736–744. doi:10.1055/s-0036-1588345
Return to citation in text: [1] -
Birepinte, M.; Liautard, V.; Chabaud, L.; Pucheault, M. Chem. – Eur. J. 2020, 26, 3236–3240. doi:10.1002/chem.201905772
Return to citation in text: [1] -
Birepinte, M.; Liautard, V.; Chabaud, L.; Pucheault, M. Org. Lett. 2020, 22, 2838–2843. doi:10.1021/acs.orglett.0c00908
Return to citation in text: [1] -
Liautard, V.; Delgado, M.; Colin, B.; Chabaud, L.; Michaud, G.; Pucheault, M. ChemRxiv 2020. doi:10.26434/chemrxiv.13274543.v1
Return to citation in text: [1] [2] [3] [4] -
Delgado, M.; Michaud, G.; Colin, B.; Simon, F.; Fouquay, S.; Pucheault, M. Procédé de polymérisation d’au moins un composé polymérisable par voie radicalaire. Fr. Pat. Appl. FR2003811, 2020.
Return to citation in text: [1] [2] -
Michaud, G.; Simon, F.; Fouquay, S.; Liautard, V.; Pucheault, M.; Colin, B. Procédé de polymérisation avec un complexe de borane-amine. Fr. Pat. Appl. FR1908159, 2019.
Return to citation in text: [1] -
Gilbert, A.; Paquin, J.-F. J. Fluorine Chem. 2019, 221, 70–74. doi:10.1016/j.jfluchem.2019.04.003
Return to citation in text: [1]
| 29. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 1. | Silvey, G. A.; Cady, G. H. J. Am. Chem. Soc. 1950, 72, 3624–3626. doi:10.1021/ja01164a084 |
| 9. | Sowaileh, M. F.; Hazlitt, R. A.; Colby, D. A. ChemMedChem 2017, 12, 1481–1490. doi:10.1002/cmdc.201700356 |
| 3. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323 |
| 4. | Bassetto, M.; Ferla, S.; Pertusati, F. Future Med. Chem. 2015, 7, 527–546. doi:10.4155/fmc.15.5 |
| 5. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 6. | Dalvit, C.; Ko, S. Y.; Vulpetti, A. J. Fluorine Chem. 2013, 152, 129–135. doi:10.1016/j.jfluchem.2013.01.017 |
| 7. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006 |
| 8. | Jackson, D. A.; Mabury, S. A. Environ. Toxicol. Chem. 2009, 28, 1866–1873. doi:10.1897/09-037.1 |
| 37. | Székely, A.; Klussmann, M. Chem. – Asian J. 2019, 14, 105–115. doi:10.1002/asia.201801636 |
| 3. | Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Org. Lett. 2015, 17, 3038–3041. doi:10.1021/acs.orglett.5b01323 |
| 4. | Bassetto, M.; Ferla, S.; Pertusati, F. Future Med. Chem. 2015, 7, 527–546. doi:10.4155/fmc.15.5 |
| 5. | Savoie, P. R.; Welch, J. T. Chem. Rev. 2015, 115, 1130–1190. doi:10.1021/cr500336u |
| 6. | Dalvit, C.; Ko, S. Y.; Vulpetti, A. J. Fluorine Chem. 2013, 152, 129–135. doi:10.1016/j.jfluchem.2013.01.017 |
| 7. | Sheppard, W. A. J. Am. Chem. Soc. 1962, 84, 3064–3072. doi:10.1021/ja00875a006 |
| 8. | Jackson, D. A.; Mabury, S. A. Environ. Toxicol. Chem. 2009, 28, 1866–1873. doi:10.1897/09-037.1 |
| 32. | Sidebottom, H. W.; Tedder, J. M.; Walton, J. C. Trans. Faraday Soc. 1969, 65, 2103–2109. doi:10.1039/tf9696502103 |
| 33. | Uematsu, R.; Saka, C.; Sumiya, Y.; Ichino, T.; Taketsugu, T.; Maeda, S. Chem. Commun. 2017, 53, 7302–7305. doi:10.1039/c7cc02541f |
| 2. | Thayer, A. M. Chem. Eng. News 2006, 84 (23), 27–32. doi:10.1021/cen-v084n023.p027 |
| 34. | Ollivier, C.; Renaud, P. Chem. Rev. 2001, 101, 3415–3434. doi:10.1021/cr010001p |
| 35. | Renaud, P.; Beauseigneur, A.; Brecht-Forster, A.; Becattini, B.; Darmency, V.; Kandhasamy, S.; Montermini, F.; Ollivier, C.; Panchaud, P.; Pozzi, D.; Scanlan, E. M.; Schaffner, A.-P.; Weber, V. Pure Appl. Chem. 2007, 79, 223–233. doi:10.1351/pac200779020223 |
| 27. | Beier, P. Pentafluorosulfanylation of Aromatics and Heteroaromatics. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Ma, J.-A.; Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 551–570. doi:10.1002/9783527824342.ch18 |
| 28. | Haufe, G. Pentafluorosulfanylation of Aliphatic Substrates. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Ma, J.-A.; Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 571–609. doi:10.1002/9783527824342.ch19 |
| 31. | Verma, R. D.; Kirchmeier, R. L.; Shreeve, J. M. Adv. Inorg. Chem. 1994, 41, 125–169. doi:10.1016/s0898-8838(08)60171-3 |
| 21. | Zhang, G.; Lee, Y.-J.; Gautam, P.; Lin, C.-C.; Liu, C.-L.; Chan, J. M. W. J. Mater. Chem. C 2019, 7, 7865–7871. doi:10.1039/c9tc00756c |
| 22. | Kirsch, P.; Bremer, M.; Heckmeier, M.; Tarumi, K. Angew. Chem., Int. Ed. 1999, 38, 1989–1992. doi:10.1002/(sici)1521-3773(19990712)38:13/14<1989::aid-anie1989>3.0.co;2-k |
| 23. | Henwood, A. F.; Webster, J.; Cordes, D.; Slawin, A. M. Z.; Jacquemin, D.; Zysman-Colman, E. RSC Adv. 2017, 7, 25566–25574. doi:10.1039/c7ra03190d |
| 24. | Iida, N.; Tanaka, K.; Tokunaga, E.; Mori, S.; Saito, N.; Shibata, N. ChemistryOpen 2015, 4, 698–702. doi:10.1002/open.201500165 |
| 25. | Chan, J. M. W. J. Mater. Chem. C 2019, 7, 12822–12834. doi:10.1039/c9tc01949a |
| 26. | Pal, A. K.; Henwood, A. F.; Cordes, D. B.; Slawin, A. M. Z.; Samuel, I. D. W.; Zysman-Colman, E. Inorg. Chem. 2017, 56, 7533–7544. doi:10.1021/acs.inorgchem.7b01075 |
| 28. | Haufe, G. Pentafluorosulfanylation of Aliphatic Substrates. In Emerging Fluorinated Motifs: Synthesis, Properties and Applications; Ma, J.-A.; Cahard, D., Eds.; Wiley-VCH: Weinheim, Germany, 2020; pp 571–609. doi:10.1002/9783527824342.ch19 |
| 17. | Banks, B. J. Parasiticidal pyrazoles. Eur. Pat. Appl. EP0933363A1, Aug 4, 1999. |
| 18. | Lim, D. S.; Choi, J. S.; Pak, C. S.; Welch, J. T. J. Pestic. Sci. (Tokyo, Jpn.) 2007, 32, 255–259. doi:10.1584/jpestics.g06-50 |
| 19. | Kay, I. T.; Barton, J. E. D.; Collins, D. J.; Kowalczyk, B.; Mitchell, G.; Shribbs, J. M.; Cox, J. M.; Barnes, N. J.; Smith, S. C. Herbicides. WO Pat. Appl. WO/94/13652, June 23, 1994. |
| 20. | Alt, G. H.; Pratt, J. K.; Phillips, W. G.; Srouji, G. H. Substituted thiazoles and their use as fungicides. Eur. Pat. Appl. EP0371950A2, June 6, 1990. |
| 10. | Welch, J. T.; Lim, D. S. Bioorg. Med. Chem. 2007, 15, 6659–6666. doi:10.1016/j.bmc.2007.08.012 |
| 11. | Wipf, P.; Mo, T.; Geib, S. J.; Caridha, D.; Dow, G. S.; Gerena, L.; Roncal, N.; Milner, E. E. Org. Biomol. Chem. 2009, 7, 4163–4165. doi:10.1039/b911483a |
| 12. | Chia, P. W.; Brennan, S. C.; Slawin, A. M. Z.; Riccardi, D.; O'Hagan, D. Org. Biomol. Chem. 2012, 10, 7922–7927. doi:10.1039/c2ob26402a |
| 13. | Zhang, Y.; Wang, Y.; He, C.; Liu, X.; Lu, Y.; Chen, T.; Pan, Q.; Xiong, J.; She, M.; Tu, Z.; Qin, X.; Li, M.; Tortorella, M. D.; Talley, J. J. J. Med. Chem. 2017, 60, 4135–4146. doi:10.1021/acs.jmedchem.6b01484 |
| 14. | Moraski, G. C.; Bristol, R.; Seeger, N.; Boshoff, H. I.; Tsang, P. S.-Y.; Miller, M. J. ChemMedChem 2017, 12, 1108–1115. doi:10.1002/cmdc.201700170 |
| 15. | Mori, S.; Tsuemoto, N.; Kasagawa, T.; Nakano, E.; Fujii, S.; Kagechika, H. Chem. Pharm. Bull. 2019, 67, 1278–1283. doi:10.1248/cpb.c19-00522 |
| 16. | Naclerio, G. A.; Abutaleb, N. S.; Onyedibe, K. I.; Seleem, M. N.; Sintim, H. O. RSC Med. Chem. 2020, 11, 102–110. doi:10.1039/c9md00391f |
| 29. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 30. | Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003 |
| 40. | Staubitz, A.; Robertson, A. P. M.; Manners, I. Chem. Rev. 2010, 110, 4079–4124. doi:10.1021/cr100088b |
| 38. | Brotherton, R. J.; Weber, C. J.; Guibert, C. R.; Little, J. L. Boron Compounds. Ullmann's Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2000. doi:10.1002/14356007.a04_309 |
| 39. | A 11B NMR spectrum of the amino-borane complex DICAB was taken over one year after the reception of the compound (see Supporting Information File 1). The 11B NMR spectrum shows a clean signal from the DICAB complex, with no sign of oxidation or degradation. This indicates that these complexes can be stored (without the need for argon) for a long time without noticeable degradation. |
| 29. | Aït-Mohand, S.; Dolbier, W. R., Jr. Org. Lett. 2002, 4, 3013–3015. doi:10.1021/ol026483o |
| 30. | Dolbier, W. R., Jr.; Aït-Mohand, S.; Schertz, T. D.; Sergeeva, T. A.; Cradlebaugh, J. A.; Mitani, A.; Gard, G. L.; Winter, R. W.; Thrasher, J. S. J. Fluorine Chem. 2006, 127, 1302–1310. doi:10.1016/j.jfluchem.2006.05.003 |
| 51. | Gilbert, A.; Paquin, J.-F. J. Fluorine Chem. 2019, 221, 70–74. doi:10.1016/j.jfluchem.2019.04.003 |
| 48. | Liautard, V.; Delgado, M.; Colin, B.; Chabaud, L.; Michaud, G.; Pucheault, M. ChemRxiv 2020. doi:10.26434/chemrxiv.13274543.v1 |
| 48. | Liautard, V.; Delgado, M.; Colin, B.; Chabaud, L.; Michaud, G.; Pucheault, M. ChemRxiv 2020. doi:10.26434/chemrxiv.13274543.v1 |
| 49. | Delgado, M.; Michaud, G.; Colin, B.; Simon, F.; Fouquay, S.; Pucheault, M. Procédé de polymérisation d’au moins un composé polymérisable par voie radicalaire. Fr. Pat. Appl. FR2003811, 2020. |
| 48. | Liautard, V.; Delgado, M.; Colin, B.; Chabaud, L.; Michaud, G.; Pucheault, M. ChemRxiv 2020. doi:10.26434/chemrxiv.13274543.v1 |
| 48. | Liautard, V.; Delgado, M.; Colin, B.; Chabaud, L.; Michaud, G.; Pucheault, M. ChemRxiv 2020. doi:10.26434/chemrxiv.13274543.v1 |
| 49. | Delgado, M.; Michaud, G.; Colin, B.; Simon, F.; Fouquay, S.; Pucheault, M. Procédé de polymérisation d’au moins un composé polymérisable par voie radicalaire. Fr. Pat. Appl. FR2003811, 2020. |
| 50. | Michaud, G.; Simon, F.; Fouquay, S.; Liautard, V.; Pucheault, M.; Colin, B. Procédé de polymérisation avec un complexe de borane-amine. Fr. Pat. Appl. FR1908159, 2019. |
| 41. | Couturier, M.; Tucker, J. L.; Andresen, B. M.; Dubé, P.; Negri, J. T. Org. Lett. 2001, 3, 465–467. doi:10.1021/ol006969+ |
| 42. | Marciasini, L. D.; Richard, J.; Cacciuttolo, B.; Sartori, G.; Birepinte, M.; Chabaud, L.; Pinet, S.; Pucheault, M. Tetrahedron 2019, 75, 164–171. doi:10.1016/j.tet.2018.11.036 |
| 43. | Guerrand, H. D. S.; Marciasini, L. D.; Gendrineau, T.; Pascu, O.; Marre, S.; Pinet, S.; Vaultier, M.; Aymonier, C.; Pucheault, M. Tetrahedron 2014, 70, 6156–6161. doi:10.1016/j.tet.2014.04.036 |
| 44. | Guerrand, H. D. S.; Vaultier, M.; Pinet, S.; Pucheault, M. Adv. Synth. Catal. 2015, 357, 1167–1174. doi:10.1002/adsc.201401153 |
| 45. | Richard, J.; Birepinte, M.; Charbonnier, J. B.; Liautard, V.; Pinet, S.; Pucheault, M. Synthesis 2017, 49, 736–744. doi:10.1055/s-0036-1588345 |
| 46. | Birepinte, M.; Liautard, V.; Chabaud, L.; Pucheault, M. Chem. – Eur. J. 2020, 26, 3236–3240. doi:10.1002/chem.201905772 |
| 47. | Birepinte, M.; Liautard, V.; Chabaud, L.; Pucheault, M. Org. Lett. 2020, 22, 2838–2843. doi:10.1021/acs.orglett.0c00908 |
© 2020 Gilbert et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)