Abstract
A series of α- and β-ethynyl-substituted BODIPY derivatives (3a, 4a, 5a, 5b, 6a, 6b) were synthesized by gold(I)-catalyzed direct C–H alkynylation reactions of dipyrromethane and BODIPY, respectively, with ethynylbenziodoxolone (EBX) in a regioselective manner. Depending on the position of the ethynyl substituent in the BODIPY skeleton, the photophysical properties of the resulting α- and β-substituted BODIPYs are notably altered. The lowest S0–S1 transition absorbance and fluorescence bands are both bathochromically shifted as the number of substituents increases, while the emission quantum yields of the β-ethynylated derivatives are significantly lower than those of α-ethynylated ones. The current method should be useful for fine-tuning of the photophysical properties of BODIPY dyes as well as for constructing BODIPY-based building cores for functional π-materials.
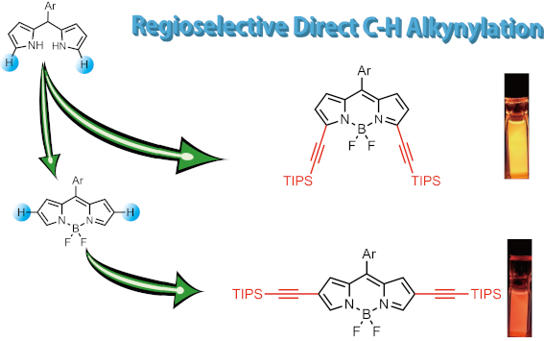
Graphical Abstract
Introduction
Boron-dipyrromethene (BODIPY, 1) and its derivatives are representative families of fluorophores that have been widely used in applications for bioimaging [1-6], photodynamic therapy [7-12], photocatalysis [13-16], optics [17-20], and so forth. The structure of BODIPY derivatives is composed of a dipyrromethene (an oxidized form of dipyrromethane 2) and a coordinated difluoroboron moiety [21]. The rigid π-conjugated scaffold of BODIPYs demonstrates fascinating optical features such as intense and narrow S0–S1 absorption and emission bands in the visible-to-near-infrared region, a high fluorescence quantum yield, and good photostability. For the applications mentioned above, various BODIPY dyes functionalized at the meso-, α-, and β-pyrrolic positions have been extensively developed to tune the optoelectronic properties [21]. Therefore, the development of more efficient and shorter step synthetic methods for the BODIPY derivatives, such as direct C–H functionalizations (e.g., arylation [22-28], annulation [29], olefination [30], styrylation [31], and borylation [32]), has been in great demand recently rather than the conventional multistep synthesis with nucleophilic substitution/cross-coupling via halogenation of BODIPYs [33] or from the activated precursors [34] via unstable pyrrolic intermediates. In particular, halogenation (e.g., bromination) of BODIPY derivatives often affords a mixture of multiply halogenated products, which is tedious to separate by column chromatography.
The introduction of one or more alkynyl groups into the BODIPY skeleton indeed produces useful building blocks for functional π-materials. For example, ethynyl-tethered BODIPY derivatives serve as a substrate in the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, which is known as “click” reaction, allowing for a biological tissue labelling [35,36]. In addition, ethynyl-substituted BODIPYs yield unique π-conjugated BODIPY-based macrocycles by Glaser-coupling reactions [37]. Conventionally, an alkynylation of the BODIPY core has been achieved by palladium-catalyzed Sonogashira cross-coupling with halogenated BODIPYs (Figure 1b) [35,37]. However, due to the coexistence of multiple C–H bonds, a regioselective direct C–H alkynylation of the BODIPY core has not yet been achieved.
![[1860-5397-16-53-1]](/bjoc/content/figures/1860-5397-16-53-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Chemical structures of BODIPY (1) and dipyrromethane (2). (b) C–C bond forming alkynylations of pyrrole and its derivatives by Sonogashira coupling and electrophilic alkynylation. (c) Peripheral alkynylated BODIPY derivatives (3–6) prepared in this work. TIPS: triisopropylsilyl group; Mes: 2,4,6-trimethylphenyl group.
Figure 1: (a) Chemical structures of BODIPY (1) and dipyrromethane (2). (b) C–C bond forming alkynylations of...
Inspired by the works of Waser and co-workers showing the gold(I)-catalyzed C–H electrophilic alkynylation of various heterocycles (e.g., pyrroles, indoles, etc.) with ethynylbenziodoxolone (EBX) as an activated ethynyl synthon [38-42], we investigated the synthesis of ethynyl-substituted BODIPY derivatives 3–6 by gold(I)-catalyzed direct C–H functionalization (Figure 1c). By taking advantage of the reactivity of β-(2 and 6)-positions of BODIPY (1), which are susceptible to electrophilic reactions, β,β'-diethynyl-substituted BODIPYs 5 and 6 were prepared regioselectively, through the C–H alkynylation of 1 without any directing groups. In addition, the corresponding dipyrromethane 2, which is a precursor of BODIPY, was first transformed into the alkynylated form under catalytic conditions, and subsequent oxidation followed by boron complexation to afford α-monoethynyl and α,α'-diethynyl-substituted BODIPYs 3 and 4, respectively. The resulting ethynylated BODIPY isomers demonstrated site-dependent photophysical properties.
Results and Discussion
Synthesis and characterization
To prepare the α,α'-diethynyl BODIPY 4a, 5-mesityldipyrromethane 2 was used as the substrate for the gold(I) catalyzed reaction (Scheme 1). Mixing five mol % of gold(I) chloride and two equivalents of TIPS-EBX with a diethyl ether solution of 2 under ambient conditions yielded a mixture of ethynyl-substituted dipyrromethanes as judged by mass spectrometry. The product mixture was treated with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) to give the α,α'-diethynyl-substituted dipyrrin 7a. Subsequent boron complexation in the presence of trimethylsilyl chloride (TMSCl) as a fluoride scavenger afforded 4a in 16% yield over three steps. Separately, α-ethynyl-monosubstituted dipyrrin 3a, was obtained via a one-pot reaction in 18% yield (in three steps from 2).
![[1860-5397-16-53-i1]](/bjoc/content/inline/1860-5397-16-53-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of α-ethynyl-substituted BODIPY derivatives 3a and 4a.
Scheme 1: Synthesis of α-ethynyl-substituted BODIPY derivatives 3a and 4a.
On the other hand, the β,β'-diethynyl-substituted BODIPY derivatives 5a and 6a, were synthesized by a similar gold-catalyzed reaction of unsubstituted BODIPY 1a in 38% and 2% yields, respectively, in the presence of zinc(II) triflate as an activator of TIPS-EBX at 100 °C [41]. In contrast to the reaction of dipyrromethane 2, the double C–H activation at the 2,6-positions of 1a required harsh conditions (Table S1, Supporting Information File 1). The representative procedure of the reaction with EBX/gold(I) catalyst at room temperature gave only traces of 5a with a large amount of unreacted starting material remaining. The addition of acid in expectation of the activation of TIPS-EBX was effective in promoting the gold–alkyne interactions [40,41]. After the investigation of various reaction conditions, such as gold catalysts (e.g., gold(I) cyanide), solvents (e.g., CH2Cl2, THF, MeCN, DMF), additives (e.g., TFA, Sc(OTf)3), and temperature, the reaction conditions as mentioned earlier were chosen for the synthesis of 5a and 6a (Table S1, Supporting Information File 1).
Because of the electron-deficient nature of BODIPY, the reactivity of the 2,6-positions is intrinsically low toward electrophiles, which hampers the β-selective functionalization. We, therefore, tested the corresponding reaction using tetramethyl-substituted BODIPY 1b that is expected to have an enhanced electron density of the BODIPY core compared with 1a (Scheme 2). Under milder conditions, the yield of the β,β'-diethynylated product 6b was indeed improved to be 19% (from 2% of 6a). These results indicate that electron-rich substrates facilitate the direct electrophilic alkynylation of the BODIPY core.
![[1860-5397-16-53-i2]](/bjoc/content/inline/1860-5397-16-53-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of β-ethynyl-substituted BODIPY derivatives 5a and 5b and β,β'-diethynyl-substituted compounds 6a and 6b.
Scheme 2: Synthesis of β-ethynyl-substituted BODIPY derivatives 5a and 5b and β,β'-diethynyl-substituted comp...
The structures of the series of TIPS-ethynyl-substituted BODIPY derivatives 3–6 were characterized by 1H and 19F NMR spectroscopy, high-resolution mass spectrometry, and X-ray crystallographic analysis. The solid-state structures of the diethynyl-substituted BODIPYs were unambiguously elucidated by X-ray diffraction analysis (3a and 6b: Figure 2 and Table S2 in Supporting Information File 1). The BODIPY cores of 3a and 6b are almost planar with smaller mean-plane deviations (defined by the 12 atoms of the tricyclic ring system) of 0.075 and 0.025 Å, respectively. The meso-mesityl groups are oriented perpendicularly to the BODIPY plane for both derivatives, indicating the rigid interlocked structures of 3a and 6b by the bulky o-methyl groups. In particular, the β-methyl substituents of 6b are sterically hindered by the neighbouring meso-aryl ring. The regioselective 2,6-diethynylation of 6a through the above alkynylation was also confirmed by its preliminary X-ray structure (Figure S20b, Supporting Information File 1).
![[1860-5397-16-53-2]](/bjoc/content/figures/1860-5397-16-53-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Top and front views of the crystal structures of (a) 4a and (b) 6b with 50% thermal ellipsoid probabilities. The isopropyl groups on the silicon atom are omitted in the front view for clarity.
Figure 2: Top and front views of the crystal structures of (a) 4a and (b) 6b with 50% thermal ellipsoid proba...
The 1H NMR spectra of the BODIPY derivatives in CDCl3 reflect the characteristic structural features. The proton signals assignable to the α-pyrrolic CHs appeared in the lowest field region (δ ca. 8.00 ppm), and the β-CH resonances are in the range of 6.5 to 6.7 ppm (Figure 3). The disappearance of the α-pyrrole proton signals of 4a is notable. In the case of 3a, only a singlet signal appeared at 7.95 ppm for the α-pyrrole CH. In contrast, 6a revealed two singlet signals at 7.99 and 6.70 ppm, assignable to the α- and β-pyrrole CHs, respectively. The 1H NMR spectrum of the mono-substituted compound 5a represents an unsymmetrical resonance pattern similar to that of 3a with two α-CH signals at 7.97 and 7.96 ppm. The proposed structures of all BODIPY compounds were elucidated based on the comparative NMR analysis and mass spectrometry (see Supporting Information File 1).
![[1860-5397-16-53-3]](/bjoc/content/figures/1860-5397-16-53-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Partial 1H NMR spectra of (a) 1a, (b) 3a, (c) 4a, (d) 5a, and (e) 6a recorded in CDCl3 at 298 K. Asterisks indicate the residual solvent peak.
Figure 3: Partial 1H NMR spectra of (a) 1a, (b) 3a, (c) 4a, (d) 5a, and (e) 6a recorded in CDCl3 at 298 K. As...
Optical properties
The α- and β-ethynyl-substituted BODIPYs exhibit large bathochromic shifts in the absorption and fluorescence spectra relative to the unsubstituted 1a with extended π-conjugation (Figure 4 and Figure 5). The molar absorption coefficients (ε) of the S0–S1 bands of the α-ethynyl-substituted BODIPYs are substantially enhanced as the number of substituted groups increases (Figure 4a). Along with the absorption spectral profiles of 3a and 4a, sharp emission bands emerge with mirror structures (Figure 5a). The fluorescence quantum yields of 3a and 4a remain high (Φf = 0.95 and 0.56, respectively) and are comparable to that of 1a (Table 1). The smaller Stokes shift values of ≈178 cm−1 and longer emission lifetimes of ≈7.83 ns indicate the rigid scaffolds of α,α-disubstituted 4a in the excited state.
![[1860-5397-16-53-4]](/bjoc/content/figures/1860-5397-16-53-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis absorption spectra of the BODIPY derivatives, (a) 1a (green), 3a (blue), 4a (red), and (b) 1a (green), 5a (blue), 6a (red) in CH2Cl2. Insets show the photo images of the solutions taken under ambient light.
Figure 4: UV–vis absorption spectra of the BODIPY derivatives, (a) 1a (green), 3a (blue), 4a (red), and (b) 1a...
![[1860-5397-16-53-5]](/bjoc/content/figures/1860-5397-16-53-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Fluorescence spectra of BODIPY derivatives. (a) 1a (green), 3a (blue), 4a (red) and (b) 1a (green), 5a (blue), and 6a (red) in CH2Cl2. Excitation was performed at 365 nm. The insets show the photo images of the solution under a UV lamp.
Figure 5: Fluorescence spectra of BODIPY derivatives. (a) 1a (green), 3a (blue), 4a (red) and (b) 1a (green), ...
Table 1: Spectral properties of BODIPYs in CH2Cl2.
|
λmax
(nm) |
εmaxa
(M−1cm−1) |
λemb
(nm) |
Stokes shift
(cm−1) |
Φfc |
τfd
(ns) |
|
| 1a | 501 | 6.3 × 104 | 513 | 467 | 0.86 | 6.60 |
| 3a | 537 | 9.2 × 104 | 546 | 307 | 0.95 | 5.81 |
| 4a | 578 | 1.4 × 105 | 584 | 178 | 0.56 | 7.83 |
| 5a | 540 | 4.0 × 104 | 569 | 944 | 0.25 | 5.70 |
| 6a | 578 | 6.3 × 104 | 597 | 551 | 0.16 | 5.52 |
| 1b | 502 | 8.8 × 104 | 510 | 312 | 0.96 | 5.35 |
| 5b | 529 | 7.8 × 104 | 547 | 622 | 0.86 | 5.59 |
| 6b | 558 | 9.7 × 104 | 574 | 500 | 0.77 | 4.97 |
aAbsorption coefficients of the S0–S1 bands in CH2Cl2. The reference values are taken from the literature [32,43]. bEmission maxima at wavelength. cEmission quantum yield determined by integrating sphere. dEmission lifetime probed at the maxima of the bands.
In the case of β-substituted BODIPYs, the S0–S1 absorption bands of 5a and 6a are significantly broadened with the full width at half maximum (fwhm) of 1827 and 1438 cm−1, respectively, compared to those of the α-substituted compounds 3a and 4a (625 and 508 cm−1, respectively, Figure 4a). Accordingly, the relatively broad emission bands were observed with lowered quantum yields, Φf of 0.25 and 0.16, respectively (Figure 5b and Table 1). The larger Stokes shift and shorter emission lifetime suggest a rapid decay of the excited species after structural relaxation (vide infra, Table 1). Also, intersystem crossing to the triplet state can be another pathway to understand the lower emission quantum yield of the BODIPYs. However, reactive singlet oxygen species (1O2) was not observed under the aerobic conditions probed by the near-infrared photoluminescence at 1270 nm. The above spectral features were likewise shown with the β-ethylene-substituted BODIPYs [32].
Similarly, the bathochromic shifts of the absorption and fluorescence bands for the tetramethyl-substituted derivatives 5b and 6b were observed in CH2Cl2 solution (Figure S21, Supporting Information File 1). However, due to the rigid structure of the highly substituted BODIPY cores, higher emission quantum yields of 0.86 and 0.77 were estimated, respectively (Table 1).
Electrochemical properties
Cyclic voltammetry and differential pulse voltammetry of the BODIPY derivatives 3a–6a were performed in dichloromethane solutions containing 0.1 M tetra-n-butylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte (Figure S23, Supporting Information File 1). All the derivatives exhibited reversible one-electron reduction waves and the irreversible oxidation ones, except for 4a. Upon installing the TIPS-ethynyl substituents into the BODIPY core, the reduction potentials are significantly shifted in the anodic direction rather than the extent of the shifts in the oxidation potentials [30]. The electron-deficient effect of the substituents is pronounced for the doubly substituted derivatives 4a and 6a. Consequently, the electrochemical energy gaps of 3a–6a are estimated to be 2.30, 2,14, 2.35, and 2.21 V, respectively, which is consistent with the calculated HOMO–LUMO energy gaps (vide infra, Figure S24, Supporting Information File 1).
Theoretical calculations
To gain insight into the substitution effect on the electronic properties of the BODIPY derivatives, density functional theory (DFT) calculations were performed at the B3LYP/6-31G(d) level of theory. The a2-symmetry of the HOMOs and b2-symmetry of the LUMOs of each, the α- and β-ethynyl-substituted BODIPYs are almost identical to those of the unsubstituted compound 1a (Figure S24, Supporting Information File 1). The HOMO energies of the α-ethynyl-substituted derivatives 3a and 4a, are destabilized with broken degeneracy upon increasing the number of ethynyl substituents. On the other hand, the corresponding energies of the β-substituted derivatives 5a and 6a are not drastically altered because of a nodal plane near the 1,7-positions in the HOMO and smaller MO coefficients at the 2,6-positions than those at the 3,5-positions. Rather, a stabilization of the LUMO energies is found in 5a and 6a even though the MO hybridization of the BODIPY core with π-orbitals of ethynyl moieties is less affected. The mesomeric effect on the ethynyl substituents at the β-position of the BODIPY core is thus different from the α-substituted ones.
Furthermore, to understand the excited-state properties (e.g., Stokes shifts) of the substituted BODIPYs, the S1-state geometries for 4a and 6a were obtained by the time-dependent (TD) DFT methods. As a result, the excited-state structure of 6a exhibits a remarkably bent distortion of the core with a plane angle of 16.1°, whereas the structure of 4a remains coplanar (Figure S25, Supporting Information File 1). The corresponding energy gap between the HOMO and LUMO of 6a is significantly decreased with the geometry relaxation at the S1 state compared to that of the S0-state structure, which could be the origin of the large Stokes shift for 6a. Therefore, in comparison with the α-substituted compounds 3a and 4a, the less emissive character of the β-ethynyl-substituted derivatives 5a and 6a agreed with the fact that the photoexcited dynamics of β-substituted BODIPYs intend to the rapid decay with structural relaxation.
Conclusion
In summary, we have synthesized novel regioselectively alkynylated BODIPY derivatives via a gold(I)-catalyzed direct C–H functionalization with TIPS-EBX reagents. This atom-economical method for the late-stage alkynylation of BODIPY dyes could be an alternate approach for functionalized fluorescent dyes without the need of preparing unstable halogenated pyrrole precursors. The resulting α- and β-ethynyl-substituted BODIPYs displayed distinct substitution-site-dependent spectral features, for instance, the extent of the bathochromic shifts of the absorption and fluorescence, variable Stokes shift and the emission quantum yields. The TIPS-protected ethynyl groups of these BODIPY dyes can be applied as substrates for the “click” CuAAC reaction toward novel functional fluorescent materials.
Supporting Information
| Supporting Information File 1: Experimental methods including detailed synthetic procedures, compound characterization data, and DFT calculations. | ||
| Format: PDF | Size: 1.9 MB | Download |
| Supporting Information File 2: Crystallographic information file of compound 3a. | ||
| Format: CIF | Size: 2.2 MB | Download |
| Supporting Information File 3: Crystallographic information file of compound 6b. | ||
| Format: CIF | Size: 2.5 MB | Download |
References
-
Ni, Y.; Wu, J. Org. Biomol. Chem. 2014, 12, 3774–3791. doi:10.1039/c3ob42554a
Return to citation in text: [1] -
Tao, J.; Sun, D.; Sun, L.; Li, Z.; Fu, B.; Liu, J.; Zhang, L.; Wang, S.; Fang, Y.; Xu, H. Dyes Pigm. 2019, 168, 166–174. doi:10.1016/j.dyepig.2019.04.054
Return to citation in text: [1] -
Kim, T.-I.; Park, S.; Choi, Y.; Kim, Y. Chem. – Asian J. 2011, 6, 1358–1361. doi:10.1002/asia.201100025
Return to citation in text: [1] -
Liu, P.; Jing, X.; Yu, F.; Lv, C.; Chen, L. Analyst 2015, 140, 4576–4583. doi:10.1039/c5an00759c
Return to citation in text: [1] -
Sharker, S. M.; Jeong, C. J.; Kim, S. M.; Lee, J.-E.; Jeong, J. H.; In, I.; Lee, H.; Park, S. Y. Chem. – Asian J. 2014, 9, 2921–2927. doi:10.1002/asia.201402399
Return to citation in text: [1] -
Gao, M.; Wang, R.; Yu, F.; Chen, L. Biomaterials 2018, 160, 1–14. doi:10.1016/j.biomaterials.2018.01.011
Return to citation in text: [1] -
Gorbe, M.; Costero, A. M.; Sancenón, F.; Martínez-Máñez, R.; Ballesteros-Cillero, R.; Ochando, L. E.; Chulvi, K.; Gotor, R.; Gil, S. Dyes Pigm. 2019, 160, 198–207. doi:10.1016/j.dyepig.2018.08.007
Return to citation in text: [1] -
Kamkaew, A.; Lim, S. H.; Lee, H. B.; Kiew, L. V.; Chung, L. Y.; Burgess, K. Chem. Soc. Rev. 2013, 42, 77–88. doi:10.1039/c2cs35216h
Return to citation in text: [1] -
Yogo, T.; Urano, Y.; Ishitsuka, Y.; Maniwa, F.; Nagano, T. J. Am. Chem. Soc. 2005, 127, 12162–12163. doi:10.1021/ja0528533
Return to citation in text: [1] -
Awuah, S. G.; You, Y. RSC Adv. 2012, 2, 11169–11183. doi:10.1039/c2ra21404k
Return to citation in text: [1] -
Huang, L.; Li, Z.; Zhao, Y.; Zhang, Y.; Wu, S.; Zhao, J.; Han, G. J. Am. Chem. Soc. 2016, 138, 14586–14591. doi:10.1021/jacs.6b05390
Return to citation in text: [1] -
Turan, I. S.; Yildiz, D.; Turksoy, A.; Gunaydin, G.; Akkaya, E. U. Angew. Chem., Int. Ed. 2016, 55, 2875–2878. doi:10.1002/anie.201511345
Return to citation in text: [1] -
Suryani, O.; Higashino, Y.; Sato, H.; Kubo, Y. ACS Appl. Energy Mater. 2019, 2, 448–458. doi:10.1021/acsaem.8b01474
Return to citation in text: [1] -
Bandyopadhyay, S.; Anil, A. G.; James, A.; Patra, A. ACS Appl. Mater. Interfaces 2016, 8, 27669–27678. doi:10.1021/acsami.6b08331
Return to citation in text: [1] -
Li, W.; Xie, Z.; Jing, X. Catal. Commun. 2011, 16, 94–97. doi:10.1016/j.catcom.2011.09.007
Return to citation in text: [1] -
Luo, G.-G.; Fang, K.; Wu, J.-H.; Mo, J. Chem. Commun. 2015, 51, 12361–12364. doi:10.1039/c5cc03897a
Return to citation in text: [1] -
Zhang, C.; Zhao, J.; Wu, S.; Wang, Z.; Wu, W.; Ma, J.; Guo, S.; Huang, L. J. Am. Chem. Soc. 2013, 135, 10566–10578. doi:10.1021/ja405170j
Return to citation in text: [1] -
Wu, W.; Zhao, J.; Sun, J.; Guo, S. J. Org. Chem. 2012, 77, 5305–5312. doi:10.1021/jo300613g
Return to citation in text: [1] -
Sun, J.; Zhong, F.; Yi, X.; Zhao, J. Inorg. Chem. 2013, 52, 6299–6310. doi:10.1021/ic302210b
Return to citation in text: [1] -
Liu, Q.; Yin, B.; Yang, T.; Yang, Y.; Shen, Z.; Yao, P.; Li, F. J. Am. Chem. Soc. 2013, 135, 5029–5037. doi:10.1021/ja3104268
Return to citation in text: [1] -
Loudet, A.; Burgess, K. Chem. Rev. 2007, 107, 4891–4932. doi:10.1021/cr078381n
Return to citation in text: [1] [2] -
Verbelen, B.; Leen, V.; Wang, L.; Boens, N.; Dehaen, W. Chem. Commun. 2012, 48, 9129–9131. doi:10.1039/c2cc34549h
Return to citation in text: [1] -
Ren, W.; Xiang, H.; Peng, C.; Musha, Z.; Chen, J.; Li, X.; Huang, R.; Hu, Y. RSC Adv. 2018, 8, 5542–5549. doi:10.1039/c7ra13070h
Return to citation in text: [1] -
Zhang, M.; Hao, E.; Zhou, J.; Yu, C.; Bai, G.; Wang, F.; Jiao, L. Org. Biomol. Chem. 2012, 10, 2139–2145. doi:10.1039/c2ob06689k
Return to citation in text: [1] -
Verbelen, B.; Boodts, S.; Hofkens, J.; Boens, N.; Dehaen, W. Angew. Chem., Int. Ed. 2015, 54, 4612–4616. doi:10.1002/anie.201410853
Return to citation in text: [1] -
Luo, L.; Wu, D.; Li, W.; Zhang, S.; Ma, Y.; Yan, S.; You, J. Org. Lett. 2014, 16, 6080–6083. doi:10.1021/ol502883x
Return to citation in text: [1] -
Zhou, X.; Wu, Q.; Yu, Y.; Yu, C.; Hao, E.; Wei, Y.; Mu, X.; Jiao, L. Org. Lett. 2016, 18, 736–739. doi:10.1021/acs.orglett.5b03706
Return to citation in text: [1] -
Wang, D.; Cheng, C.; Wu, Q.; Wang, J.; Kang, Z.; Guo, X.; Wu, H.; Hao, E.; Jiao, L. Org. Lett. 2019, 21, 5121–5125. doi:10.1021/acs.orglett.9b01722
Return to citation in text: [1] -
Yang, X.; Jiang, L.; Yang, M.; Zhang, H.; Lan, J.; Zhou, F.; Chen, X.; Wu, D.; You, J. J. Org. Chem. 2018, 83, 9538–9546. doi:10.1021/acs.joc.8b01239
Return to citation in text: [1] -
Wang, J.; Li, Y.; Gong, Q.; Wang, H.; Hao, E.; Lo, P.-C.; Jiao, L. J. Org. Chem. 2019, 84, 5078–5090. doi:10.1021/acs.joc.9b00020
Return to citation in text: [1] [2] -
Wang, J.; Wu, Q.; Gong, Q.; Cheng, K.; Liu, Q.; Yu, C.; Hao, E.; Jiao, L. Adv. Synth. Catal. 2019, 361, 769–777. doi:10.1002/adsc.201801338
Return to citation in text: [1] -
Chen, J.; Mizumura, M.; Shinokubo, H.; Osuka, A. Chem. – Eur. J. 2009, 15, 5942–5949. doi:10.1002/chem.200802541
Return to citation in text: [1] [2] [3] -
Feng, Z.; Jiao, L.; Feng, Y.; Yu, C.; Chen, N.; Wei, Y.; Mu, X.; Hao, E. J. Org. Chem. 2016, 81, 6281–6291. doi:10.1021/acs.joc.6b00858
Return to citation in text: [1] -
Kowada, T.; Yamaguchi, S.; Fujinaga, H.; Ohe, K. Tetrahedron 2011, 67, 3105–3110. doi:10.1016/j.tet.2011.02.073
Return to citation in text: [1] -
Wirtz, M.; Grüter, A.; Rebmann, P.; Dier, T.; Volmer, D. A.; Huch, V.; Jung, G. Chem. Commun. 2014, 50, 12694–12697. doi:10.1039/c4cc05288a
Return to citation in text: [1] [2] -
Albrecht, M.; Lippach, A.; Exner, M. P.; Jerbi, J.; Springborg, M.; Budisa, N.; Wenz, G. Org. Biomol. Chem. 2015, 13, 6728–6736. doi:10.1039/c5ob00505a
Return to citation in text: [1] -
Sakida, T.; Yamaguchi, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2011, 50, 2280–2283. doi:10.1002/anie.201006314
Return to citation in text: [1] [2] -
Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Org. Lett. 2012, 14, 744–747. doi:10.1021/ol203289v
Return to citation in text: [1] -
Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304–7307. doi:10.1002/anie.201003179
Return to citation in text: [1] [2] -
Li, Y.; Waser, J. Beilstein J. Org. Chem. 2013, 9, 1763–1767. doi:10.3762/bjoc.9.204
Return to citation in text: [1] [2] [3] -
Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073
Return to citation in text: [1] -
Nepomnyashchii, A. B.; Bröring, M.; Ahrens, J.; Bard, A. J. J. Am. Chem. Soc. 2011, 133, 8633–8645. doi:10.1021/ja2010219
Return to citation in text: [1]
| 40. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304–7307. doi:10.1002/anie.201003179 |
| 41. | Li, Y.; Waser, J. Beilstein J. Org. Chem. 2013, 9, 1763–1767. doi:10.3762/bjoc.9.204 |
| 38. | Brand, J. P.; Charpentier, J.; Waser, J. Angew. Chem., Int. Ed. 2009, 48, 9346–9349. doi:10.1002/anie.200905419 |
| 39. | Brand, J. P.; Waser, J. Org. Lett. 2012, 14, 744–747. doi:10.1021/ol203289v |
| 40. | Brand, J. P.; Waser, J. Angew. Chem., Int. Ed. 2010, 49, 7304–7307. doi:10.1002/anie.201003179 |
| 41. | Li, Y.; Waser, J. Beilstein J. Org. Chem. 2013, 9, 1763–1767. doi:10.3762/bjoc.9.204 |
| 42. | Li, Y.; Hari, D. P.; Vita, M. V.; Waser, J. Angew. Chem., Int. Ed. 2016, 55, 4436–4454. doi:10.1002/anie.201509073 |
| 41. | Li, Y.; Waser, J. Beilstein J. Org. Chem. 2013, 9, 1763–1767. doi:10.3762/bjoc.9.204 |
| 1. | Ni, Y.; Wu, J. Org. Biomol. Chem. 2014, 12, 3774–3791. doi:10.1039/c3ob42554a |
| 2. | Tao, J.; Sun, D.; Sun, L.; Li, Z.; Fu, B.; Liu, J.; Zhang, L.; Wang, S.; Fang, Y.; Xu, H. Dyes Pigm. 2019, 168, 166–174. doi:10.1016/j.dyepig.2019.04.054 |
| 3. | Kim, T.-I.; Park, S.; Choi, Y.; Kim, Y. Chem. – Asian J. 2011, 6, 1358–1361. doi:10.1002/asia.201100025 |
| 4. | Liu, P.; Jing, X.; Yu, F.; Lv, C.; Chen, L. Analyst 2015, 140, 4576–4583. doi:10.1039/c5an00759c |
| 5. | Sharker, S. M.; Jeong, C. J.; Kim, S. M.; Lee, J.-E.; Jeong, J. H.; In, I.; Lee, H.; Park, S. Y. Chem. – Asian J. 2014, 9, 2921–2927. doi:10.1002/asia.201402399 |
| 6. | Gao, M.; Wang, R.; Yu, F.; Chen, L. Biomaterials 2018, 160, 1–14. doi:10.1016/j.biomaterials.2018.01.011 |
| 21. | Loudet, A.; Burgess, K. Chem. Rev. 2007, 107, 4891–4932. doi:10.1021/cr078381n |
| 37. | Sakida, T.; Yamaguchi, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2011, 50, 2280–2283. doi:10.1002/anie.201006314 |
| 17. | Zhang, C.; Zhao, J.; Wu, S.; Wang, Z.; Wu, W.; Ma, J.; Guo, S.; Huang, L. J. Am. Chem. Soc. 2013, 135, 10566–10578. doi:10.1021/ja405170j |
| 18. | Wu, W.; Zhao, J.; Sun, J.; Guo, S. J. Org. Chem. 2012, 77, 5305–5312. doi:10.1021/jo300613g |
| 19. | Sun, J.; Zhong, F.; Yi, X.; Zhao, J. Inorg. Chem. 2013, 52, 6299–6310. doi:10.1021/ic302210b |
| 20. | Liu, Q.; Yin, B.; Yang, T.; Yang, Y.; Shen, Z.; Yao, P.; Li, F. J. Am. Chem. Soc. 2013, 135, 5029–5037. doi:10.1021/ja3104268 |
| 35. | Wirtz, M.; Grüter, A.; Rebmann, P.; Dier, T.; Volmer, D. A.; Huch, V.; Jung, G. Chem. Commun. 2014, 50, 12694–12697. doi:10.1039/c4cc05288a |
| 37. | Sakida, T.; Yamaguchi, S.; Shinokubo, H. Angew. Chem., Int. Ed. 2011, 50, 2280–2283. doi:10.1002/anie.201006314 |
| 13. | Suryani, O.; Higashino, Y.; Sato, H.; Kubo, Y. ACS Appl. Energy Mater. 2019, 2, 448–458. doi:10.1021/acsaem.8b01474 |
| 14. | Bandyopadhyay, S.; Anil, A. G.; James, A.; Patra, A. ACS Appl. Mater. Interfaces 2016, 8, 27669–27678. doi:10.1021/acsami.6b08331 |
| 15. | Li, W.; Xie, Z.; Jing, X. Catal. Commun. 2011, 16, 94–97. doi:10.1016/j.catcom.2011.09.007 |
| 16. | Luo, G.-G.; Fang, K.; Wu, J.-H.; Mo, J. Chem. Commun. 2015, 51, 12361–12364. doi:10.1039/c5cc03897a |
| 34. | Kowada, T.; Yamaguchi, S.; Fujinaga, H.; Ohe, K. Tetrahedron 2011, 67, 3105–3110. doi:10.1016/j.tet.2011.02.073 |
| 7. | Gorbe, M.; Costero, A. M.; Sancenón, F.; Martínez-Máñez, R.; Ballesteros-Cillero, R.; Ochando, L. E.; Chulvi, K.; Gotor, R.; Gil, S. Dyes Pigm. 2019, 160, 198–207. doi:10.1016/j.dyepig.2018.08.007 |
| 8. | Kamkaew, A.; Lim, S. H.; Lee, H. B.; Kiew, L. V.; Chung, L. Y.; Burgess, K. Chem. Soc. Rev. 2013, 42, 77–88. doi:10.1039/c2cs35216h |
| 9. | Yogo, T.; Urano, Y.; Ishitsuka, Y.; Maniwa, F.; Nagano, T. J. Am. Chem. Soc. 2005, 127, 12162–12163. doi:10.1021/ja0528533 |
| 10. | Awuah, S. G.; You, Y. RSC Adv. 2012, 2, 11169–11183. doi:10.1039/c2ra21404k |
| 11. | Huang, L.; Li, Z.; Zhao, Y.; Zhang, Y.; Wu, S.; Zhao, J.; Han, G. J. Am. Chem. Soc. 2016, 138, 14586–14591. doi:10.1021/jacs.6b05390 |
| 12. | Turan, I. S.; Yildiz, D.; Turksoy, A.; Gunaydin, G.; Akkaya, E. U. Angew. Chem., Int. Ed. 2016, 55, 2875–2878. doi:10.1002/anie.201511345 |
| 35. | Wirtz, M.; Grüter, A.; Rebmann, P.; Dier, T.; Volmer, D. A.; Huch, V.; Jung, G. Chem. Commun. 2014, 50, 12694–12697. doi:10.1039/c4cc05288a |
| 36. | Albrecht, M.; Lippach, A.; Exner, M. P.; Jerbi, J.; Springborg, M.; Budisa, N.; Wenz, G. Org. Biomol. Chem. 2015, 13, 6728–6736. doi:10.1039/c5ob00505a |
| 30. | Wang, J.; Li, Y.; Gong, Q.; Wang, H.; Hao, E.; Lo, P.-C.; Jiao, L. J. Org. Chem. 2019, 84, 5078–5090. doi:10.1021/acs.joc.9b00020 |
| 32. | Chen, J.; Mizumura, M.; Shinokubo, H.; Osuka, A. Chem. – Eur. J. 2009, 15, 5942–5949. doi:10.1002/chem.200802541 |
| 30. | Wang, J.; Li, Y.; Gong, Q.; Wang, H.; Hao, E.; Lo, P.-C.; Jiao, L. J. Org. Chem. 2019, 84, 5078–5090. doi:10.1021/acs.joc.9b00020 |
| 29. | Yang, X.; Jiang, L.; Yang, M.; Zhang, H.; Lan, J.; Zhou, F.; Chen, X.; Wu, D.; You, J. J. Org. Chem. 2018, 83, 9538–9546. doi:10.1021/acs.joc.8b01239 |
| 33. | Feng, Z.; Jiao, L.; Feng, Y.; Yu, C.; Chen, N.; Wei, Y.; Mu, X.; Hao, E. J. Org. Chem. 2016, 81, 6281–6291. doi:10.1021/acs.joc.6b00858 |
| 22. | Verbelen, B.; Leen, V.; Wang, L.; Boens, N.; Dehaen, W. Chem. Commun. 2012, 48, 9129–9131. doi:10.1039/c2cc34549h |
| 23. | Ren, W.; Xiang, H.; Peng, C.; Musha, Z.; Chen, J.; Li, X.; Huang, R.; Hu, Y. RSC Adv. 2018, 8, 5542–5549. doi:10.1039/c7ra13070h |
| 24. | Zhang, M.; Hao, E.; Zhou, J.; Yu, C.; Bai, G.; Wang, F.; Jiao, L. Org. Biomol. Chem. 2012, 10, 2139–2145. doi:10.1039/c2ob06689k |
| 25. | Verbelen, B.; Boodts, S.; Hofkens, J.; Boens, N.; Dehaen, W. Angew. Chem., Int. Ed. 2015, 54, 4612–4616. doi:10.1002/anie.201410853 |
| 26. | Luo, L.; Wu, D.; Li, W.; Zhang, S.; Ma, Y.; Yan, S.; You, J. Org. Lett. 2014, 16, 6080–6083. doi:10.1021/ol502883x |
| 27. | Zhou, X.; Wu, Q.; Yu, Y.; Yu, C.; Hao, E.; Wei, Y.; Mu, X.; Jiao, L. Org. Lett. 2016, 18, 736–739. doi:10.1021/acs.orglett.5b03706 |
| 28. | Wang, D.; Cheng, C.; Wu, Q.; Wang, J.; Kang, Z.; Guo, X.; Wu, H.; Hao, E.; Jiao, L. Org. Lett. 2019, 21, 5121–5125. doi:10.1021/acs.orglett.9b01722 |
| 32. | Chen, J.; Mizumura, M.; Shinokubo, H.; Osuka, A. Chem. – Eur. J. 2009, 15, 5942–5949. doi:10.1002/chem.200802541 |
| 43. | Nepomnyashchii, A. B.; Bröring, M.; Ahrens, J.; Bard, A. J. J. Am. Chem. Soc. 2011, 133, 8633–8645. doi:10.1021/ja2010219 |
| 21. | Loudet, A.; Burgess, K. Chem. Rev. 2007, 107, 4891–4932. doi:10.1021/cr078381n |
| 31. | Wang, J.; Wu, Q.; Gong, Q.; Cheng, K.; Liu, Q.; Yu, C.; Hao, E.; Jiao, L. Adv. Synth. Catal. 2019, 361, 769–777. doi:10.1002/adsc.201801338 |
| 32. | Chen, J.; Mizumura, M.; Shinokubo, H.; Osuka, A. Chem. – Eur. J. 2009, 15, 5942–5949. doi:10.1002/chem.200802541 |
© 2020 Shimada et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)