Abstract
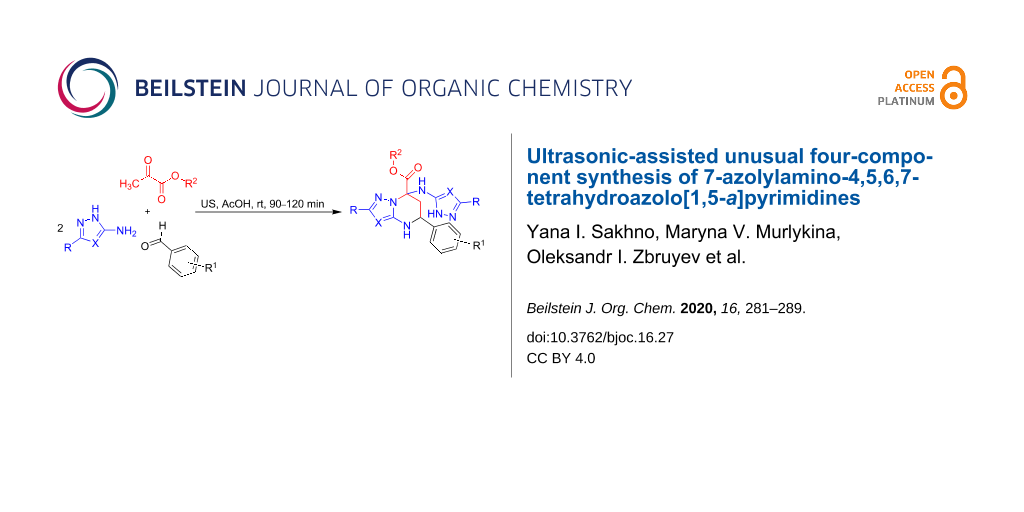
Four-component reactions of 3-amino-1,2,4-triazole or 5-amino-1H-pyrazole-4-carbonitrile with aromatic aldehydes and pyruvic acid or its esters under ultrasonication were studied. Unusual for such a reaction type, a cascade of elementary stages led to the formation of 7-azolylaminotetrahydroazolo[1,5-a]pyrimidines.

Graphical Abstract
Introduction
Tetrahydropyrimidines are heterocycles of high pharmacological importance, and they have attracted the attention of medicinal chemists because of their various biological activities [1]. Furthermore, tetrahydropyrimidines containing fused azole rings are a privileged class of heterocycles due to their antiviral [2], antitubercular, antitumor [3], antibacterial [4,5], and bone-anabolic activities [6,7]. Previously, some tetrahydroazolopyrimidines II (Scheme 1) were synthesized using sequential synthetic routes [8-15], in most cases involving the reduction of the dihydroazolopyrimidines I obtained via well-known reactions of aminoazoles with α,β-unsaturated ketones [8-11] or via their multicomponent analogues. On the other hand, multicomponent reactions (MCRs) directly leading to tetrahydroazolopyrimidine heterocyclic systems have also been published [4,5,16-24]. In particular, multicomponent approaches were described for the synthesis of 7-hydroxytetrahydroazolopyrimidines by the reaction of aminoazoles, aromatic aldehydes, and some carbonyl compounds (Scheme 1, IV and Scheme 2, VI and VIII) [4,5,16-23]. Oxygen-bridged tetrahydroazolopyrimidines III were also obtained via similar MCRs when using salicylic aldehydes, allowing further intermolecular cyclization [23,24].
![[1860-5397-16-27-i1]](/bjoc/content/inline/1860-5397-16-27-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of tetrahydroazolopyrimidine derivatives.
Scheme 1: Synthesis of tetrahydroazolopyrimidine derivatives.
![[1860-5397-16-27-i2]](/bjoc/content/inline/1860-5397-16-27-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Various multicomponent reactions involving pyruvic acids (pyruvates) and different α-aminoazoles.
Scheme 2: Various multicomponent reactions involving pyruvic acids (pyruvates) and different α-aminoazoles.
One of the promising reagents for the synthesis of diverse types of azoloazines by multicomponent and sequential reactions is pyruvic acid, along with its derivatives (Scheme 2). The selectivity of such interactions can be tuned, for example, by application of a condition-based divergence strategy [25], which is based on the variation of solvents, catalysts, and activation methods [16,26-29].
Thus, using nonclassical activation methods such as ultrasonication and microwave irradiation enabled us to develop highly selective procedures for obtaining triazolo-, tetrazolo-, and pyrazolopyrimidines [4,5,16-20,27-29], as well as oxygen-bridged tetrahydropyrazolopyrimidines [4,19], furanone [17,30], and pyrrolone derivatives [5,16-18].
Besides the presence of an OH group in the tetrahydropyrimidine moiety (e.g., Scheme 2, VI and VIII), the formation of heterocycles that are amino-substituted in a similar position is also possible and has been described [31,32]. In particular, the reaction of methyl pyruvate and anilines led to the formation of 4-arylamino-substituted tetrahydroquinolines (Scheme 3). To the best of our knowledge, an analogous reaction has not been described for aminoazoles.
![[1860-5397-16-27-i3]](/bjoc/content/inline/1860-5397-16-27-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 4-arylamino-substituted tetrahydroquinolines.
Scheme 3: Synthesis of 4-arylamino-substituted tetrahydroquinolines.
We were interested to study reactions similar to the ones shown in Scheme 3, involving aromatic aldehydes and pyruvic acid but using aminoazoles instead of anilines. Therefore, the present work is devoted to the study of multicomponent reactions involving 3-amino-1,2,4-triazole or 5-aminopyrazoles, aromatic aldehydes, and pyruvic acid or its esters under ultrasonication.
Results and Discussion
It was discovered that MCRs involving 3-amino-1,2,4-triazole or 5-amino-1H-pyrazole-4-carbonitrile with aromatic aldehydes and pyruvic acid or its esters under ultrasonication led to the formation of 4,5,6,7-tetrahydroazolo[1,5-a]pyrimidines 4a–u (Scheme 4) containing an azolylamino substituent in the 7-position via an unusual pseudo four-component reaction, rather than two heterocycles of the types V–X (Scheme 2). It should be noted that such a type of MCR, giving previously undescribed 7-azolylamino-substituted tetrahydroazolopyrimidines, is reported for the first time herein.
![[1860-5397-16-27-i4]](/bjoc/content/inline/1860-5397-16-27-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Ultrasound-assisted multicomponent reactions of 3-amino-1,2,4-triazole or 5-amino-1H-pyrazole-4-carbonitrile, aldehydes, and pyruvic acid/ethyl pyruvate.
Scheme 4: Ultrasound-assisted multicomponent reactions of 3-amino-1,2,4-triazole or 5-amino-1H-pyrazole-4-car...
Thus, using 2 equivalents of 5-aminopyrazole-4-carbonitrile 1a/b in MCRs with aromatic aldehydes 2a–c and pyruvic acid (3a) in acetic acid at room temperature under ultrasonication for 90 min gave 3-cyano-7-((4-cyano-1H-pyrazol-5-yl)amino)-5-aryl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-7-carboxylic acids 4a–c. There is also the possibility of applying ethyl pyruvate (3b) instead of pyruvic acid as a reactant in the reaction with 3-substituted-5-aminopyrazole-4-carbonitriles (R = H, CH3) 1a/b and aromatic aldehydes 2a–f (ultrasonication in acetic acid at room temperature for 120 min). In this case, the corresponding ethyl 3-cyano-7-((4-cyano-3-substituted-1H-pyrazol-5-yl)amino)-2-substituted-5-aryl-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-7-carboxylates (substituents = H, CH3) 4d–o were isolated in 45–80% yields (Scheme 4, Table 1).
Table 1: Synthesis of compounds 4a–u.
| starting material | US reaction time (min) | product | yield (%) | ||||||
| 1 | X | R | R1 | 2 | R2 | 3 | 4 | ||
| 1a |
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-27-i7.svg?max-width=637&scale=1.0)
|
H | H | 2a | H | 3a | 90 | 4a | 76 |
| 1a | „ | H | 4-CH3O | 2b | H | 3a | 90 | 4b | 86 |
| 1a | „ | H | 4-Cl | 2c | H | 3a | 90 | 4c | 75 |
| 1a | „ | H | 4-H | 2a | C2H5 | 3b | 120 | 4d | 60 |
| 1a | „ | H | 4-CH3O | 2b | C2H5 | 3b | 120 | 4e | 73 |
| 1a | „ | H | 4-Cl | 2c | C2H5 | 3b | 120 | 4f | 55 |
| 1a | „ | H | 4-Br | 2d | C2H5 | 3b | 120 | 4g | 80 |
| 1a | „ | H | 4-COOCH3 | 2e | C2H5 | 3b | 120 | 4h | 64 |
| 1a | „ | H | 4-CN | 2f | C2H5 | 3b | 120 | 4i | 53 |
| 1b | „ | CH3 | H | 2a | C2H5 | 3b | 120 | 4j | 45 |
| 1b | „ | CH3 | 4-CH3O | 2b | C2H5 | 3b | 120 | 4k | 68 |
| 1b | „ | CH3 | 4-Cl | 2c | C2H5 | 3b | 120 | 4l | 55 |
| 1b | „ | CH3 | 4-Br | 2d | C2H5 | 3b | 120 | 4m | 63 |
| 1b | „ | CH3 | 4-COOCH3 | 2e | C2H5 | 3b | 120 | 4n | 55 |
| 1b | „ | CH3 | 4-CN | 2f | C2H5 | 3b | 120 | 4o | 47 |
| 1c | N | H | 4-CH3O | 2b | H | 3a | 120 | 4p | 60 |
| 1c | N | H | 3-CH3O | 2g | H | 3a | 120 | 4q | 65 |
| 1c | N | H | 2-CH3O | 2h | H | 3a | 120 | 4r | 70 |
| 1c | N | H | 4-OH | 2k | H | 3a | 120 | 4s | 76 |
| 1c | N | H | 3-OH | 2j | H | 3a | 120 | 4t | 34 |
| 1c | N | H | 2-OH | 2i | H | 3a | 120 | 4u | 49 |
The same products were isolated while carrying out this reaction in acetic acid at room temperature with intensive stirring instead of ultrasonic irradiation. However, the reaction time had to be increased to 24 h, and the yields and purity of the compounds 4 decreased (as seen via TLC and NMR analysis), obviously due to the worse homogenization and mass transfer compared to ultrasonication.
Literature data [17,26,27,33-35] indicates that 5-aminopyrazoles bearing an electron-withdrawing substituent in the 4-position, such as a carbonitrile group, often behave similar to 3-amino-1,2,4-triazole; therefore, we studied the latter under the same reaction conditions. We showed that the pseudo four-component heterocyclization of two equivalents of 3-amino-1,2,4-triazole (1c) with aromatic aldehydes 2a–f and pyruvic acid (3a) carried out in acetic acid at a room temperature under ultrasonication for 120 min gave 7-((1H-1,2,4-triazol-5-yl)amino)-5-aryl-4,5,6,7-tetrahydro[1,2,4]triazolo[1,5-a]pyrimidine-7-carboxylic acids 4p–u in 34–76% yields (Scheme 4). In contrast to pyrazolyl-substituted pyrazolo[1,5-a]pyrimidine-7-carboxylic acids 4d–o, triazolyl-substituted derivatives 4p–u were unstable in protic solvents in the presence of water, and especially upon increasing the temperature gradually transformed into 7-hydroxytriazolo[1,5-a]pyrimidines 5a–f, which had been obtained earlier [5] by the three-component reaction of starting materials 1c, 2a–f, and 3a (HOAc, 65 °C, 48 h). This instability complicated the isolation and characterization of the heterocycles 4p–u. The condensation of compounds 1c, 2a–f, and 3a in dry DMF under otherwise identical conditions was a way to avoid the rapid conversion of triazolyl derivatives 4p–u into 7-hydroxytriazolo[1,5-a]pyrimidines 5a–f and to isolate the heterocycles 4p–u, but in lower yields in comparison to the reaction in acetic acid.
We consequently screened various condensation conditions, specifically by applying different temperatures in the range of 0–110 °C (both with the help of conventional heating and microwave irradiation) and by using different solvents and catalysts, such as HOAc, DMSO, primary alcohols with and without the presence of HCl, Yb(OTf)3, or Et3N. In all these cases, mixtures of triazolo[1,5-a]pyrimidines 4p–u and 5a–f in different ratios (the content of compounds 5a–f increased with the elevation of temperature and time) with impurities of starting reagents and unidentified compounds were obtained.
In contrast to tetrahydropyrimidines 4p–u, containing triazolyl fragments, our attempts to obtain compounds 6 from pyrazolyl derivatives 4b/c/e/f were unsuccessful. In particular, under the same conditions (prolonged stirring in acetic acid at temperatures up to 70 °C), the starting tetrahydropyrazolopyrimidines 4b/c/e/f remained unchanged. Then, it was established that compound 4e remained stable under refluxing in acetic acid for 60 min, while heating for 120–180 min led to its decomposition. At the same time, compound 4b, after refluxing in acetic acid for ca. 120 min, was converted into 3-cyano-7-(4-methoxyphenyl)-4,7-dihydropyrazolo[1,5-a]pyrimidine-5-carboxylic acid (7, yield 35%). The same carboxylic acid 7 was obtained by counter synthesis through a three-component reaction involving 5-aminopyrazole-4-carbonitrile (1a), 4-methoxybenzaldehyde (2b), and pyruvic acid (3a) under heating at reflux in acetic acid for 240 min (yield 28%) or through two-component heterocyclization of 5-aminopyrazole 1a and p-methoxybenzylidenepyruvic acid (8, obtained from p-methoxybenzaldehyde and pyruvic acid) under the same conditions in a much shorter reaction time (10 min) with 66% yield (Scheme 5).
![[1860-5397-16-27-i5]](/bjoc/content/inline/1860-5397-16-27-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 3-cyano-7-(4-methoxyphenyl)-4,7-dihydropyrazolo[1,5-a]pyrimidine-5-carboxylic acid (7).
Scheme 5: Synthesis of 3-cyano-7-(4-methoxyphenyl)-4,7-dihydropyrazolo[1,5-a]pyrimidine-5-carboxylic acid (7)....
The results presented above indicate that the reaction possibly proceeded under kinetic and thermodynamic control, depending on the conditions. In particular, ultrasonication of the starting materials 1a, 2b, and 3a for 90 min at room temperature provided the kinetically controlled azolyltetrahydropyrimidine derivative 4b (Scheme 4), while high-temperature treatment of the same starting materials (HOAc, refluxing at 118 °C for 240 min) yielded the thermodynamically preferred dihydropyrazolopyrimidine-5-carboxylic acid 7 (Scheme 5).
The most probable pathway A (Scheme 6) to compounds 4 includes initial formation of the corresponding azomethines 9 and 10. A similar pathway was previously described and discussed in publications [31,32]. We cannot exclude other mechanisms, for example, via arylidenepyruvic acids (esters) 11 formed by water elimination from appropriate aldols (pathway B). However, our attempts to synthesize compound 4 by direct reaction of unsaturated acid 8 and aminoazole 1a under different conditions (ultrasonic activation and conventional heating) were unsuccessful, and only dihydropyrimidine 7 was obtained.
![[1860-5397-16-27-i6]](/bjoc/content/inline/1860-5397-16-27-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Structure elucidation
The purity and structures of the synthesized heterocycles were determined using elemental analysis, mass spectrometry, NMR spectroscopy (1D and 2D NMR experiments), and X-ray diffraction analysis. The signals in the 1H and 13C NMR spectra of some of the compounds 4 were duplicated. This could have been caused by two reasons, namely by free rotation of the azolyl fragment in the position 7 or by the presence of a second diastereomer. To establish the reason of duplication, NMR spectra were recorded at different temperatures. As the temperature was increased from 25 °C to 130 °C, the peaks were still duplicated, proving the presence of two diastereomers.
The 1H NMR spectra of 4a–o exhibited three signals of an AMX system for the tetrahydropyrimidine ring (about 2.27–2.48 ppm, 3.03–3.25 ppm, and 4.52–4.83 ppm), three signals for protons of the NH groups (ca. 7.85–8.13 ppm for the pyrimidine NH, 6.90–7.21 ppm for the NH pyrazolyl moiety, and 11.79–12.07 ppm for the aminopyrazole moiety), a CH signal of the proton at position 2 of the pyrazolopyrimidine, signals of the aryl ring at 7.54–8.34 ppm, and signals corresponding to the alkyl substituent.
The 1H NMR spectra of 4a–c/j/k/o showed a double set of signals for the AMX system of the tetrahydropyrimidine ring, the NH protons, and the protons of the CH2CH3 moiety (in the case of esters) due to the presence of two isomers of 4.
The 1H NMR and 13C NMR spectra of triazolyl derivatives 4p–u were more complicated, both due to the duplication of the signals and the fast decomposition of the compounds 4p–u in solutions. Therefore, 2D NMR and, in some cases, 13C NMR spectra were overcrowded and uninformative. Particularly, tetrahydropyrimidines 4s–u with hydroxy substituent in the aryl ring happened to be less stable than the derivatives 4p–r with the methoxy group; this explains the absence of 13C NMR spectra for compounds 4t/u in our work.
The 1H NMR spectra of the products 4p–u exhibited a broad singlet for the triazole NH and carboxyl groups at ca. 13.10–13.30 ppm, a broad singlet for the phenolic OH group at 9.40–9.70 ppm (for compounds 4s–u), singlets of pyrimidine NH and CH groups of the triazolyl and triazole fragments in the interval of 7.31–7.91 ppm, a singlet of the triazolylamino group at 7.23–7.35 ppm, a multiplet for the CH proton in position 5 at 5.10–5.25 ppm (for the derivatives 4r/u with ortho-substituent in the aryl ring) or at 4.60–4.90 ppm for the compounds 4p/q/s/t, multiplets for the CH2 group in position 6 at 2.90–3.60 ppm and 2.11–2.50 ppm, and multiplets of aromatic protons at 6.65–7.45 ppm.
The spectral data obtained for the tetrahydropyrimidines 4 most likely corresponded to the possible regioisomers A and B (Figure 1). Additional NOESY experiments, in particular for compound 4k, showed the presence of cross-peaks between the CH proton at position 5 with the pyrimidine NH group and cross-peaks of the NH group with protons of the aromatic system, thus excluding structure B (Figure 1).
![[1860-5397-16-27-1]](/bjoc/content/figures/1860-5397-16-27-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Alternative structures A and B for the tetrahydroazolopyrimidines 4.
Figure 1: Alternative structures A and B for the tetrahydroazolopyrimidines 4.
Eventually, the structure of tetrahydropyrimidines 4 was proven by X-ray analysis carried out for a single crystal of one diastereomer of compound 4g, which allowed assignment of the structure as ethyl 5-(4-bromophenyl)-3-cyano-7-((4-cyano-1H-pyrazol-5-yl)amino)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-7-carboxylate (Figure 2).
![[1860-5397-16-27-2]](/bjoc/content/figures/1860-5397-16-27-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Molecular structure of ethyl 5-(4-bromophenyl)-3-cyano-7-((4-cyano-1H-pyrazol-5-yl)amino)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-7-carboxylate (4g) obtained from X-ray diffraction data.
Figure 2: Molecular structure of ethyl 5-(4-bromophenyl)-3-cyano-7-((4-cyano-1H-pyrazol-5-yl)amino)-4,5,6,7-t...
Compound 4g existed as a solvate with acetonitrile in the crystal lattice. The tetrahydropyrimidine ring adopted a half-chair conformation (the puckering parameters [36] were: S = 0.70, Θ = 25.6°, Ψ = 21.6°). Deviations of the C2 and C3 atoms from the mean-plane of the remaining atoms of this ring were −0.26 Å and 0.35 Å, respectively. The phenyl substituent was located in an equatorial position and was slightly turned in relation to the N1–C3 endocyclic bond (the C4–N1–C3–C7 and N1–C3–C7–C12 torsion angles were −163.6(4)° and 19.7(7)°, respectively). Short intramolecular contacts appeared: the H1(N)···H12 distance was 2.10 Å and the van der Waals radii sum [37] was 2.34 Å, the H···N1 distance was 2.55 Å (and the van der Waals radii sum was 2.67 Å), while the H1···C12 distance was 2.60 Å, with a van der Waals radii sum of 2.87 Å. The two vicinal substituents at the C1 atom had different orientations in relation to the partially saturated cycle: the ester substituent was in an axial position, while the other substituent was found in an equatorial position (the C4–N3–C1–C18 and C4–N3–C1–N5 torsion angles were −104.0(5)° and 139.6(5)°, respectively). The carboxylic acid fragment was turned in relation to the N3–C1 endocyclic bond (the N3–C1–C18–O1 torsion angle was −131.4(5)°). The ethyl group was located in an ap position to the C1–C18 bond and was almost orthogonal to the C18–O2 bond (the C1–C18–O2–C19 and C18–O2–C19–C20 torsion angles were −175.0(4)° and 85.1(7)°, respectively). The planar cyanopyrazolimino substituent was turned significantly to the N3–C1 endocyclic bond (the N3–C1–N5–C14 torsion angle was −57.5(6)°).
In the crystal phase, molecules 4g formed centrosymmetric dimers due to the N1–H···N4 intermolecular hydrogen bonds (1 x, 1 y, 2 z; H···N 2.22 Å, N–H···N 164°). The dimers were bound by a N6–H···O1 intermolecular hydrogen bond (x 1, y, z; H···O 2.16 Å, N–H···O 162°), forming chains along the [100] crystallographic direction (Figure 3).
![[1860-5397-16-27-3]](/bjoc/content/figures/1860-5397-16-27-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Chains of 4g molecules in the crystal phase.
Figure 3: Chains of 4g molecules in the crystal phase.
Conclusion
In this study we disclosed a new direction for the multicomponent reaction of 5-aminopyrazole-4-carbonitriles or 3-amino-1,2,4-triazole with pyruvic acid or ethyl pyruvate and aromatic aldehydes under ultrasonication, leading to 7-azolylamino-4,5,6,7-tetrahydroazolo[1,5-a]pyrimidines via a cascade of elementary stages that is unusual for such transformations. Thus, we extended the molecular diversity of the compounds obtained by introducing an additional azolyl substituent to the pyrimidine ring.
Funding
The authors thank the National Academy of Sciences of Ukraine for financial support in the frame of the projects "Creation of modern bases for obtaining and analyzing substances and components of materials for pharmaceutical purposes" (0119U100727) and "Investigation of structural features of nitrogen containing heterocycles with potential biological activity" (0119U100716). M. Murlykina was supported by the Grant of National Academy of Sciences of Ukraine for young scientists’ research laboratories and groups (0118U100275).
References
-
Sepehri, S.; Sanchez, H. P.; Fassihi, A. J. Pharm. Pharm. Sci. 2015, 18, 1–52. doi:10.18433/j3q01v
Return to citation in text: [1] -
Yu, W.; Goddard, C.; Clearfield, E.; Mills, C.; Xiao, T.; Guo, H.; Morrey, J. D.; Motter, N. E.; Zhao, K.; Block, T. M.; Cuconati, A.; Xu, X. J. Med. Chem. 2011, 54, 5660–5670. doi:10.1021/jm200696v
Return to citation in text: [1] -
Hassan, A. Y.; Sarg, M. T.; Bayoumi, A. H.; El-Deeb, M. A. J. Heterocycl. Chem. 2018, 55, 1450–1478. doi:10.1002/jhet.3184
Return to citation in text: [1] -
Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Konovalova, I. S.; Shishkin, O. V.; Sysoiev, D. A.; Kornet, M. N.; Chebanov, V. A. Tetrahedron 2013, 69, 9261–9269. doi:10.1016/j.tet.2013.08.055
Return to citation in text: [1] [2] [3] [4] [5] -
Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Yoshida, M.; Mori, A.; Morimoto, S.; Kotani, E.; Oka, M.; Notoya, K.; Makino, H.; Ono, M.; Shirasaki, M.; Tada, N.; Fujita, H.; Ban, J.; Ikeda, Y.; Kawamoto, T.; Goto, M.; Kimura, H.; Baba, A.; Yasuma, T. Bioorg. Med. Chem. 2011, 19, 1881–1894. doi:10.1016/j.bmc.2011.02.001
Return to citation in text: [1] -
Yoshida, M.; Mori, A.; Kotani, E.; Oka, M.; Makino, H.; Fujita, H.; Ban, J.; Ikeda, Y.; Kawamoto, T.; Goto, M.; Kimura, H.; Baba, A.; Yasuma, T. J. Med. Chem. 2011, 54, 1430–1440. doi:10.1021/jm101452x
Return to citation in text: [1] -
Desenko, S. M.; Shishkin, O. V.; Orlov, V. D.; Lipson, V. V.; Lindeman, S. V.; Struchkov, Y. T. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1994, 30, 851–855. doi:10.1007/bf01169645
Return to citation in text: [1] [2] -
Kolosov, M. A.; Shvets, E. H.; Manuenkov, D. A.; Vlasenko, S. A.; Omelchenko, I. V.; Shishkina, S. V.; Orlov, V. D. Tetrahedron Lett. 2017, 58, 1207–1210. doi:10.1016/j.tetlet.2017.02.035
Return to citation in text: [1] [2] -
Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2001, 37, 747–754. doi:10.1023/a:1011925631511
Return to citation in text: [1] [2] -
Desenko, S. M.; Orlov, V. D.; Lipson, V. V. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1990, 26, 1362–1366. doi:10.1007/bf00473964
Return to citation in text: [1] [2] -
Bellec, C.; Lhommet, G. J. Heterocycl. Chem. 1995, 32, 1793–1800. doi:10.1002/jhet.5570320621
Return to citation in text: [1] -
Zemlyanaya, N. I.; Karnozhitskaya, T. M.; Musatov, V. I.; Konovalova, I. S.; Shishkina, S. V.; Lipson, V. V. Russ. J. Org. Chem. 2018, 54, 1241–1249. doi:10.1134/s1070428018080201
Return to citation in text: [1] -
Lipson, V. V.; Karnozhitskaya, T. M.; Shishkina, S. V.; Shishkin, O. V.; Turov, A. V. Russ. Chem. Bull. 2009, 58, 1441–1444. doi:10.1007/s11172-009-0193-1
Return to citation in text: [1] -
Bartashevich, E. V.; Plekhanov, P. V.; Rusinov, G. L.; Potemkin, V. A.; Belik, A. V.; Chupakhin, O. N. Russ. Chem. Bull. 1999, 48, 1553–1557. doi:10.1007/bf02496411
Return to citation in text: [1] -
Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041–11049. doi:10.1016/j.tet.2008.09.089
Return to citation in text: [1] [2] [3] [4] [5] -
Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Murlykina, M. V.; Sakhno, Ya. I.; Desenko, S. M.; Chebanov, V. A. In Heterocyclic compounds chemistry. Recent aspects; Kartsev, V. G., Ed.; ICSPF Press: Moscow, Russia, 2014; pp 318–324.
Return to citation in text: [1] [2] [3] [4] -
Sakhno, Y. I.; Murlykina, M. V.; Morozova, A. D.; Kozyryev, A. V.; Chebanov, V. A. Fr.-Ukr. J. Chem. 2015, 3, 1–20. doi:10.17721/fujcv3i2p1-20
Return to citation in text: [1] [2] [3] [4] -
Sakhno, Y. I.; Kozyryev, A. V.; Desenko, S. M.; Shishkina, S. V.; Musatov, V. I.; Sysoiev, D. O.; Chebanov, V. A. Tetrahedron 2018, 74, 564–571. doi:10.1016/j.tet.2017.12.031
Return to citation in text: [1] [2] [3] -
Komykhov, S. A.; Bondarenko, A. A.; Musatov, V. I.; Diachkov, M. V.; Gorobets, N. Y.; Desenko, S. M. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2017, 53, 378–380. doi:10.1007/s10593-017-2059-z
Return to citation in text: [1] [2] -
Světlík, J.; Kettmann, V. Tetrahedron Lett. 2011, 52, 1062–1066. doi:10.1016/j.tetlet.2010.12.051
Return to citation in text: [1] [2] -
Gorobets, N. Y.; Sedash, Y. V.; Ostras, K. S.; Zaremba, O. V.; Shishkina, S. V.; Baumer, V. N.; Shishkin, O. V.; Kovalenko, S. M.; Desenko, S. M.; Van der Eycken, E. V. Tetrahedron Lett. 2010, 51, 2095–2098. doi:10.1016/j.tetlet.2010.02.045
Return to citation in text: [1] [2] [3] -
Gümüş, M. K.; Gorobets, N. Y.; Sedash, Y. V.; Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2017, 53, 1261–1267. doi:10.1007/s10593-018-2204-3
Return to citation in text: [1] [2] -
Ruijter, E.; Scheffelaar, R.; Orru, R. V. A. Angew. Chem., Int. Ed. 2011, 50, 6234–6246. doi:10.1002/anie.201006515
Return to citation in text: [1] -
Chebanov, V.; Gura, K.; Desenko, S. Aminoazoles as Key Reagents in Multicomponent Heterocyclizations. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R.; Ruijter, E., Eds.; Top. Heterocycl. Chem., Vol. 23; 2010; pp 41–84. doi:10.1007/7081_2009_21
Return to citation in text: [1] [2] -
Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229–1242. doi:10.1016/j.tet.2006.11.048
Return to citation in text: [1] [2] [3] -
Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M. Ultrason. Sonochem. 2012, 19, 707–709. doi:10.1016/j.ultsonch.2011.08.003
Return to citation in text: [1] [2] -
Murlykina, M. V.; Morozova, A. D.; Zviagin, I. M.; Sakhno, Y. I.; Desenko, S. M.; Chebanov, V. A. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 527. doi:10.3389/fchem.2018.00527
Return to citation in text: [1] [2] -
Morozova, A. D.; Muravyova, E. A.; Shishkina, S. V.; Sysoiev, D.; Glasnov, T.; Musatov, V. I.; Desenko, S. M.; Chebanov, V. A. Chem. Heterocycl. Compd. 2019, 55, 78–89. doi:10.1007/s10593-019-02422-8
Return to citation in text: [1] -
Luo, C.; Huang, Y. J. Am. Chem. Soc. 2013, 135, 8193–8196. doi:10.1021/ja4040945
Return to citation in text: [1] [2] -
Tapia, I.; Alcazar, V.; Grande, M.; Moran, J. R. Tetrahedron 1988, 44, 5113–5116. doi:10.1016/s0040-4020(01)86016-6
Return to citation in text: [1] [2] -
Chebanov, V. A.; Desenko, S. M. Diversity-Oriented Synth. 2014, 1, 43–63. doi:10.2478/dos-2014-0003
Return to citation in text: [1] -
Chebanov, V. A.; Saraev, V. E.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Desenko, S. M. Eur. J. Org. Chem. 2012, 5515–5524. doi:10.1002/ejoc.201200669
Return to citation in text: [1] -
Muravyova, E. A.; Desenko, S. M.; Rudenko, R. V.; Shishkina, S. V.; Shishkin, O. V.; Sen’ko, Y. V.; Vashchenko, E. V.; Chebanov, V. A. Tetrahedron 2011, 67, 9389–9400. doi:10.1016/j.tet.2011.09.138
Return to citation in text: [1] -
Zefirov, N. S.; Palyulin, V. A.; Dashevskaya, E. E. J. Phys. Org. Chem. 1990, 3, 147–158. doi:10.1002/poc.610030304
Return to citation in text: [1] -
Zefirov, Yu. V. Kristallographiya 1997, 42, 936–958.
Return to citation in text: [1]
| 5. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469 |
| 31. | Luo, C.; Huang, Y. J. Am. Chem. Soc. 2013, 135, 8193–8196. doi:10.1021/ja4040945 |
| 32. | Tapia, I.; Alcazar, V.; Grande, M.; Moran, J. R. Tetrahedron 1988, 44, 5113–5116. doi:10.1016/s0040-4020(01)86016-6 |
| 17. | Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9 |
| 26. | Chebanov, V.; Gura, K.; Desenko, S. Aminoazoles as Key Reagents in Multicomponent Heterocyclizations. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R.; Ruijter, E., Eds.; Top. Heterocycl. Chem., Vol. 23; 2010; pp 41–84. doi:10.1007/7081_2009_21 |
| 27. | Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229–1242. doi:10.1016/j.tet.2006.11.048 |
| 33. | Chebanov, V. A.; Desenko, S. M. Diversity-Oriented Synth. 2014, 1, 43–63. doi:10.2478/dos-2014-0003 |
| 34. | Chebanov, V. A.; Saraev, V. E.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Desenko, S. M. Eur. J. Org. Chem. 2012, 5515–5524. doi:10.1002/ejoc.201200669 |
| 35. | Muravyova, E. A.; Desenko, S. M.; Rudenko, R. V.; Shishkina, S. V.; Shishkin, O. V.; Sen’ko, Y. V.; Vashchenko, E. V.; Chebanov, V. A. Tetrahedron 2011, 67, 9389–9400. doi:10.1016/j.tet.2011.09.138 |
| 1. | Sepehri, S.; Sanchez, H. P.; Fassihi, A. J. Pharm. Pharm. Sci. 2015, 18, 1–52. doi:10.18433/j3q01v |
| 6. | Yoshida, M.; Mori, A.; Morimoto, S.; Kotani, E.; Oka, M.; Notoya, K.; Makino, H.; Ono, M.; Shirasaki, M.; Tada, N.; Fujita, H.; Ban, J.; Ikeda, Y.; Kawamoto, T.; Goto, M.; Kimura, H.; Baba, A.; Yasuma, T. Bioorg. Med. Chem. 2011, 19, 1881–1894. doi:10.1016/j.bmc.2011.02.001 |
| 7. | Yoshida, M.; Mori, A.; Kotani, E.; Oka, M.; Makino, H.; Fujita, H.; Ban, J.; Ikeda, Y.; Kawamoto, T.; Goto, M.; Kimura, H.; Baba, A.; Yasuma, T. J. Med. Chem. 2011, 54, 1430–1440. doi:10.1021/jm101452x |
| 17. | Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9 |
| 30. | Morozova, A. D.; Muravyova, E. A.; Shishkina, S. V.; Sysoiev, D.; Glasnov, T.; Musatov, V. I.; Desenko, S. M.; Chebanov, V. A. Chem. Heterocycl. Compd. 2019, 55, 78–89. doi:10.1007/s10593-019-02422-8 |
| 4. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Konovalova, I. S.; Shishkin, O. V.; Sysoiev, D. A.; Kornet, M. N.; Chebanov, V. A. Tetrahedron 2013, 69, 9261–9269. doi:10.1016/j.tet.2013.08.055 |
| 5. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469 |
| 5. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469 |
| 16. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041–11049. doi:10.1016/j.tet.2008.09.089 |
| 17. | Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9 |
| 18. | Murlykina, M. V.; Sakhno, Ya. I.; Desenko, S. M.; Chebanov, V. A. In Heterocyclic compounds chemistry. Recent aspects; Kartsev, V. G., Ed.; ICSPF Press: Moscow, Russia, 2014; pp 318–324. |
| 3. | Hassan, A. Y.; Sarg, M. T.; Bayoumi, A. H.; El-Deeb, M. A. J. Heterocycl. Chem. 2018, 55, 1450–1478. doi:10.1002/jhet.3184 |
| 4. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Konovalova, I. S.; Shishkin, O. V.; Sysoiev, D. A.; Kornet, M. N.; Chebanov, V. A. Tetrahedron 2013, 69, 9261–9269. doi:10.1016/j.tet.2013.08.055 |
| 5. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469 |
| 16. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041–11049. doi:10.1016/j.tet.2008.09.089 |
| 17. | Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9 |
| 18. | Murlykina, M. V.; Sakhno, Ya. I.; Desenko, S. M.; Chebanov, V. A. In Heterocyclic compounds chemistry. Recent aspects; Kartsev, V. G., Ed.; ICSPF Press: Moscow, Russia, 2014; pp 318–324. |
| 19. | Sakhno, Y. I.; Murlykina, M. V.; Morozova, A. D.; Kozyryev, A. V.; Chebanov, V. A. Fr.-Ukr. J. Chem. 2015, 3, 1–20. doi:10.17721/fujcv3i2p1-20 |
| 20. | Sakhno, Y. I.; Kozyryev, A. V.; Desenko, S. M.; Shishkina, S. V.; Musatov, V. I.; Sysoiev, D. O.; Chebanov, V. A. Tetrahedron 2018, 74, 564–571. doi:10.1016/j.tet.2017.12.031 |
| 27. | Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229–1242. doi:10.1016/j.tet.2006.11.048 |
| 28. | Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M. Ultrason. Sonochem. 2012, 19, 707–709. doi:10.1016/j.ultsonch.2011.08.003 |
| 29. | Murlykina, M. V.; Morozova, A. D.; Zviagin, I. M.; Sakhno, Y. I.; Desenko, S. M.; Chebanov, V. A. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 527. doi:10.3389/fchem.2018.00527 |
| 2. | Yu, W.; Goddard, C.; Clearfield, E.; Mills, C.; Xiao, T.; Guo, H.; Morrey, J. D.; Motter, N. E.; Zhao, K.; Block, T. M.; Cuconati, A.; Xu, X. J. Med. Chem. 2011, 54, 5660–5670. doi:10.1021/jm200696v |
| 4. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Konovalova, I. S.; Shishkin, O. V.; Sysoiev, D. A.; Kornet, M. N.; Chebanov, V. A. Tetrahedron 2013, 69, 9261–9269. doi:10.1016/j.tet.2013.08.055 |
| 19. | Sakhno, Y. I.; Murlykina, M. V.; Morozova, A. D.; Kozyryev, A. V.; Chebanov, V. A. Fr.-Ukr. J. Chem. 2015, 3, 1–20. doi:10.17721/fujcv3i2p1-20 |
| 4. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Konovalova, I. S.; Shishkin, O. V.; Sysoiev, D. A.; Kornet, M. N.; Chebanov, V. A. Tetrahedron 2013, 69, 9261–9269. doi:10.1016/j.tet.2013.08.055 |
| 5. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469 |
| 16. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041–11049. doi:10.1016/j.tet.2008.09.089 |
| 17. | Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9 |
| 18. | Murlykina, M. V.; Sakhno, Ya. I.; Desenko, S. M.; Chebanov, V. A. In Heterocyclic compounds chemistry. Recent aspects; Kartsev, V. G., Ed.; ICSPF Press: Moscow, Russia, 2014; pp 318–324. |
| 19. | Sakhno, Y. I.; Murlykina, M. V.; Morozova, A. D.; Kozyryev, A. V.; Chebanov, V. A. Fr.-Ukr. J. Chem. 2015, 3, 1–20. doi:10.17721/fujcv3i2p1-20 |
| 20. | Sakhno, Y. I.; Kozyryev, A. V.; Desenko, S. M.; Shishkina, S. V.; Musatov, V. I.; Sysoiev, D. O.; Chebanov, V. A. Tetrahedron 2018, 74, 564–571. doi:10.1016/j.tet.2017.12.031 |
| 21. | Komykhov, S. A.; Bondarenko, A. A.; Musatov, V. I.; Diachkov, M. V.; Gorobets, N. Y.; Desenko, S. M. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2017, 53, 378–380. doi:10.1007/s10593-017-2059-z |
| 22. | Světlík, J.; Kettmann, V. Tetrahedron Lett. 2011, 52, 1062–1066. doi:10.1016/j.tetlet.2010.12.051 |
| 23. | Gorobets, N. Y.; Sedash, Y. V.; Ostras, K. S.; Zaremba, O. V.; Shishkina, S. V.; Baumer, V. N.; Shishkin, O. V.; Kovalenko, S. M.; Desenko, S. M.; Van der Eycken, E. V. Tetrahedron Lett. 2010, 51, 2095–2098. doi:10.1016/j.tetlet.2010.02.045 |
| 25. | Ruijter, E.; Scheffelaar, R.; Orru, R. V. A. Angew. Chem., Int. Ed. 2011, 50, 6234–6246. doi:10.1002/anie.201006515 |
| 4. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Konovalova, I. S.; Shishkin, O. V.; Sysoiev, D. A.; Kornet, M. N.; Chebanov, V. A. Tetrahedron 2013, 69, 9261–9269. doi:10.1016/j.tet.2013.08.055 |
| 5. | Murlykina, M. V.; Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoiev, D. O.; Kornet, M. N.; Schols, D.; Goeman, J. L.; Van der Eycken, J.; Van der Eycken, E. V.; Chebanov, V. A. Eur. J. Org. Chem. 2015, 4481–4492. doi:10.1002/ejoc.201500469 |
| 16. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041–11049. doi:10.1016/j.tet.2008.09.089 |
| 17. | Sakhno, Y. I.; Shishkina, S. V.; Shishkin, O. V.; Musatov, V. I.; Vashchenko, E. V.; Desenko, S. M.; Chebanov, V. A. Mol. Diversity 2010, 14, 523–531. doi:10.1007/s11030-010-9226-9 |
| 18. | Murlykina, M. V.; Sakhno, Ya. I.; Desenko, S. M.; Chebanov, V. A. In Heterocyclic compounds chemistry. Recent aspects; Kartsev, V. G., Ed.; ICSPF Press: Moscow, Russia, 2014; pp 318–324. |
| 19. | Sakhno, Y. I.; Murlykina, M. V.; Morozova, A. D.; Kozyryev, A. V.; Chebanov, V. A. Fr.-Ukr. J. Chem. 2015, 3, 1–20. doi:10.17721/fujcv3i2p1-20 |
| 20. | Sakhno, Y. I.; Kozyryev, A. V.; Desenko, S. M.; Shishkina, S. V.; Musatov, V. I.; Sysoiev, D. O.; Chebanov, V. A. Tetrahedron 2018, 74, 564–571. doi:10.1016/j.tet.2017.12.031 |
| 21. | Komykhov, S. A.; Bondarenko, A. A.; Musatov, V. I.; Diachkov, M. V.; Gorobets, N. Y.; Desenko, S. M. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2017, 53, 378–380. doi:10.1007/s10593-017-2059-z |
| 22. | Světlík, J.; Kettmann, V. Tetrahedron Lett. 2011, 52, 1062–1066. doi:10.1016/j.tetlet.2010.12.051 |
| 23. | Gorobets, N. Y.; Sedash, Y. V.; Ostras, K. S.; Zaremba, O. V.; Shishkina, S. V.; Baumer, V. N.; Shishkin, O. V.; Kovalenko, S. M.; Desenko, S. M.; Van der Eycken, E. V. Tetrahedron Lett. 2010, 51, 2095–2098. doi:10.1016/j.tetlet.2010.02.045 |
| 24. | Gümüş, M. K.; Gorobets, N. Y.; Sedash, Y. V.; Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2017, 53, 1261–1267. doi:10.1007/s10593-018-2204-3 |
| 16. | Sakhno, Y. I.; Desenko, S. M.; Shishkina, S. V.; Shishkin, O. V.; Sysoyev, D. O.; Groth, U.; Kappe, C. O.; Chebanov, V. A. Tetrahedron 2008, 64, 11041–11049. doi:10.1016/j.tet.2008.09.089 |
| 26. | Chebanov, V.; Gura, K.; Desenko, S. Aminoazoles as Key Reagents in Multicomponent Heterocyclizations. In Synthesis of Heterocycles via Multicomponent Reactions I; Orru, R.; Ruijter, E., Eds.; Top. Heterocycl. Chem., Vol. 23; 2010; pp 41–84. doi:10.1007/7081_2009_21 |
| 27. | Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M.; Chernenko, V. N.; Musatov, V. I.; Shishkina, S. V.; Shishkin, O. V.; Kappe, C. O. Tetrahedron 2007, 63, 1229–1242. doi:10.1016/j.tet.2006.11.048 |
| 28. | Chebanov, V. A.; Sakhno, Y. I.; Desenko, S. M. Ultrason. Sonochem. 2012, 19, 707–709. doi:10.1016/j.ultsonch.2011.08.003 |
| 29. | Murlykina, M. V.; Morozova, A. D.; Zviagin, I. M.; Sakhno, Y. I.; Desenko, S. M.; Chebanov, V. A. Front. Chem. (Lausanne, Switz.) 2018, 6, No. 527. doi:10.3389/fchem.2018.00527 |
| 8. | Desenko, S. M.; Shishkin, O. V.; Orlov, V. D.; Lipson, V. V.; Lindeman, S. V.; Struchkov, Y. T. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1994, 30, 851–855. doi:10.1007/bf01169645 |
| 9. | Kolosov, M. A.; Shvets, E. H.; Manuenkov, D. A.; Vlasenko, S. A.; Omelchenko, I. V.; Shishkina, S. V.; Orlov, V. D. Tetrahedron Lett. 2017, 58, 1207–1210. doi:10.1016/j.tetlet.2017.02.035 |
| 10. | Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2001, 37, 747–754. doi:10.1023/a:1011925631511 |
| 11. | Desenko, S. M.; Orlov, V. D.; Lipson, V. V. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1990, 26, 1362–1366. doi:10.1007/bf00473964 |
| 31. | Luo, C.; Huang, Y. J. Am. Chem. Soc. 2013, 135, 8193–8196. doi:10.1021/ja4040945 |
| 32. | Tapia, I.; Alcazar, V.; Grande, M.; Moran, J. R. Tetrahedron 1988, 44, 5113–5116. doi:10.1016/s0040-4020(01)86016-6 |
| 8. | Desenko, S. M.; Shishkin, O. V.; Orlov, V. D.; Lipson, V. V.; Lindeman, S. V.; Struchkov, Y. T. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1994, 30, 851–855. doi:10.1007/bf01169645 |
| 9. | Kolosov, M. A.; Shvets, E. H.; Manuenkov, D. A.; Vlasenko, S. A.; Omelchenko, I. V.; Shishkina, S. V.; Orlov, V. D. Tetrahedron Lett. 2017, 58, 1207–1210. doi:10.1016/j.tetlet.2017.02.035 |
| 10. | Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2001, 37, 747–754. doi:10.1023/a:1011925631511 |
| 11. | Desenko, S. M.; Orlov, V. D.; Lipson, V. V. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 1990, 26, 1362–1366. doi:10.1007/bf00473964 |
| 12. | Bellec, C.; Lhommet, G. J. Heterocycl. Chem. 1995, 32, 1793–1800. doi:10.1002/jhet.5570320621 |
| 13. | Zemlyanaya, N. I.; Karnozhitskaya, T. M.; Musatov, V. I.; Konovalova, I. S.; Shishkina, S. V.; Lipson, V. V. Russ. J. Org. Chem. 2018, 54, 1241–1249. doi:10.1134/s1070428018080201 |
| 14. | Lipson, V. V.; Karnozhitskaya, T. M.; Shishkina, S. V.; Shishkin, O. V.; Turov, A. V. Russ. Chem. Bull. 2009, 58, 1441–1444. doi:10.1007/s11172-009-0193-1 |
| 15. | Bartashevich, E. V.; Plekhanov, P. V.; Rusinov, G. L.; Potemkin, V. A.; Belik, A. V.; Chupakhin, O. N. Russ. Chem. Bull. 1999, 48, 1553–1557. doi:10.1007/bf02496411 |
| 23. | Gorobets, N. Y.; Sedash, Y. V.; Ostras, K. S.; Zaremba, O. V.; Shishkina, S. V.; Baumer, V. N.; Shishkin, O. V.; Kovalenko, S. M.; Desenko, S. M.; Van der Eycken, E. V. Tetrahedron Lett. 2010, 51, 2095–2098. doi:10.1016/j.tetlet.2010.02.045 |
| 24. | Gümüş, M. K.; Gorobets, N. Y.; Sedash, Y. V.; Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. (N. Y., NY, U. S.) 2017, 53, 1261–1267. doi:10.1007/s10593-018-2204-3 |
| 36. | Zefirov, N. S.; Palyulin, V. A.; Dashevskaya, E. E. J. Phys. Org. Chem. 1990, 3, 147–158. doi:10.1002/poc.610030304 |
© 2020 Sakhno et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)