Abstract
Herein, we present an efficient synthesis of dipeptide analogues of α-fluorinated β-aminophosphonates. Each step of the synthesis was optimized to provide excellent yields. Moreover, the absolute configuration of the obtained compounds was determined by X-ray analysis, which proved the stereochemistry that was proposed based on NMR studies.

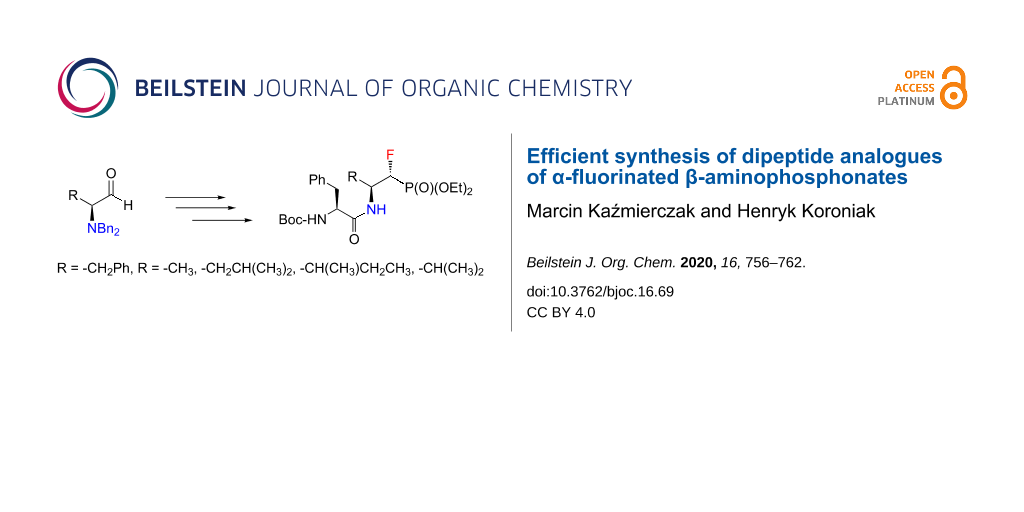
Graphical Abstract
Introduction
The chemistry of fluorinated aminophosphonates is constantly being developed, mainly due to their wide spectra of applications. What is more, they are valuable targets for biomedical investigations. Due to the strong interest in this type of compounds, several marvelous reviews about the synthesis and application of fluorinated aminophosphonates have been published in recent years [1-3]. To the best of our knowledge, among the many applications of fluorinated aminophosphonates and aminophosphonic acid derivatives, they exhibit antiviral [4,5], antibacterial [6] and antifungal [7] activities. Moreover, α-fluorinated phosphonates can be considered as hydrolytically stable mimics of naturally occurring phosphates [8]. Due to the fact that enzyme binding may depend on the C–F stereochemistry, the synthesis of such compounds with a specific configuration is very important [9-11]. α-Fluorinated phosphonates can be prepared by many different protocols [12-16].
Nucleophilic fluorination is one of the fundamental reactions in organic chemistry in the field of the synthesis of building blocks with a potential biological activity. To date, scientists have developed many nucleophilic fluorinating reagents [17-19]. Among others, we can distinguish a family of such chemicals having in their structure a sulfonyl fluoride system. For example 2-pyridinesulfonyl fluoride (PyFluor, 1) [20,21] or perfluorobutanesulfonyl fluoride (PBSF, 2) [22,23] (Figure 1). On the other hand, Hu just recently presented a novel deoxyfluorination reagent with a similar structure to 1 and 2, containing a sulfonimidoyl instead of sulfonyl group. 4-Chloro-N-tosylbenzene-1-sulfonimidoyl fluoride (SulfoxFluor, 3, Figure 1) may be an interesting alternative to 1 or 2. It is not commercially available yet, however, it can be obtained from inexpensive materials [24].
![[1860-5397-16-69-1]](/bjoc/content/figures/1860-5397-16-69-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Chemical structure of PyFluor, PBSF and SulfoxFluor.
Figure 1: Chemical structure of PyFluor, PBSF and SulfoxFluor.
Very often small organic molecules containing a fluorine atom are transformed into biologically active compounds. As an example, we can consider peptide bond formation between an amine and a carboxylic acid. This transformation is a very important reaction in organic synthesis and therefore, many coupling reagents are available on the market [25-29].
Our previous studies have shown that the regioselectivity of the nucleophilic fluorination of amino alcohols can be controlled depending on the fluorinating reagent used [30,31]. This work presents the optimization of nucleophilic fluorination conditions. What is more, we discuss the effect of the base used on the regioselectivity of the fluorination reaction. The absolute configuration of the obtained compounds was determined and confirmed by X-ray analysis. Furthermore, we present the use of α-fluorinated β-aminophosphonates as building blocks in the synthesis of their dipeptide analogues. In addition, we show the results of the use of several coupling reagents in the synthesis of amide bonds.
Results and Discussion
Our goal was to increase the efficiency of the synthesis of α-fluorinated β-aminophosphonate dipeptide analogues 15. In the first stage, we optimized Pudovik's reaction (Scheme 1) [32].
![[1860-5397-16-69-i1]](/bjoc/content/inline/1860-5397-16-69-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
In our previous studies, the Pudovik reaction worked with satisfactory yield and diastereoselectivity [30]. Unfortunately, we were not able to isolate the pure anti-isomer in every case. The latest research shows that this reaction can successfully be carried out under milder conditions, without any solvent and in the presence of a weak base [33]. After testing many modifications, it turned out that the use of triethylamine (1 equiv) as a base, and without a solvent, gave the best results with aldehydes 4 (1 equiv) and diethyl phosphite (1 equiv). These conditions not only increased the diastereoselectivity of the Pudovik reaction, but also improved its yield drastically (Table 1). The implementation of this allowed us to obtain the anti-isomer (1R,2S) in very good yields.
Table 1: Optimization of reaction conditions.
| entry | product | anti/syna | anti/synb | yieldc |
| 1 | 5a | 91:9 | 99:1 | 86% |
| 2 | 5b | 86:14 | 99:1 | 71% |
| 3 | 5c | 90:10 | 99:1 | 75% |
| 4 | 5d | 92:8 | 99:1 | 88% |
| 5 | 5e | 90:10 | 99:1 | 79% |
aCrude reaction mixture ratio based on 31P NMR. bRatio after isolation based on 31P NMR. cYield of the amino alcohol 5 after isolation.
There are not many literature examples of the regioselective fluorination of amino alcohols using by PyFluor (1) or PBSF (2). In general, the fluorination reactions with these reagents require the use of a base as an activator [20]. Doyle demonstrated the combination between PyFluor or PBSF and bases such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 6), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD, 7), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG) 8, tert-butylimino-tri(pyrrolidino)phosphorane (BTPP, 9, Figure 2) [21]. We screened the alcohols 5 against sulfonyl fluoride reagents 1 and 2 and a combination of selected bases 6–9 (Scheme 2). All reactions during the optimization step were carried out on a 0.1 mmol scale under an argon atmosphere while the reaction medium was toluene.
![[1860-5397-16-69-2]](/bjoc/content/figures/1860-5397-16-69-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-69-i2]](/bjoc/content/inline/1860-5397-16-69-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the first stage of the optimization three representative amino alcohols were chosen. Each bearing a different group in the side chain: an aromatic (5a), a small (5b) and a large aliphatic substituent (5c). Unfortunately, under the tested conditions, we did not observe a complete regioselectivity of the fluorination reaction. Nevertheless, in each case, the use of PyFluor as a nucleophilic fluorinating reagent and MTBD as an activator improved the regioselectivity of this reaction, as well as, the yield of the α-isomer 11 (best results are shown in Table 2, for all tested conditions see Table S1 in Supporting Information File 1). In our experience, there was no significant correlation between steric bulkiness of the base (from DBU to BTPP) used, and the yield of α-isomer 11 formation. It is worth mentioning that application of PBSF gives slightly worse results in each case. We assume it is associated with a much greater reactivity of this reagent in relation to PyFluor.
Table 2: Optimization of reaction conditions.
| entry | product | reagent | base | 11/12 ratioa | yieldb |
| 1 | 11a | PyFluor | MTBD | 73:27 | 61% |
| 2 | 11b | PyFluor | MTBD | 60:40 | 47% |
| 3 | 11c | PyFluor | MTBD | 87:13 | 70% |
| 4 | 11d | PyFluor | MTBD | 68:32 | 59% |
| 5 | 11e | PyFluor | MTBD | 82:18 | 69% |
aCrude reaction mixture ratio based on 31P NMR and 19F NMR; bYield of α-fluorides 11 after isolation.
The mechanism of the PyFluor-mediated deoxyfluorination of the α-hydroxy-β-aminophosphonates 5 was previously proposed. Based on spectroscopic studies (19F,1H-HOESY, 1H,1H-NOESY, as well as J-couplings) a relative configuration of N,N-dibenzyl-protected α-fluoro-β-aminophosphonates 11 was established as (1R,2S), but we failed to crystallize these compounds [31]. That is why the removal of the benzyl protecting group from fluorides 11 and the transfer of the free amines 13 into the salts 14 was applied. The salts 14 could be then crystallized and subjected to X-ray studies.
A standard N−debenzylation protocol was employed to remove the benzyl protecting group. The hydrogenolysis reaction was catalyzed by palladium on carbon (Pd/C), and was carried out in trifluoroethanol (TFE) as a solvent [34,35]. The free amines 13 were converted into stable oxalate salts 14 with quantitative yields. The precipitation reactions proceeded in the presence of 1 equiv of oxalic acid in diethyl ether (Scheme 3) [36]. Unlike amines 13, salts 14 are very stable and can be stored for months at room temperature.
![[1860-5397-16-69-i3]](/bjoc/content/inline/1860-5397-16-69-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
What is more, we attempted to crystallize at least one derivative to confirm the absolute stereochemistry of the obtained compounds. In order to obtain a single crystal suitable for X-ray diffraction studies, various crystallization techniques were tested [37,38]. Crystallization of derivative 14c from D2O brought the expected results. The analysis confirmed the structure of the resulting compound 14c. The absolute configuration of 14c is consistent with the stereochemistry we proposed based on NMR studies. The crystals of 14c contained the (1R,2S)-diastereoisomer (Figure 3). The correct absolute configuration was determined on the basis of Flack and Parson’s parameters, as well as of slightly better R factors for the correct model.
![[1860-5397-16-69-3]](/bjoc/content/figures/1860-5397-16-69-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structure of compound (1R,2S)-14c (ORTEP image).
Figure 3: Molecular structure of compound (1R,2S)-14c (ORTEP image).
Moreover, amines 13 were used as substrates in the formation of peptide bonds with N-Boc-phenylalanine (Boc-Phe-OH, Scheme 4). For this purpose we examined several coupling reagents available on the market. We chose amine 13a as a model substrate. In each case, the amine–acid coupling reaction proceeded with good to excellent yields (Table 3). At the beginning a mixture of N,N′-dicyclohexylcarbodiimide (DCC) [39] and 1-hydroxybenzotriazole (HOBt) [40] was used. The reaction took place “only” in good yield (63%). Crude products required very careful purification due to the byproduct formed during the reaction – N,N′-dicyclohexylurea (DCU), which is insoluble in the solvents used in the synthesis (CH2Cl2, DMF). Replacing DCC with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCl) [41] significantly increased the reaction yield (86%). What is more, the reaction byproduct is water-soluble and easy to remove by extraction. Nevertheless, HOBt was still used as an additive. This reagent has recently been reported to exhibit explosive properties [42]. Fortunately, stable 1-hydroxybenzotriazole substitutes are available on the market, and can be used in the reaction of peptide bond formation [43]. One of them is OxymaPure [44] which was successfully employed as a replacement for HOBt. Using EDCI/OxymaPure conditions, an even greater yield of the product formation (88%) was observed, however, the reaction still required a long reation time. Finally, COMU [45] as a coupling reagent was applied. The model reaction took place with a yield of 92% after just two hours. COMU allowed to obtain the dipeptide analogue 15a not only with an excellent yield, but also in a very short time. Under these conditions, the remaining amines 13 were reacted, to receive a series of dipeptide analogues of α-fluorinated β-aminophosphonates 15.
![[1860-5397-16-69-i4]](/bjoc/content/inline/1860-5397-16-69-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 15. aConditions are given in the Experimental section.
Scheme 4: Synthesis of 15. aConditions are given in the Experimental section.
Table 3: Optimization of reaction conditions.
| entry | product | reagent | additive | base | time | yielda |
| 1 | 15a | DCC | HOBt | Et3N | 24 h | 63% |
| 2 | 15a | EDCI | HOBt | Et3N | 18 h | 86% |
| 3 | 15a | EDCI | OxymaPure | DIPEA | 18 h | 88% |
| 4 | 15a | COMU | – | DIPEA | 2 h | 92% |
| 5 | 15b | COMU | – | DIPEA | 3 h | 91% |
| 6 | 15c | COMU | – | DIPEA | 3 h | 94% |
| 7 | 15d | COMU | – | DIPEA | 3 h | 92% |
| 8 | 15e | COMU | – | DIPEA | 3 h | 92% |
aYield of dipeptide analogues 15 after isolation.
Conclusion
In summary, an efficient approach to the synthesis of dipeptide analogues of α-fluorinated β-aminophosphonates 15 was presented. These compounds were prepared in a four-step sequence starting from N,N-dibenzylamino aldehydes 5. Each step of the synthesis has been optimized to provide very good yields (Pudovik, deoxyfluorination and amine–acid coupling reactions). Furthermore, the absolute configuration of the obtained compounds has been confirmed by X-ray analysis, which is consistent with the stereochemistry proposed based on NMR studies. Dipeptide analogues of α-fluorinated β-aminophosphonates 15 presented in this publication, after deprotection of the amine function appear to be attractive compounds with potential biological activity.
Experimental
General methods
1H NMR, 13C NMR, 19F NMR and 31P NMR spectra were obtained on a Bruker ASCEND 400 (400 MHz) and a Bruker ASCEND 600 (600 MHz) spectrometer. All 2D spectra were recorded on a Bruker ASCEND 600 (600 MHz) spectrometer. 1H NMR chemical shifts were expressed in parts per million downfield from tetramethylsilane (TMS) as an internal standard (δ = 0) in CDCl3 or CD3CN. 13C NMR chemical shifts were expressed in parts per million downfield from CDCl3 (δ = 77.0) or CD3CN (δ = 1.39) as internal standards. 19F NMR chemical shifts were expressed in parts per million upfield from CFCl3 as an internal standard (δ = 0) in CDCl3 or CD3CN. 31P NMR Chemical shifts were expressed in parts per million in CDCl3 or CD3CN. High-resolution mass spectra were recorded by electron spray (ESIMS) techniques using a QToF Impact HD Bruker spectrometer. Reagent grade chemicals were used. Solvents were dried with CaH2 (CH2Cl2), NaH (Et2O), P2O5 (PhMe), and distilled under argon atmosphere. All moisture sensitive reactions were carried out under an argon atmosphere using ovendried glassware. TLC was performed on Merck Kieselgel 60-F254 with EtOAc/hexane and MeOH/CHCl3 as developing systems, and products were detected by inspection under UV light (254 nm) and with a solution of potassium permanganate. Merck Kieselgel 60 (0.063–0.200 μm), Merck Kieselgel 60 (0.040–0.063 μm), and Merck Kieselgel 60 (0.015–0.004 μm), were used for column chromatography.
X-ray diffraction data for 14c were collected at 100(1) K by the ω-scan technique on Rigaku four-circle Xcalibur diffractometer (Eos detector) with graphite-monochromatized Mo Kα radiation (λ = 0.71073 Å). The data were corrected for Lorentz-polarization and absorption effects [46]. Accurate unit cell parameters were determined by a least-squares fit of 4300 reflections of highest intensity, chosen from the whole experiment. The structure was solved with SHELXT and refined with the full-matrix least-squares procedure on F2 by SHELXL [47]. All non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed in the calculated positions and refined as ‘riding model’ with the isotropic displacement parameters set at 1.2 (1.5 for methyl and hydroxy groups) times the Ueq value for appropriate non-hydrogen atom. As the crystals were obtained from D2O solution, this solvent was used for final model (although, no difference between refinements with H2O and D2O could be found). The correct absolute configuration was determined on the basis of Flack and Parson’s parameters, as well as of slightly better R factors for the correct model. The crystallographic data for the structural analysis has been deposited with the Cambridge Crystallographic Data Centre, CCDC No. 1977323 (14c). Copies of this information may be obtained free of charge from http://www.ccdc.cam.ac.uk.
Procedure for the synthesis of amino alcohols 5
TEA (1,0 equiv) was added under an argon atmosphere to a mixture of a stirred solution of diethyl phosphite (1,0 equiv) and the appropriate aldehyde 4 (1,0 equiv). The reaction mixture was stirred at room temperature overnight, diluted with 20 mL of water and extracted with ethyl acetate (3 × 15 mL). The organic layers were washed with NaClsat., dried over MgSO4 or Na2SO4, filtrated and concentrated under reduced pressure. The crude products were isolated using column chromatography (chloroform → chloroform/methanol 100:0.5 v/v). The NMR spectroscopic data for amino alcohols 5 were in good agreement with our previous research [30,31,48].
Procedure for the synthesis of fluorides 11
PyFluor (1,2 equiv) or PBSF (1,2 equiv) and base (2 equiv) were added under an argon atmosphere to a stirred solution of amino alcohol 5 (1mmol) in 2,5 mL PhMe, under an argon atmosphere. The reaction mixture was stirred at room temperature until completion of the reaction monitored by TLC (3–18 h). The reaction mixture was then diluted with 20 mL of water and extracted with ethyl acetate (3 × 15 mL). The organic layers were dried over MgSO4 or Na2SO4, filtrated and concentrated under reduced pressure. The crude products were isolated using column chromatography (n-hexane/ethyl acetate 90:10, v/v → n-hexane/ethyl acetate 60:40, v/v). The NMR spectroscopic data for fluorides 11 were in good agreement with our previous research [31].
Procedure for the synthesis of amines 13
The amines 13 were prepared in a similar manner as described in [35]. Fluoride 11 (1 equiv) was dissolved in 5 mL of TFE and then 10% Pd/C (20% v/v) was added. The solution was stirred under an atmosphere of hydrogen at room temperature for 3 days. After this time the catalyst was filtered off (Celite), the solvent was evaporated and the crude product was purified using flash chromatography (chloroform/methanol 100:0, v/v chloroform/methanol → 50:0.5 v/v).
Procedure for the synthesis of oxalates 14
The oxalates 14 were prepared in a similar manner as described in [35]. A solution of amine 13 (1 equiv) was dissolved in anhydrous diethyl ether and was added dropwise to a vigorously stirred solution of oxalic acid (1 equiv) in diethyl ether under an argon atmosphere. The mixture was left overnight in a freezer. The next day, the precipitate was filtered off to give a white solid.
Procedure for the synthesis of dipeptide analogues 15
DCC/HOBt conditions: The analogue 15a was prepared in a similar manner as described in [35]. To a cooled (0 °C) solution of amine 13 (1.05 equiv) in methylene chloride a solution of a Boc-Phe-OH (1 equiv), HOBt (1.25 equiv) in a 3:7 CH2Cl2/DMF solution and DCC (1.25 equiv) under an argon atmosphere were added. Stirring was continued at this temperature for 30 min and then the solution was stirred for 24 h at room temperature. The DCU was filtered off, and the filtrate was washed with cold solutions of 1 M NaOH, water, 1 M citric acid, water, 1 M NaHCO3, and water. The organic extract was dried over anhydrous MgSO4. The drying agent was filtered off and the solvent was evaporated. The crude product was purified by flash chromatography (n-hexane/ethyl acetate 60:40 → 50:50).
EDCI/HOBt conditions: To a cooled (0 °C) solution of amine 13 (1.05 equiv) in DMF, Boc-Phe-OH (1 equiv), TEA (1 equiv), HOBt (1.3 equiv) and EDCI (1.25 equiv) were added. Stirring was continued at this temperature for 30 min and then the solution was stirred for 18 h at room temperature. The reaction mixture was then diluted with 20 mL of ethyl acetate and washed with NaHCO3sat, 1 M HCl, and NaClsat. The organic layer was dried over MgSO4 or Na2SO4, filtrated and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-hexane/ethyl acetate 60:40 → 50:50).
EDCI/OxymaPure conditions: Boc-Phe-OH (1 equiv), OxymaPure (1.1 equiv), and EDCI (1.1 equiv) were mixed in DMF at 0 °C under an argon atmosphere. The reaction mixture was stirred for 5 min at 0 °C to preactivate the acid and generate the active ester, and then DIPEA (1 equiv) followed by amine 13 (1 equiv) were added. Stirring was continued at this temperature for 30 min and then the solution was stirred for 18 h at room temperature. The reaction mixture was then diluted with 20 mL of ethyl acetate and washed with NaHCO3sat, 1 M HCl, and NaClsat. The organic layer was dried over MgSO4 or Na2SO4, filtrated and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-hexane/ethyl acetate 60:40 → 50:50).
COMU conditions: COMU (1 equiv) was added at 0 °C to a mixture of Boc-Phe-OH (1 equiv) and DIPEA (2 equiv) in DMF under an argon atmosphere. The reaction mixture was activated for 5 min, followed by the addition of amine 13 (1 equiv) in DMF. Stirring was continued at this temperature for 30 min and then the solution was stirred for 2–3 h at room temperature (monitored by 31P or 19F NMR). The reaction mixture was then diluted with 20 mL of ethyl acetate and washed with NaHCO3sat, 1 M HCl, and NaClsat. The organic layer was dried over MgSO4 or Na2SO4, filtrated and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-hexane/ethyl acetate 60:40 → 50:50).
Supporting Information
| Supporting Information File 1: Optimization of reaction conditions data, characterization data, copies of NMR spectra for compounds 13–15, single crystal X-ray data for compound 14c. | ||
| Format: PDF | Size: 5.7 MB | Download |
| Supporting Information File 2: CIF file of 14c. | ||
| Format: CIF | Size: 380.9 KB | Download |
References
-
Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q
Return to citation in text: [1] -
Turcheniuk, K. V.; Kukhar, V. P.; Röschenthaler, G.-V.; Aceña, J. L.; Soloshonok, V. A.; Sorochinsky, A. E. RSC Adv. 2013, 3, 6693–6716. doi:10.1039/c3ra22891f
Return to citation in text: [1] -
Cytlak, T.; Kaźmierczak, M.; Skibińska, M.; Koroniak, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 602–620. doi:10.1080/10426507.2017.1287706
Return to citation in text: [1] -
Song, B.-A.; Wu, Y.-L.; Yang, S.; Hu, D.-Y.; He, X.-Q.; Jin, L.-H. Molecules 2003, 8, 186–192. doi:10.3390/80100186
Return to citation in text: [1] -
Rao, X.; Song, Z.; He, L. Heteroat. Chem. 2008, 19, 512–516. doi:10.1002/hc.20471
Return to citation in text: [1] -
Herczegh, P.; Buxton, T. B.; McPherson, J. C.; Kovács-Kulyassa, Á.; Brewer, P. D.; Sztaricskai, F.; Stroebel, G. G.; Plowman, K. M.; Farcasiu, D.; Hartmann, J. F. J. Med. Chem. 2002, 45, 2338–2341. doi:10.1021/jm0105326
Return to citation in text: [1] -
Yang, S.; Gao, X.-W.; Diao, C.-L.; Song, B.-A.; Jin, L.-H.; Xu, G.-F.; Zhang, G.-P.; Wang, W.; Hu, D.-Y.; Xue, W.; Zhou, X.; Lu, P. Chin. J. Chem. 2006, 24, 1581–1588. doi:10.1002/cjoc.200690296
Return to citation in text: [1] -
Berkowitz, D. B.; Bose, M. J. Fluorine Chem. 2001, 112, 13–33. doi:10.1016/s0022-1139(01)00478-x
Return to citation in text: [1] -
Berkowitz, D. B.; Bose, M.; Pfannenstiel, T. J.; Doukov, T. J. Org. Chem. 2000, 65, 4498–4508. doi:10.1021/jo000220v
Return to citation in text: [1] -
Nieschalk, J.; Batsanov, A. S.; O'Hagan, D.; Howard, J. Tetrahedron 1996, 52, 165–176. doi:10.1016/0040-4020(95)00890-k
Return to citation in text: [1] -
Nieschalk, J.; O'Hagan, D. J. Chem. Soc., Chem. Commun. 1995, 719–720. doi:10.1039/c39950000719
Return to citation in text: [1] -
Tarasenko, K. V.; Romanenko, V. D.; Sorochinsky, A. E. J. Fluorine Chem. 2018, 211, 124–128. doi:10.1016/j.jfluchem.2018.04.014
Return to citation in text: [1] -
Berkowitz, D. B.; Bose, M.; Asher, N. G. Org. Lett. 2001, 3, 2009–2012. doi:10.1021/ol015983z
Return to citation in text: [1] -
McKenna, C. E.; Shen, P.-D. J. Org. Chem. 1981, 46, 4573–4576. doi:10.1021/jo00335a053
Return to citation in text: [1] -
Blackburn, G. M.; Kent, D. E. J. Chem. Soc., Chem. Commun. 1981, 511–513. doi:10.1039/c39810000511
Return to citation in text: [1] -
Blackburn, G. M.; England, D. A.; Kolkmann, F. J. Chem. Soc., Chem. Commun. 1981, 930–932. doi:10.1039/c39810000930
Return to citation in text: [1] -
Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386
Return to citation in text: [1] -
Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a
Return to citation in text: [1] -
Hu, W.-L.; Hu, X.-G.; Hunter, L. Synthesis 2017, 49, 4917–4930. doi:10.1055/s-0036-1590881
Return to citation in text: [1] -
Nielsen, M. K.; Ugaz, C. R.; Li, W.; Doyle, A. G. J. Am. Chem. Soc. 2015, 137, 9571–9574. doi:10.1021/jacs.5b06307
Return to citation in text: [1] [2] -
Nielsen, M. K.; Ahneman, D. T.; Riera, O.; Doyle, A. G. J. Am. Chem. Soc. 2018, 140, 5004–5008. doi:10.1021/jacs.8b01523
Return to citation in text: [1] [2] -
Bennua-Skalmowski, B.; Vorbrüggen, H. Tetrahedron Lett. 1995, 36, 2611–2614. doi:10.1016/0040-4039(95)00355-g
Return to citation in text: [1] -
Vorbrüggen, H. Synthesis 2008, 1165–1174. doi:10.1055/s-2008-1067006
Return to citation in text: [1] -
Guo, J.; Kuang, C.; Rong, J.; Li, L.; Ni, C.; Hu, J. Chem. – Eur. J. 2019, 25, 7259–7264. doi:10.1002/chem.201901176
Return to citation in text: [1] -
Montalbetti, C. A. G. N.; Falque, V. Tetrahedron 2005, 61, 10827–10852. doi:10.1016/j.tet.2005.08.031
Return to citation in text: [1] -
Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606–631. doi:10.1039/b701677h
Return to citation in text: [1] -
Pattabiraman, V. R.; Bode, J. W. Nature 2011, 480, 471–479. doi:10.1038/nature10702
Return to citation in text: [1] -
El-Faham, A.; Albericio, F. Chem. Rev. 2011, 111, 6557–6602. doi:10.1021/cr100048w
Return to citation in text: [1] -
Albericio, F.; El-Faham, A. Org. Process Res. Dev. 2018, 22, 760–772. doi:10.1021/acs.oprd.8b00159
Return to citation in text: [1] -
Kaźmierczak, M.; Koroniak, H. J. Fluorine Chem. 2012, 139, 23–27. doi:10.1016/j.jfluchem.2012.03.016
Return to citation in text: [1] [2] [3] -
Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631
Return to citation in text: [1] [2] [3] [4] -
Pudovik, A. N.; Konovalova, I. V. Synthesis 1979, 81–96. doi:10.1055/s-1979-28566
Return to citation in text: [1] -
Rádai, Z.; Keglevich, G. Molecules 2018, 23, 1493. doi:10.3390/molecules23061493
Return to citation in text: [1] -
Bailey, P. D.; Beard, M. A.; Dang, H. P. T.; Phillips, T. R.; Price, R. A.; Whittaker, J. H. Tetrahedron Lett. 2008, 49, 2150–2153. doi:10.1016/j.tetlet.2008.01.104
Return to citation in text: [1] -
Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008
Return to citation in text: [1] [2] [3] [4] -
Kafarski, P.; Lejczak, B. Synthesis 1988, 307–310. doi:10.1055/s-1988-27550
Return to citation in text: [1] -
Vaidhyanathan, R.; Natarajan, S.; Rao, C. N. R. J. Mol. Struct. 2002, 608, 123–133. doi:10.1016/s0022-2860(01)00944-9
Return to citation in text: [1] -
Spingler, B.; Schnidrig, S.; Todorova, T.; Wild, F. CrystEngComm 2012, 14, 751–757. doi:10.1039/c1ce05624g
Return to citation in text: [1] -
Sheehan, J. C.; Hess, G. P. J. Am. Chem. Soc. 1955, 77, 1067–1068. doi:10.1021/ja01609a099
Return to citation in text: [1] -
König, W.; Geiger, R. Chem. Ber. 1970, 103, 788–798. doi:10.1002/cber.19701030319
Return to citation in text: [1] -
Sheehan, J.; Cruickshank, P.; Boshart, G. J. Org. Chem. 1961, 26, 2525–2528. doi:10.1021/jo01351a600
Return to citation in text: [1] -
Wehrstedt, K. D.; Wandrey, P. A.; Heitkamp, D. J. Hazard. Mater. 2005, 126, 1–7. doi:10.1016/j.jhazmat.2005.05.044
Return to citation in text: [1] -
Jad, Y. E.; Khattab, S. N.; de la Torre, B. G.; Govender, T.; Kruger, H. G.; El-Faham, A.; Albericio, F. Eur. J. Org. Chem. 2015, 3116–3120. doi:10.1002/ejoc.201500142
Return to citation in text: [1] -
Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Chem. – Eur. J. 2009, 15, 9394–9403. doi:10.1002/chem.200900614
Return to citation in text: [1] -
El-Faham, A.; Funosas, R. S.; Prohens, R.; Albericio, F. Chem. – Eur. J. 2009, 15, 9404–9416. doi:10.1002/chem.200900615
Return to citation in text: [1] -
CrysAlis PRO, version 1.171.38.41; Rigaku Oxford Diffraction, 2015.
Return to citation in text: [1] -
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930
Return to citation in text: [1] -
Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191, 459–468. doi:10.1080/10426507.2015.1091832
Return to citation in text: [1]
| 44. | Subirós-Funosas, R.; Prohens, R.; Barbas, R.; El-Faham, A.; Albericio, F. Chem. – Eur. J. 2009, 15, 9394–9403. doi:10.1002/chem.200900614 |
| 45. | El-Faham, A.; Funosas, R. S.; Prohens, R.; Albericio, F. Chem. – Eur. J. 2009, 15, 9404–9416. doi:10.1002/chem.200900615 |
| 1. | Romanenko, V. D.; Kukhar, V. P. Chem. Rev. 2006, 106, 3868–3935. doi:10.1021/cr051000q |
| 2. | Turcheniuk, K. V.; Kukhar, V. P.; Röschenthaler, G.-V.; Aceña, J. L.; Soloshonok, V. A.; Sorochinsky, A. E. RSC Adv. 2013, 3, 6693–6716. doi:10.1039/c3ra22891f |
| 3. | Cytlak, T.; Kaźmierczak, M.; Skibińska, M.; Koroniak, H. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 602–620. doi:10.1080/10426507.2017.1287706 |
| 8. | Berkowitz, D. B.; Bose, M. J. Fluorine Chem. 2001, 112, 13–33. doi:10.1016/s0022-1139(01)00478-x |
| 30. | Kaźmierczak, M.; Koroniak, H. J. Fluorine Chem. 2012, 139, 23–27. doi:10.1016/j.jfluchem.2012.03.016 |
| 7. | Yang, S.; Gao, X.-W.; Diao, C.-L.; Song, B.-A.; Jin, L.-H.; Xu, G.-F.; Zhang, G.-P.; Wang, W.; Hu, D.-Y.; Xue, W.; Zhou, X.; Lu, P. Chin. J. Chem. 2006, 24, 1581–1588. doi:10.1002/cjoc.200690296 |
| 33. | Rádai, Z.; Keglevich, G. Molecules 2018, 23, 1493. doi:10.3390/molecules23061493 |
| 6. | Herczegh, P.; Buxton, T. B.; McPherson, J. C.; Kovács-Kulyassa, Á.; Brewer, P. D.; Sztaricskai, F.; Stroebel, G. G.; Plowman, K. M.; Farcasiu, D.; Hartmann, J. F. J. Med. Chem. 2002, 45, 2338–2341. doi:10.1021/jm0105326 |
| 30. | Kaźmierczak, M.; Koroniak, H. J. Fluorine Chem. 2012, 139, 23–27. doi:10.1016/j.jfluchem.2012.03.016 |
| 31. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 35. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 4. | Song, B.-A.; Wu, Y.-L.; Yang, S.; Hu, D.-Y.; He, X.-Q.; Jin, L.-H. Molecules 2003, 8, 186–192. doi:10.3390/80100186 |
| 5. | Rao, X.; Song, Z.; He, L. Heteroat. Chem. 2008, 19, 512–516. doi:10.1002/hc.20471 |
| 32. | Pudovik, A. N.; Konovalova, I. V. Synthesis 1979, 81–96. doi:10.1055/s-1979-28566 |
| 35. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 20. | Nielsen, M. K.; Ugaz, C. R.; Li, W.; Doyle, A. G. J. Am. Chem. Soc. 2015, 137, 9571–9574. doi:10.1021/jacs.5b06307 |
| 21. | Nielsen, M. K.; Ahneman, D. T.; Riera, O.; Doyle, A. G. J. Am. Chem. Soc. 2018, 140, 5004–5008. doi:10.1021/jacs.8b01523 |
| 24. | Guo, J.; Kuang, C.; Rong, J.; Li, L.; Ni, C.; Hu, J. Chem. – Eur. J. 2019, 25, 7259–7264. doi:10.1002/chem.201901176 |
| 31. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 17. | Ni, C.; Hu, M.; Hu, J. Chem. Rev. 2015, 115, 765–825. doi:10.1021/cr5002386 |
| 18. | Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a |
| 19. | Hu, W.-L.; Hu, X.-G.; Hunter, L. Synthesis 2017, 49, 4917–4930. doi:10.1055/s-0036-1590881 |
| 25. | Montalbetti, C. A. G. N.; Falque, V. Tetrahedron 2005, 61, 10827–10852. doi:10.1016/j.tet.2005.08.031 |
| 26. | Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606–631. doi:10.1039/b701677h |
| 27. | Pattabiraman, V. R.; Bode, J. W. Nature 2011, 480, 471–479. doi:10.1038/nature10702 |
| 28. | El-Faham, A.; Albericio, F. Chem. Rev. 2011, 111, 6557–6602. doi:10.1021/cr100048w |
| 29. | Albericio, F.; El-Faham, A. Org. Process Res. Dev. 2018, 22, 760–772. doi:10.1021/acs.oprd.8b00159 |
| 35. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
| 12. | Tarasenko, K. V.; Romanenko, V. D.; Sorochinsky, A. E. J. Fluorine Chem. 2018, 211, 124–128. doi:10.1016/j.jfluchem.2018.04.014 |
| 13. | Berkowitz, D. B.; Bose, M.; Asher, N. G. Org. Lett. 2001, 3, 2009–2012. doi:10.1021/ol015983z |
| 14. | McKenna, C. E.; Shen, P.-D. J. Org. Chem. 1981, 46, 4573–4576. doi:10.1021/jo00335a053 |
| 15. | Blackburn, G. M.; Kent, D. E. J. Chem. Soc., Chem. Commun. 1981, 511–513. doi:10.1039/c39810000511 |
| 16. | Blackburn, G. M.; England, D. A.; Kolkmann, F. J. Chem. Soc., Chem. Commun. 1981, 930–932. doi:10.1039/c39810000930 |
| 47. | Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. doi:10.1107/s0108767307043930 |
| 9. | Berkowitz, D. B.; Bose, M.; Pfannenstiel, T. J.; Doukov, T. J. Org. Chem. 2000, 65, 4498–4508. doi:10.1021/jo000220v |
| 10. | Nieschalk, J.; Batsanov, A. S.; O'Hagan, D.; Howard, J. Tetrahedron 1996, 52, 165–176. doi:10.1016/0040-4020(95)00890-k |
| 11. | Nieschalk, J.; O'Hagan, D. J. Chem. Soc., Chem. Commun. 1995, 719–720. doi:10.1039/c39950000719 |
| 22. | Bennua-Skalmowski, B.; Vorbrüggen, H. Tetrahedron Lett. 1995, 36, 2611–2614. doi:10.1016/0040-4039(95)00355-g |
| 23. | Vorbrüggen, H. Synthesis 2008, 1165–1174. doi:10.1055/s-2008-1067006 |
| 30. | Kaźmierczak, M.; Koroniak, H. J. Fluorine Chem. 2012, 139, 23–27. doi:10.1016/j.jfluchem.2012.03.016 |
| 31. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 48. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Phosphorus, Sulfur Silicon Relat. Elem. 2016, 191, 459–468. doi:10.1080/10426507.2015.1091832 |
| 31. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. Eur. J. Org. Chem. 2018, 3844–3852. doi:10.1002/ejoc.201800631 |
| 20. | Nielsen, M. K.; Ugaz, C. R.; Li, W.; Doyle, A. G. J. Am. Chem. Soc. 2015, 137, 9571–9574. doi:10.1021/jacs.5b06307 |
| 21. | Nielsen, M. K.; Ahneman, D. T.; Riera, O.; Doyle, A. G. J. Am. Chem. Soc. 2018, 140, 5004–5008. doi:10.1021/jacs.8b01523 |
| 42. | Wehrstedt, K. D.; Wandrey, P. A.; Heitkamp, D. J. Hazard. Mater. 2005, 126, 1–7. doi:10.1016/j.jhazmat.2005.05.044 |
| 43. | Jad, Y. E.; Khattab, S. N.; de la Torre, B. G.; Govender, T.; Kruger, H. G.; El-Faham, A.; Albericio, F. Eur. J. Org. Chem. 2015, 3116–3120. doi:10.1002/ejoc.201500142 |
| 40. | König, W.; Geiger, R. Chem. Ber. 1970, 103, 788–798. doi:10.1002/cber.19701030319 |
| 41. | Sheehan, J.; Cruickshank, P.; Boshart, G. J. Org. Chem. 1961, 26, 2525–2528. doi:10.1021/jo01351a600 |
| 37. | Vaidhyanathan, R.; Natarajan, S.; Rao, C. N. R. J. Mol. Struct. 2002, 608, 123–133. doi:10.1016/s0022-2860(01)00944-9 |
| 38. | Spingler, B.; Schnidrig, S.; Todorova, T.; Wild, F. CrystEngComm 2012, 14, 751–757. doi:10.1039/c1ce05624g |
| 39. | Sheehan, J. C.; Hess, G. P. J. Am. Chem. Soc. 1955, 77, 1067–1068. doi:10.1021/ja01609a099 |
| 34. | Bailey, P. D.; Beard, M. A.; Dang, H. P. T.; Phillips, T. R.; Price, R. A.; Whittaker, J. H. Tetrahedron Lett. 2008, 49, 2150–2153. doi:10.1016/j.tetlet.2008.01.104 |
| 35. | Kaźmierczak, M.; Kubicki, M.; Koroniak, H. J. Fluorine Chem. 2014, 167, 128–134. doi:10.1016/j.jfluchem.2014.06.008 |
© 2020 Kaźmierczak and Koroniak; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)