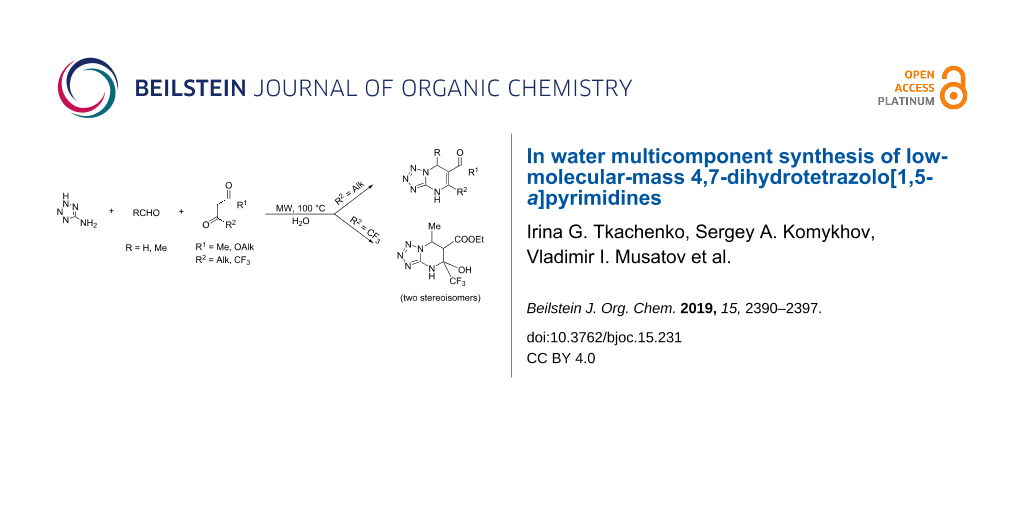
Abstract
The three-component reaction of 5-aminotetrazole with aliphatic aldehydes (formaldehyde, acetaldehyde) and acetoacetic ester derivatives in water under microwave irradiation leads to the selective formation of 4,7-dihydrotetrazolo[1,5-a]pyrimidine derivatives. Under similar conditions using 4,4,4-trifluoroacetoacetic ester 5-hydroxy-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidines are obtained. The analogous reaction with acetylacetone requires scandium(III) triflate as catalyst. The antioxidant activity of selected compounds was assayed with 1,1-diphenyl-2-picrylhydrazyl.

Graphical Abstract
Introduction
Tetrazolo[1,5-a]pyrimidines and their partially hydrogenated derivatives are known for their interesting biological properties. They have been reported to have anticancer [1], antimicrobial [2,3] and antioxidant [3] activities and to act as inhibitors of hepatitis B virus [4]. The dihydro derivatives of tetrazolo[1,5-a]pyrimidines belong to a bit special kind of dihydropyrimidines due to the strong electron-withdrawing properties of the tetrazole ring, which makes them useful for studying various theoretical issues, e.g., tautomerism [5], intramolecular transformations [6], etc.
There are several approaches to the synthesis of 4,7-dihydrotetrazolo[1,5-a]pyrimidines. Two of them make use of 5-aminotetrazole as a starting material and subsequent dihydropyrimidine ring formation. The first approach [6,7] represents a two-component cyclocondensation of 5-aminotetrazole (1) as binucleophilic component and bielectrophilic α,β-unsaturated carbonyl compounds 2 (Scheme 1, reaction 1). The second method [8-12] comprises the three-component reaction of amine 1 with the synthetic precursors of the unsaturated ketone 2 (Scheme 1, reaction 2), i.e., carbonyl compound 4 and a methylene-active compound 5. These approaches are general and useful for the preparation of variety of dihydroazolopyrimidines which is easily accessible by variation of the binucleophilic component 1 (instead of 1, 3-amino-1,2,4-triazole, 2-aminobenzimidazole, 3-aminopyrazoles, 4-amino-1,2,3-triazoles, etc. can be used [13]). However, a relatively low reactivity of amine 1 due to the electron deficiency of the tetrazole ring has been reported several times [5,6]. A third approach (Scheme 1, reaction 3) is completely different and consists of the tetrazole ring formation through cyclization of dihydropyrimidinethiones with sodium azide [14].
![[1860-5397-15-231-i1]](/bjoc/content/inline/1860-5397-15-231-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Three synthetic approaches to dihydrotetrazolo[1,5-a]pyrimidines.
Scheme 1: Three synthetic approaches to dihydrotetrazolo[1,5-a]pyrimidines.
Generally, all three approaches allow for the preparation of a broad range of compounds 3 with a wide variety of substituents R1–R3. However, the vast majority of reported data [5-12,14-18] comprises aryl-substituted azolopyrimidines (with either R1 or R3 or both being aromatic substituents), and there are only a view examples of compounds 3 having no aryl substituent [19,20]. The reason for this relatively low synthetic availability might be due to their higher solubility (compared to aryl-substituted analogues) and difficulties associated with their isolation.
Further, to address green chemistry principles [21-24] for the synthesis of dihydrotetrazolopyrimidines one may concern carrying out these reactions under solvent-free conditions [15-17] or using water as a “green solvent” [18].
Results and Discussion
Synthesis
Due to the small number of reports available for dihydroazolopyrimidines with aliphatic substituents, we aimed by this work to synthesize a range of dihydrotetrazolo[1,5-a]pyrimidines containing no aromatic substituents. The second important reason for our interest in these compounds is based on the Lipinski rules for orally active drugs [25]. According to one of them, drug-like molecules should have molecular masses lower than 500 Da. Therefore, the minimization of molecular masses for the targeted tetrazolo[1,5-a]pyrimidines can be achieved by exclusion of large aryl substituents from their structures. Thus, we selected aliphatic aldehydes, i.e., formaldehyde and acetaldehyde, as the starting material and chose a multicomponent approach to minimize the number of reaction steps according to green chemistry principles as well. Based on our recent research [26-28], where water proved to be an effective solvent for the multicomponent synthesis of low-molecular-mass dihydroazolopyrimidines, we decided to use water also in this case.
The three-component reactions of 5-aminotetrazole (1) with aldehydes 7a,b (paraformaldehyde, acetaldehyde) and a set of acetoacetic ester derivatives 8a–d in water under microwave irradiation at 100 °C led to the formation of the corresponding 5,6,7-trisubstituted 4,7-dihydrotetrazolo[1,5-a]pyrimidines 9a–g as single products. No formation of isomeric compounds 9’ was observed in any case (Scheme 2). Increasing the temperature (to 130 °C) and prolongation of the reaction time did not improve the yields of 9, while decreasing the temperature to 80 °C resulted in a reduced yield.
![[1860-5397-15-231-i2]](/bjoc/content/inline/1860-5397-15-231-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Three-component reaction of 1, 7a,b and 8a–d in water.
Scheme 2: Three-component reaction of 1, 7a,b and 8a–d in water.
The structures of all products 9a–g were proven by their spectral data. For example, the 1H NMR spectra of 9a–c contained singlets for NH (10.84–10.90 ppm) and CH2 (5.06–5.08 ppm) protons that corresponded to the 4,7-dihydro structure of the tetrazolo[1,5-a]pyrimidines. Further, five basic signals were observed in the 13C NMR spectrum for the dihydrotetrazolopyrimidine bicycle: one signal was in the aliphatic area, three signals appeared at lower field, and one signal (C-6 of the bicycle) was observed between aliphatic and unsaturated carbons (92.5–92.7 ppm) which is typical for these compounds. Although the NMR data corresponded quite well to the proposed structure 9, they did not allow the complete rejection of the isomeric structure 9' for the reaction product. The final confirmation of structures 9a–g was achieved by X-ray analysis of 9a (Figure 1).
![[1860-5397-15-231-1]](/bjoc/content/figures/1860-5397-15-231-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structure of 9a according to X-ray data. Displacement ellipsoids are shown at the 50% probability level.
Figure 1: Molecular structure of 9a according to X-ray data. Displacement ellipsoids are shown at the 50% pro...
A specific behavior of acetylacetone (10) as the 1,3-dicarbonyl compound was observed in the three-component reaction. Reacting it with amine 1 and aldehyde 7a under conditions similar to those for acetoacetic esters (8a–d) led to formation of a mixture of compounds 11 and 12 (Scheme 3). Heteroaromatic tetrazolopyrimidine 11 was obtained as a single product from the three-component process performed at room temperature. Its structure based on NMR spectral data corresponded to a two-component condensation of amine 1 and acetylacetone (10). The selective formation of the target compound 12 was achieved in the presence of scandium(III) triflate as a catalyst.
![[1860-5397-15-231-i3]](/bjoc/content/inline/1860-5397-15-231-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Three-component reaction of 5-aminotetrazole (1) with formaldehyde (7a) and acetylacetone (10).
Scheme 3: Three-component reaction of 5-aminotetrazole (1) with formaldehyde (7a) and acetylacetone (10).
Another example of a 1,3-dicarbonyl compound that often exhibits unusual behavior in the three-component synthesis of azolopyrimidines [27,29] is ethyl 4,4,4-trifluoroacetoacetate (13). In these transformations, the last reaction step, a water elimination, sometimes does not occur, and the final product is the 5-hydroxy-containing tetrahydro derivative. In our experiments, the three-component reaction of amine 1 with aldehyde 7b and compound 13 in water under microwave irradiation afforded tetrahydro derivative 14 as a mixture of two stereoisomers. Inspection of the mixture by 1H NMR revealed isomer A as the major component and isomer B as minor component in a ratio of ≈55:45. The 1H NMR spectrum of 14 contained a double set of signals for two methyl and one methylene group, two signals for methine protons and two singlets for NH and OH protons. The NOESY experiment allowed us to accomplish the final confirmation of the structure and to establish the relative stereochemistry of both stereoisomers. NOESY cross-peaks between signals of NH and OH protons revealed that A and B are in fact stereoisomers, rejecting the regioisomeric structure 14’. The different 3J values between 6-H and 7-H for both isomers (4.4 Hz for A and 11.2 Hz for B) are indicative of different relative orientations of these protons and suggested that these protons in isomer B have trans-orientation, whereas those in isomer A have cis-orientation. Further, NOE between methyl and hydroxy groups for B isomer and a merely weak NOE for those groups in isomer A further allowed to propose a cis-orientation of these substituents for isomer B and the opposite one for A (Scheme 4).
![[1860-5397-15-231-i4]](/bjoc/content/inline/1860-5397-15-231-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: a) Three-component reaction of 5-aminotetrazole (1) with acetaldehyde (7b) and ethyl 4,4,4-trifluoroacetoacetate (13); b) structure investigation of 14 by NMR.
Scheme 4: a) Three-component reaction of 5-aminotetrazole (1) with acetaldehyde (7b) and ethyl 4,4,4-trifluor...
Antioxidant properties of 9, 11, 14
Among the methods for the antioxidant activity (AOA) estimation, using 1,1-diphenyl-2-picrylhydrazyl (DPPH) is one of the most common and widespread [30-33]. The free radical scavenging activity of tetrazolo[1,5-a]pyrimidines 9d,f, 11, and 14 was measured spectrophotometrically as percentage of reducing the free-radical concentration in the presence of a test compound in methanol/dimethyl sulfoxide solutions. Ascorbic acid served as positive control. Among the tested compounds 9f showed the best results at 10−3 mol/L concentration and compound 9d exhibited the highest AOA at the other tested concentrations. The results of the radical scavenging experiments are collected in Table 1 and are promising for further detailed studies.
Conclusion
The three-component method which is based on the in-water reaction of 5-aminotetrazole with 1,3-dicarbonyl compounds and aliphatic aldehydes, like formaldehyde or acetaldehyde, under microwave activation, was applied for the preparation of low-molecular-mass 4,7-dihydrotetrazolo[1,5-a]pyrimidines. The use of acetoacetic ester derivatives as 1,3-dicarbonyl compounds in the reaction showed high selectivity under catalyst-free conditions, whereas in the case of acetylacetone the formation of a side product was observed. The desired product could be obtained with high selectivity by performing the reaction in the presence of scandium(III) triflate as a catalyst. 4,4,4-Trifluoromethylacetoacetic ester showed high reactivity in the current reaction forming the corresponding 5-hydroxy-5-trifluoromethyl-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidine as a mixture of isomers. Some of the prepared tetrazolopyrimidines showed free-radical scavenging activity towards DPPH.
Experimental
General. The melting points were determined with a Gallenkamp melting point apparatus. The NMR spectra were recorded at 400 MHz with a Varian MR-400 spectrometer. The EIMS spectra were measured on a GC–MS Varian 1200L (ionizing voltage 70 eV, direct input of the sample) instrument. Elemental analysis was realized on a EuroVector EA-3000. Analytical samples of the compounds were obtained by crystallization from water and further drying at room temperature in air. Microwave experiments were performed using septum-sealed reaction vials in an Emrys Creator EXP from Biotage AB (Uppsala, Sweden) possessing a single-mode microwave cavity producing controlled irradiation at 2.45 GHz. Solvents and all reagents were commercially available and used without additional purification.
Crystal data: The crystal structure of compound 9a was measured on an Xcalibur-3 diffractometer (graphite monochromated Mo Kα radiation, CCD detector, ω-scanning). The structure was solved by the direct method using the SHELXTL package [34,35]. Full-matrix least-squares refinement against F2 in anisotropic approximation was used for non-hydrogen atoms. Positions of hydrogen atoms were determined from electron density difference maps and refined by ‘‘riding” model with Uiso = nUeq of the carrier atom (n = 1.5 for methyl groups and n = 1.2 for other hydrogen atoms). Crystal data for C8H11N5O2 (9a) (M = 209.22 g/mol): triclinic, space group P−1, a = 4.2983(5) Å, b = 9.4739(8) Å, c = 13.1398(14) Å, α = 73.252(8)°, β = 88.290(9)°, γ = 79.628(8)°, V = 503.87(9) Å3, Z = 2, μ(Mo Kα) = 0.104 mm−1, Dcalc = 1.379 g/cm3. Intensities were measured at 293 K within 2θmax ≤ 54.992° (3905 reflections total, 2320 unique reflections, Rint = 0.031, Rsigma = 0.068). The final R1 = 0.062, wR2 = 0.136 for 2504 observed reflections with I ≥ 2σ(I) and R1 = 0.135, wR2 = 0.171 for all data, S = 1.024. Crystallographic data (excluding structure factors) for the structure of 9a have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 1942287. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK, (fax: +44-(0)1223-336033 or by email: deposit@ccdc.cam.ac.uk).
Free radical scavenging activity determination. A solution of 0.1 mmol/L DPPH (2 mL) in methanol was added to 2 mL of a solution of the investigated substance (9, 11, 14) in dimethyl sulfoxide (DMSO) at different concentrations (10−3, 10−5, 10−7 mol/L). The control solution was prepared by mixing 2 mL of DMSO and 0.1 mmol/L DPPH solution (2 mL). The mixture was shaken vigorously and allowed to stand at room temperature for 30 min in the dark. The absorbance was measured at 517 nm using a spectrophotometer Specord-200. The percentage of DPPH scavenging was calculated as (AOA%) = [A0 − A1/A0] × 100, where A0 is the absorbance of the control solution, A1 is the absorbance of the test solution. Ascorbic acid dissolved in DMSO served as the reference compound.
General procedure for the synthesis of 9a–g. In a septum-sealed reaction vial, a solution of 5-aminotetrazole (1, 1.7 mmol), aldehyde 7 (paraformaldehyde 7a or acetaldehyde 7b, 0.15 g, ≈2 mmol) and acetoacetic ester derivative 8a–d (1.77 mmol) in water (3.5 mL) was irradiated in a microwave reactor at 100 °C for 25–30 minutes. The crystalline product started to separate either during the course of reaction or just after cooling. The precipitate was filtered off, washed with water and air-dried.
Ethyl 5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9a). White solid, yield 85%; mp 200–202 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.22 (t, 3JHH = 6.8 Hz, 3H, CH3), 2.32 (s, 3Н, СН3), 4.11 (q, 3JHH = 7.2 Hz, 2Н, CH2), 5.07 (s, 2Н, CН2), 10.87 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 14.2 (CH3), 18.1 (CH3), 44.1 (C-7), 59.8 (CH2), 92.7 (C-6), 146.7 (C-5), 148.9 (C-3a), 164.8 (CO); EIMS (70 eV, m/z, (%)): 209 (13) [M+], 181 (21), 109 (10); Anal. calcd for С8Н11N5O2 (209.09): C, 45.93; H, 5.30; N, 33.48%; found: C, 45.63; H, 5.07; N, 33.71%.
Ethyl 5-propyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9b). White solid, yield 75%; mp 191–193 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.92 (t, 3JHH = 7.2 Hz, 3H, CH3), 1.22 (t, 3JHH = 7.2 Hz, 3H, СН3), 1.57 (q, 3JHH = 7.2 Hz, 2Н, CH2), 2.71 (t, 3JHH = 7.6 Hz, 2Н, CН2), 4.12 (q, 3JHH = 7.2 Hz, 2H, CH2), 5.08 (s, 2H, CH2), 10.84 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 13.6 (CH3), 14.1 (CH3), 21.6 (CH2), 32.7 (CH2), 44.2 (C-7), 59.8 (CH2), 92.5 (C-6), 149.0 (C-5), 150.6 (C-3a), 164.6 (CO); EIMS (70 eV, m/z (%)): 237 (2) [M+], 137 (13), 111 (10), 109 (19); Anal. calcd for С10Н15N5O2 (237.12): C, 50.62; H, 6.37; N, 29.52%; found: C, 50.42; H, 6.54; N, 29.38%.
2-Methoxyethyl 5-methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9c) White solid, yield 88%; mp 107–109 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.33 (s, 3Н, СН3), 3.27 (s, 3Н, CH3), 3.56 (t, 3JHH = 4.8 Hz, 2Н, CН2), 4.20 (t, 3JHH = 4.8 Hz, 2H, CH2), 5.07 (s, 2H, CH2), 10.89 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 18.1 (CH3), 44.1 (C-7), 58.1 (CH3) , 62.8 (CH2), 69.8 (CH2), 92.5 (C-6), 147.0 (C-5), 148.9 (C-3a), 164.8 (CO); EIMS (70 eV, m/z (%)): 239 (12) [M+], 238 (100); Anal. calcd for С9Н13N5O3 (239.23): C, 45.18; H, 5.48; N, 29.27%; found: C, 45.27; H, 5.65; N, 29.09%.
Ethyl 5,7-dimethyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9d). White solid, yield 87%; mp 173–175 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.24 (t, 3JHH = 7.2 Hz, 3H, CH3), 1.47 (d, 3JHH = 6.4 Hz, 3Н, СН3), 2.32 (s, 3Н, CH3), 4.15 (m, 2Н, CН2), 5.66 (q, 3JHH = 6.0 Hz, 1Н, Н); 10.96 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 14.1 (CH3), 18.4 (CH3), 23.0 (CH3), 51.5 (C-7), 59.8 (CH2), 98.4 (C-6), 146.3 (C-5), 148.6 (C-3a), 164.8 (CO); EIMS (70 eV, m/z (%)): 223 (10) [M+], 222 (100); Anal. calcd for С9Н13N5O2 (223.11): C, 48.42; H, 5.87; N, 31.37%; found: C, 48.60; H, 5.65; N, 31.52%.
Ethyl 7-methyl-5-propyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9e). Colorless solid, yield 78%; mp 137–139 °C; 1H NMR (400 MHz, DMSO-d6) δ 0.91 (t, 3JHH = 7.2 Hz, 3H, CH3), 1.24 (t, 3JHH = 7.2 Hz, 3Н, СН3), 1.46 (d, 3JHH = 6.4 Hz, 3H, CH3), 1.57 (m, 2Н, CH2), 2.69 (m, 2Н, CН2), 4.15 (m, 2H, CH2), 5.67 (q, 3JHH = 6.0 Hz, 1H, CH), 10.96 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 13.6 (CH3), 14.1 (CH3), 21.6 (CH3), 23.1 (CH2), 32.9 (CH2), 51.5 (C-7), 59.9 (CH2), 98.3 (C-6), 148.7 (C-5), 150.2 (C-3a), 164.6 (CO); EIMS (70 eV, m/z (%)): 251 (18) [ M+], 250 (100); Anal. calcd for С11Н17N5O2 (251.14): C, 52.58; H, 6.82; N, 27.87%; found: C, 52.69; H, 6.74; N, 27.96%.
2-Methoxyethyl 5,7-dimethyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9f) Colorless solid, yield 85%; mp 138–140 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.48 (d, 3JHH = 6.4 Hz, 3Н, СН3), 2.32 (s, 3Н, CH3), 3.27 (s, 3Н, CН3), 3.57 (t, 3JHH = 4.8 Hz, 2H, CH2), 4.15 (m, 1H, CH2), 4.29 (m, 1H, CH2), 5.65 (q, 3JHH = 6.0 Hz, 1H, CH), 10.99 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 18.5 (CH3), 23.0 (CH3), 51.6 (C-7), 58.1 (CH3), 62.8 (CH2), 69.9 (CH2), 98.2 (C-6), 146.8 (C-5), 148.6 (C-3a), 164.8 (CO); EIMS (70 eV, m/z (%)): 253 (13) [M+], 252 (100); Anal. calcd for С10Н15N5O3 (253.26): C, 47.42; H, 5.97; N, 27.65%; found: C, 47.53; H, 5.88; N, 27.76%.
tert-Butyl 5,7-dimethyl-4,7-dihydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (9g) Colorless solid, yield 85%; mp 174–175 °C; 1H NMR (400 MHz, DMSO-d6) δ 1.43 (s, 9Н, 3СН3), 1.44 (d, 3JHH = 1.6 Hz, 3Н, CH3), 2.26 (s, 3Н, CН3), 5.57 (q, 3JHH = 6.0 Hz, 1H, CH), 10.82 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 18.4 (CH3), 22.9 (CH3), 27.9(3CH3-t-Bu), 51.6 (C-7), 80.2 (C-t-Bu), 99.6 (C-6), 145.3 (C-5), 148.6 (C-3a), 164.2 (CO); EIMS (70 eV, m/z (%)): 251 (19) [M+], 250 (100); Anal. calcd for С11Н17N5O2 (251.28): C, 52.58; H, 6.82; N, 27.87%; found: C, 52.69; H, 6.92; N, 27.78%.
1-(5-Methyl-4,7-dihydrotetrazolo[1,5-a]pyrimidin-6-yl)ethanone (12). In a vial, a solution of 5-aminotetrazole (1, 1.2 mmol, 0.1 g), paraformaldehyde (7a, 1.26 mmol, 0.038 g), acetylacetone (10, 1.2 mmol, 0.122 ml) and 0.012 g scandium(III) triflate (Sc(OTf)3·nH2O) in water (3.2 mL) was irradiated in a microwave reactor at 100 °C for 20 min. The crystalline product started to separate after cooling to 5 °C. The resulting precipitate was filtered off, washed with water and air-dried. Colorless solid, yield 78%; mp 165–167 °C; 1H NMR (400 MHz, DMSO-d6) δ 2.25 (s, 3H, СН3), 2.32 (s, 3Н, СН3), 5.2 (s, 2Н, CН2), 10.84 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ 19.2 (CH3), 30.3 (CH3), 44.6 (C-7), 102.9 (C-6), 145.5 (C-5), 148.8 (C-3a), 194.6 (CO); EIMS (70 eV, m/z (%)): 179 (15) [ M+], 178 (100), 122 (32); Anal. calcd for С7Н9N5O (179.08): C, 46.92; H, 5.06; N, 39.09%; found: C, 47.20; H, 5.35; N, 39.36%.
Ethyl (5RS,6RS,7SR)-5-hydroxy-7-methyl-5-(trifluoromethyl)-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidine-6-carboxylate and ethyl (5SR,6SR,7SR)-5-hydroxy-7-methyl-5-(trifluoromethyl)-4,5,6,7-tetrahydrotetrazolo[1,5-a]pyrimidine-6-carboxylate (14). In a vial, a solution of 5-aminotetrazole (1, 1.7 mmol), acetaldehyde (7b, 0.15 g, ≈2 mmol) and ethyl 4,4,4-trifluoroacetoacetate (13, 1.77 mmol) in water (3.5 mL) was irradiated in a microwave reactor at 100 °C for 25–30 min. The crystalline product started to separate either during the course of reaction or just after cooling. The precipitate was filtered, washed with water and air-dried. White solid, yield 90%; mp 139–140 °C; 1H NMR (400 MHz, DMSO-d6) isomer A: δ 1.08 (t, 3JHH = 7.2 Hz, 3H, CH3), 1.59 (d, 3JHH = 6.4 Hz, 3Н, CH3), 3.28 (d, 3JHH = 4.4 Hz, 1Н, CН), 4.02 (q, 3JHH = 7.2 Hz, 2H, CH2), 4.76 (m, 1H, CH), 7.77 (s, 1H, OH), 9.49 (s, 1H, NH); isomer B: 1.19 (t, 3JHH = 7.2 Hz, 3H, CH3), 1.59 (d, 3JHH = 6.4 Hz, 3Н, CH3, 3.17 (d, 3JHH = 11,2 Hz, 1Н, CН), 4.17 (q, 3JHH = 7.2 Hz, 2H, CH2), 4,63 (m, 1H, CH), 7,89 (s, 1H, OH), 9.34 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) isomer A: δ 13.7 (CH3), 16.2 (7-CH3) , 46.9 (C-7), 49.3 (C-6), 61.4 (CH2) , 80.8 (C-5, q, 2JCC= 31.0 Hz), 123.0 (q, 1JCF = 286.0 Hz), 151.6 (C-3a), 167.0 (CO); isomer B: δ 13.7 (CH3), 14.3 (7-CH3), 48.2 (C-7), 49.0 (C-6), 61.0 (CH2), 80.8 (C-5, q, 2JCC= 32.0 Hz), 123.0 (q, 1JCF = 286.0 Hz), 152.3 (C-3a), 166.3 (CO); EIMS (70 eV, m/z (%)): 295 (15) [M+], 294 (100), 277 (12), 276 (60), 180 (25); Anal. calcd for С9Н12F3N5O3 (295.23): C, 36.62; H, 4.10; F, 19.31; N, 23.72%; found: C, 36.53; H, 4.21; F, 19.20; N, 23.84%.
Acknowledgements
The authors thank the National Academy of Sciences of Ukraine for financial support in the frame of the projects "Creation of modern bases for obtaining and analyzing substances and components of materials for pharmaceutical purposes" (0119U100727) and "Investigation of structural features of nitrogen-containing heterocycles with potential biological activity" (0119U100716) and the Ministry of Education and Science of Ukraine for financial support in the frame of project “Molecular docking for express identification of new potential drugs” (0119U002550).
References
-
Łakomska, I.; Fandzloch, M. Coord. Chem. Rev. 2016, 327–328, 221–241. doi:10.1016/j.ccr.2016.04.014
Return to citation in text: [1] -
Gein, V. L.; Mishunin, V. V.; Tsyplyakova, E. P.; Vinokurova, O. V.; Vakhrin, M. I. Pharm. Chem. J. 2009, 43, 652–654. doi:10.1007/s11094-010-0373-1
Khim. Farm. Zh. 2009, 43, 12, 10-12.
Return to citation in text: [1] -
Raju, C.; Madhaiyan, K.; Uma, R.; Sridhar, R.; Ramakrishna, S. RSC Adv. 2012, 2, 11657–11663. doi:10.1039/c2ra21330c
Return to citation in text: [1] [2] -
Dougherty, A. M.; Guo, H.; Westby, G.; Liu, Y.; Simsek, E.; Guo, J.-T.; Mehta, A.; Norton, P.; Gu, B.; Block, T.; Cuconati, A. Antimicrob. Agents Chemother. 2007, 51, 4427–4437. doi:10.1128/aac.00541-07
Return to citation in text: [1] -
Orlov, V. D.; Desenko, S. M.; Pivnenko, N. S. Chem. Heterocycl. Compd. 1988, 24, 1233–1237. doi:10.1007/bf00633502
Khim. Geterotsikl. Soedin. 1988, 11, 1489-1493.
Return to citation in text: [1] [2] [3] -
Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. 2001, 37, 747–754. doi:10.1023/a:1011925631511
Return to citation in text: [1] [2] [3] [4] -
Scheuermann, T. H.; Stroud, D.; Sleet, C. E.; Bayeh, L.; Shokri, C.; Wang, H.; Caldwell, C. G.; Longgood, J.; MacMillan, J. B.; Bruick, R. K.; Gardner, K. H.; Tambar, U. K. J. Med. Chem. 2015, 58, 5930–5941. doi:10.1021/acs.jmedchem.5b00529
Return to citation in text: [1] [2] -
Chebanov, V.; Sakhno, Y.; Desenko, S.; Shishkina, S.; Musatov, V.; Shishkin, O.; Knyazeva, I. Synthesis 2005, 2597–2601. doi:10.1055/s-2005-872073
Return to citation in text: [1] [2] -
Pryadeina, M. V.; Burgart, Y. V.; Saloutin, V. I.; Kodess, M. I.; Ulomskii, E. N.; Rusinov, V. L. Russ. J. Org. Chem. 2004, 40, 902–907. doi:10.1023/b:rujo.0000044558.47152.65
Return to citation in text: [1] [2] -
Abelman, M.; Jiang, R.; Zablocki, J. Optionally condensed dihydropyridine, dihydropyrimidine and dihydropyrane derivatives acting as late sodium channel blockers. Int. Patent Appl. WO2009/006580 A1, Jan 8, 2009.
Return to citation in text: [1] [2] -
Kour, P.; Singh, V. P.; Khajuria, B.; Singh, T.; Kumar, A. Tetrahedron Lett. 2017, 58, 4179–4185. doi:10.1016/j.tetlet.2017.09.052
Return to citation in text: [1] [2] -
Maleki, A.; Ravaghi, P.; Aghaei, M.; Movahed, H. Res. Chem. Intermed. 2017, 43, 5485–5494. doi:10.1007/s11164-017-2941-4
Return to citation in text: [1] [2] -
Chebanov, V. A.; Desenko, S. M.; Gurley, T. W. Six-Membered Azaheterocycles Based on 1,3-Binucleophiles. Azaheterocycles Based on α,β-Unsaturated Carbonyls; Springer-Verlag: Heidelberg, Germany, 2008; pp 61–147. doi:10.1007/978-3-540-68367-4_4
Return to citation in text: [1] -
Wang, X.-C.; Wei, Y.; Da, Y.-X.; Zhang, Z.; Quan, Z.-J. Heterocycles 2011, 83, 2811–2822. doi:10.3987/com-11-12351
Return to citation in text: [1] [2] -
Ghorbani-Vaghei, R.; Toghraei-Semiromi, Z.; Amiri, M.; Karimi-Nami, R. Mol. Diversity 2013, 17, 307–318. doi:10.1007/s11030-013-9435-0
Return to citation in text: [1] [2] -
Gein, V. L.; Vladimirov, I. N.; Fedorova, O. V.; Kurbatova, A. A.; Nosova, N. V.; Krylova, I. V.; Vakhrin, M. I. Russ. J. Org. Chem. 2010, 46, 699–705. doi:10.1134/s1070428010050180
Return to citation in text: [1] [2] -
Yao, C.; Lei, S.; Wang, C.; Yu, C.; Tu, S. J. Heterocycl. Chem. 2008, 45, 1609–1613. doi:10.1002/jhet.5570450609
Return to citation in text: [1] [2] -
Zeng, L.-Y.; Cai, C. J. Comb. Chem. 2010, 12, 35–40. doi:10.1021/cc9000983
Return to citation in text: [1] [2] -
Fedorova, O. V.; Zhidovinova, M. S.; Rusinov, G. L.; Ovchinnikova, I. G. Russ. Chem. Bull. 2003, 52, 1768–1769. doi:10.1023/a:1026052603951
Return to citation in text: [1] -
Kolosov, M. A.; Shvets, E. H.; Manuenkov, D. A.; Kulyk, O. G.; Mazepa, A. V.; Orlov, V. D. Synth. Commun. 2019, 49, 611–615. doi:10.1080/00397911.2019.1566476
Return to citation in text: [1] -
Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, U.S.A., 1998.
Return to citation in text: [1] -
Odell, L. R.; Larhed, M. Microwave‐Accelerated Homogeneous Catalysis in Water. In Handbook of Green Chemistry; Crabtree, R. H.; Anastas, P. T., Eds.; Green Catalysis – Homogeneous Catalysis, Vol. 1; Wiley-VCH: Weinheim, Germany, 2009; pp 79–99. doi:10.1002/9783527628698.hgc004
Return to citation in text: [1] -
Dallinger, D.; Kappe, C. O. Chem. Rev. 2007, 107, 2563–2591. doi:10.1021/cr0509410
Return to citation in text: [1] -
Wu, X.; Larhed, M. Org. Lett. 2005, 7, 3327–3329. doi:10.1021/ol0512031
Return to citation in text: [1] -
Leeson, P. Nature 2012, 481, 455–456. doi:10.1038/481455a
Return to citation in text: [1] -
Komykhov, S. A.; Tkachenko, I. G.; Musatov, V. I.; Diachkov, M. V.; Chebanov, V. A.; Desenko, S. M. ARKIVOC 2016, No. iv, 277–287. doi:10.3998/ark.5550190.p009.610
Return to citation in text: [1] -
Tkachenko, I. G.; Komykhov, S. A.; Gladkov, E. S.; Musatov, V. I.; Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2019, 55, 392–396. doi:10.1007/s10593-019-02470-0
Khim. Geterotsikl. Soedin. 2019, 55, 392-396.
Return to citation in text: [1] [2] -
Tkachenko, I. G.; Komykhov, S. A.; Musatov, V. I.; Chebanov, V. A.; Desenko, S. M. Fr.-Ukr. J. Chem. 2019, 7, 90–95.
Return to citation in text: [1] -
Desenko, S. M.; Gladkov, E. S.; Nenaidenko, V. G.; Shishkin, O. V.; Shishkina, S. V. Chem. Heterocycl. Compd. 2004, 40, 65–69. doi:10.1023/b:cohc.0000023769.66759.7d
Khim. Geterotsikl. Soedin. 2004, 71-76.
Return to citation in text: [1] -
Blois, M. S. Nature 1958, 181, 1199–1200. doi:10.1038/1811199a0
Return to citation in text: [1] -
Li, B.; Pratt, D. A. Free Radical Biol. Med. 2015, 82, 187–202. doi:10.1016/j.freeradbiomed.2015.01.020
Return to citation in text: [1] -
Kaur, I.; Geetha, T. Mini-Rev. Med. Chem. 2006, 6, 305–312. doi:10.2174/138955706776073448
Return to citation in text: [1] -
Sherma, J. J. AOAC Int. 2018, 101, 1285–1294. doi:10.5740/jaoacint.18-0116
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370
Return to citation in text: [1]
| 27. |
Tkachenko, I. G.; Komykhov, S. A.; Gladkov, E. S.; Musatov, V. I.; Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2019, 55, 392–396. doi:10.1007/s10593-019-02470-0
Khim. Geterotsikl. Soedin. 2019, 55, 392-396. |
| 29. |
Desenko, S. M.; Gladkov, E. S.; Nenaidenko, V. G.; Shishkin, O. V.; Shishkina, S. V. Chem. Heterocycl. Compd. 2004, 40, 65–69. doi:10.1023/b:cohc.0000023769.66759.7d
Khim. Geterotsikl. Soedin. 2004, 71-76. |
| 26. | Komykhov, S. A.; Tkachenko, I. G.; Musatov, V. I.; Diachkov, M. V.; Chebanov, V. A.; Desenko, S. M. ARKIVOC 2016, No. iv, 277–287. doi:10.3998/ark.5550190.p009.610 |
| 27. |
Tkachenko, I. G.; Komykhov, S. A.; Gladkov, E. S.; Musatov, V. I.; Chebanov, V. A.; Desenko, S. M. Chem. Heterocycl. Compd. 2019, 55, 392–396. doi:10.1007/s10593-019-02470-0
Khim. Geterotsikl. Soedin. 2019, 55, 392-396. |
| 28. | Tkachenko, I. G.; Komykhov, S. A.; Musatov, V. I.; Chebanov, V. A.; Desenko, S. M. Fr.-Ukr. J. Chem. 2019, 7, 90–95. |
| 1. | Łakomska, I.; Fandzloch, M. Coord. Chem. Rev. 2016, 327–328, 221–241. doi:10.1016/j.ccr.2016.04.014 |
| 5. |
Orlov, V. D.; Desenko, S. M.; Pivnenko, N. S. Chem. Heterocycl. Compd. 1988, 24, 1233–1237. doi:10.1007/bf00633502
Khim. Geterotsikl. Soedin. 1988, 11, 1489-1493. |
| 15. | Ghorbani-Vaghei, R.; Toghraei-Semiromi, Z.; Amiri, M.; Karimi-Nami, R. Mol. Diversity 2013, 17, 307–318. doi:10.1007/s11030-013-9435-0 |
| 16. | Gein, V. L.; Vladimirov, I. N.; Fedorova, O. V.; Kurbatova, A. A.; Nosova, N. V.; Krylova, I. V.; Vakhrin, M. I. Russ. J. Org. Chem. 2010, 46, 699–705. doi:10.1134/s1070428010050180 |
| 17. | Yao, C.; Lei, S.; Wang, C.; Yu, C.; Tu, S. J. Heterocycl. Chem. 2008, 45, 1609–1613. doi:10.1002/jhet.5570450609 |
| 4. | Dougherty, A. M.; Guo, H.; Westby, G.; Liu, Y.; Simsek, E.; Guo, J.-T.; Mehta, A.; Norton, P.; Gu, B.; Block, T.; Cuconati, A. Antimicrob. Agents Chemother. 2007, 51, 4427–4437. doi:10.1128/aac.00541-07 |
| 3. | Raju, C.; Madhaiyan, K.; Uma, R.; Sridhar, R.; Ramakrishna, S. RSC Adv. 2012, 2, 11657–11663. doi:10.1039/c2ra21330c |
| 19. | Fedorova, O. V.; Zhidovinova, M. S.; Rusinov, G. L.; Ovchinnikova, I. G. Russ. Chem. Bull. 2003, 52, 1768–1769. doi:10.1023/a:1026052603951 |
| 20. | Kolosov, M. A.; Shvets, E. H.; Manuenkov, D. A.; Kulyk, O. G.; Mazepa, A. V.; Orlov, V. D. Synth. Commun. 2019, 49, 611–615. doi:10.1080/00397911.2019.1566476 |
| 2. |
Gein, V. L.; Mishunin, V. V.; Tsyplyakova, E. P.; Vinokurova, O. V.; Vakhrin, M. I. Pharm. Chem. J. 2009, 43, 652–654. doi:10.1007/s11094-010-0373-1
Khim. Farm. Zh. 2009, 43, 12, 10-12. |
| 3. | Raju, C.; Madhaiyan, K.; Uma, R.; Sridhar, R.; Ramakrishna, S. RSC Adv. 2012, 2, 11657–11663. doi:10.1039/c2ra21330c |
| 21. | Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, U.S.A., 1998. |
| 22. | Odell, L. R.; Larhed, M. Microwave‐Accelerated Homogeneous Catalysis in Water. In Handbook of Green Chemistry; Crabtree, R. H.; Anastas, P. T., Eds.; Green Catalysis – Homogeneous Catalysis, Vol. 1; Wiley-VCH: Weinheim, Germany, 2009; pp 79–99. doi:10.1002/9783527628698.hgc004 |
| 23. | Dallinger, D.; Kappe, C. O. Chem. Rev. 2007, 107, 2563–2591. doi:10.1021/cr0509410 |
| 24. | Wu, X.; Larhed, M. Org. Lett. 2005, 7, 3327–3329. doi:10.1021/ol0512031 |
| 13. | Chebanov, V. A.; Desenko, S. M.; Gurley, T. W. Six-Membered Azaheterocycles Based on 1,3-Binucleophiles. Azaheterocycles Based on α,β-Unsaturated Carbonyls; Springer-Verlag: Heidelberg, Germany, 2008; pp 61–147. doi:10.1007/978-3-540-68367-4_4 |
| 14. | Wang, X.-C.; Wei, Y.; Da, Y.-X.; Zhang, Z.; Quan, Z.-J. Heterocycles 2011, 83, 2811–2822. doi:10.3987/com-11-12351 |
| 8. | Chebanov, V.; Sakhno, Y.; Desenko, S.; Shishkina, S.; Musatov, V.; Shishkin, O.; Knyazeva, I. Synthesis 2005, 2597–2601. doi:10.1055/s-2005-872073 |
| 9. | Pryadeina, M. V.; Burgart, Y. V.; Saloutin, V. I.; Kodess, M. I.; Ulomskii, E. N.; Rusinov, V. L. Russ. J. Org. Chem. 2004, 40, 902–907. doi:10.1023/b:rujo.0000044558.47152.65 |
| 10. | Abelman, M.; Jiang, R.; Zablocki, J. Optionally condensed dihydropyridine, dihydropyrimidine and dihydropyrane derivatives acting as late sodium channel blockers. Int. Patent Appl. WO2009/006580 A1, Jan 8, 2009. |
| 11. | Kour, P.; Singh, V. P.; Khajuria, B.; Singh, T.; Kumar, A. Tetrahedron Lett. 2017, 58, 4179–4185. doi:10.1016/j.tetlet.2017.09.052 |
| 12. | Maleki, A.; Ravaghi, P.; Aghaei, M.; Movahed, H. Res. Chem. Intermed. 2017, 43, 5485–5494. doi:10.1007/s11164-017-2941-4 |
| 5. |
Orlov, V. D.; Desenko, S. M.; Pivnenko, N. S. Chem. Heterocycl. Compd. 1988, 24, 1233–1237. doi:10.1007/bf00633502
Khim. Geterotsikl. Soedin. 1988, 11, 1489-1493. |
| 6. | Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. 2001, 37, 747–754. doi:10.1023/a:1011925631511 |
| 7. | Scheuermann, T. H.; Stroud, D.; Sleet, C. E.; Bayeh, L.; Shokri, C.; Wang, H.; Caldwell, C. G.; Longgood, J.; MacMillan, J. B.; Bruick, R. K.; Gardner, K. H.; Tambar, U. K. J. Med. Chem. 2015, 58, 5930–5941. doi:10.1021/acs.jmedchem.5b00529 |
| 8. | Chebanov, V.; Sakhno, Y.; Desenko, S.; Shishkina, S.; Musatov, V.; Shishkin, O.; Knyazeva, I. Synthesis 2005, 2597–2601. doi:10.1055/s-2005-872073 |
| 9. | Pryadeina, M. V.; Burgart, Y. V.; Saloutin, V. I.; Kodess, M. I.; Ulomskii, E. N.; Rusinov, V. L. Russ. J. Org. Chem. 2004, 40, 902–907. doi:10.1023/b:rujo.0000044558.47152.65 |
| 10. | Abelman, M.; Jiang, R.; Zablocki, J. Optionally condensed dihydropyridine, dihydropyrimidine and dihydropyrane derivatives acting as late sodium channel blockers. Int. Patent Appl. WO2009/006580 A1, Jan 8, 2009. |
| 11. | Kour, P.; Singh, V. P.; Khajuria, B.; Singh, T.; Kumar, A. Tetrahedron Lett. 2017, 58, 4179–4185. doi:10.1016/j.tetlet.2017.09.052 |
| 12. | Maleki, A.; Ravaghi, P.; Aghaei, M.; Movahed, H. Res. Chem. Intermed. 2017, 43, 5485–5494. doi:10.1007/s11164-017-2941-4 |
| 14. | Wang, X.-C.; Wei, Y.; Da, Y.-X.; Zhang, Z.; Quan, Z.-J. Heterocycles 2011, 83, 2811–2822. doi:10.3987/com-11-12351 |
| 15. | Ghorbani-Vaghei, R.; Toghraei-Semiromi, Z.; Amiri, M.; Karimi-Nami, R. Mol. Diversity 2013, 17, 307–318. doi:10.1007/s11030-013-9435-0 |
| 16. | Gein, V. L.; Vladimirov, I. N.; Fedorova, O. V.; Kurbatova, A. A.; Nosova, N. V.; Krylova, I. V.; Vakhrin, M. I. Russ. J. Org. Chem. 2010, 46, 699–705. doi:10.1134/s1070428010050180 |
| 17. | Yao, C.; Lei, S.; Wang, C.; Yu, C.; Tu, S. J. Heterocycl. Chem. 2008, 45, 1609–1613. doi:10.1002/jhet.5570450609 |
| 18. | Zeng, L.-Y.; Cai, C. J. Comb. Chem. 2010, 12, 35–40. doi:10.1021/cc9000983 |
| 6. | Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. 2001, 37, 747–754. doi:10.1023/a:1011925631511 |
| 7. | Scheuermann, T. H.; Stroud, D.; Sleet, C. E.; Bayeh, L.; Shokri, C.; Wang, H.; Caldwell, C. G.; Longgood, J.; MacMillan, J. B.; Bruick, R. K.; Gardner, K. H.; Tambar, U. K. J. Med. Chem. 2015, 58, 5930–5941. doi:10.1021/acs.jmedchem.5b00529 |
| 30. | Blois, M. S. Nature 1958, 181, 1199–1200. doi:10.1038/1811199a0 |
| 31. | Li, B.; Pratt, D. A. Free Radical Biol. Med. 2015, 82, 187–202. doi:10.1016/j.freeradbiomed.2015.01.020 |
| 32. | Kaur, I.; Geetha, T. Mini-Rev. Med. Chem. 2006, 6, 305–312. doi:10.2174/138955706776073448 |
| 33. | Sherma, J. J. AOAC Int. 2018, 101, 1285–1294. doi:10.5740/jaoacint.18-0116 |
| 6. | Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. 2001, 37, 747–754. doi:10.1023/a:1011925631511 |
| 5. |
Orlov, V. D.; Desenko, S. M.; Pivnenko, N. S. Chem. Heterocycl. Compd. 1988, 24, 1233–1237. doi:10.1007/bf00633502
Khim. Geterotsikl. Soedin. 1988, 11, 1489-1493. |
| 6. | Desenko, S. M.; Gladkov, E. S.; Komykhov, S. A.; Shishkin, O. V.; Orlov, V. D. Chem. Heterocycl. Compd. 2001, 37, 747–754. doi:10.1023/a:1011925631511 |
| 34. | Sheldrick, G. M. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. doi:10.1107/s2053229614024218 |
| 35. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. doi:10.1107/s2053273314026370 |
© 2019 Tkachenko et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)