Abstract
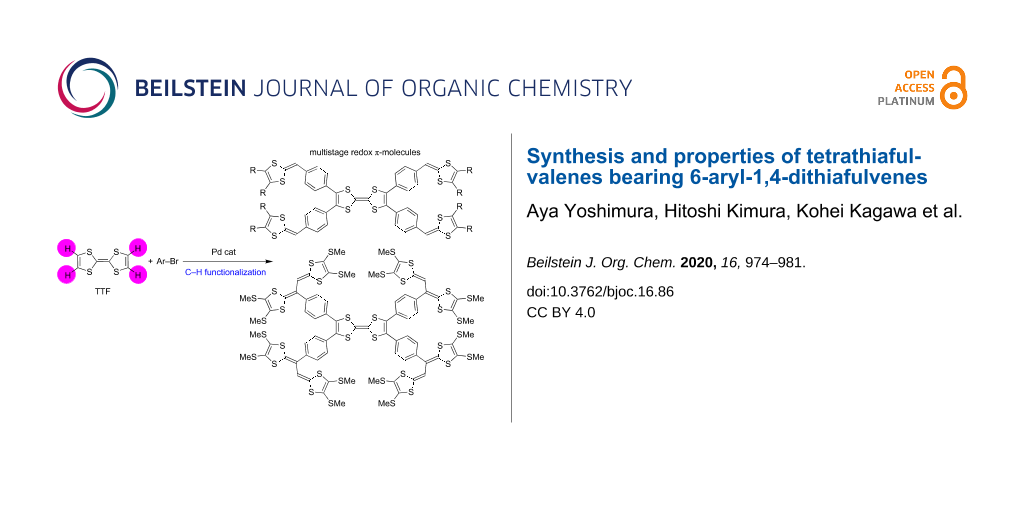
Novel multistage redox tetrathiafulvalenes (TTFs) bearing 6-aryl-1,4-dithiafulvene moieties were synthesized by palladium-catalyzed direct C–H arylation. In the presence of a catalytic amount of Pd(OAc)2, P(t-Bu3)·HBF4, and an excess of Cs2CO3, the C–H arylation of TTF with several aryl bromides bearing 1,3-dithiol-2-ylidenes took place efficiently to produce the corresponding π-conjugated molecules. We also succeeded in the estimation of the oxidation potentials and number of electrons involved in each oxidation step of the obtained compounds by digital simulations.

Graphical Abstract
Introduction
Tetrathiafulvalenes (TTFs) with extended π-conjugation have attracted attention as possible components of functional materials, such as molecular conductors, field-effect transistors (FETs), and positive electrode materials for rechargeable batteries because the TTF moiety has strong electron-donating properties attributed to the formation of stable aromatic 1,3-dithiol-2-ylidenes (1,3-dithiole rings) by one- and two-electron oxidation [1-16]. Considerable efforts have been devoted to the development of peripherally benzene- or thiophene-substituted TTFs. As for peripherally benzene-functionalized TTFs, some synthetic approaches, crystal and electronic structures, electrochemical and optical properties, and the nature of radical ion complexes with DDQ or iodine were reported [17-24]. Peripherally thiophene-functionalized TTFs, as potential precursors to conducting polymers, and organic metals were also prepared and characterized [25-29]. To design more tempting molecules, the attachment of 1,3-dithiole rings to aromatic rings appears very appealing since these allow to produce novel multistage redox systems. However, such molecules could formerly not be synthesized by conventional approaches. In 2011, a breakthrough synthesis of arylated TTF derivatives by a palladium-catalyzed direct C–H arylation was reported, and the structural and electrochemical properties of the products were clarified [30]. This motivated us to synthesize novel multistage redox-TTFs bearing 1,3-dithiole rings on aromatic rings, 1–3 (Figure 1). In addition, we focused on cross-conjugated systems with 1,3-dithiole rings, which are of interest as novel multistage redox systems as well as donor components for organic conductors [1,31-41]. The palladium-catalyzed C–H arylation might offer access to new cross-conjugated molecules bearing vinyl-extended TTF moieties (EBDTs), such as 4 (Figure 1), and the electrochemical properties of these representatives should be brought to light. Herein, we report the synthesis and electrochemical properties of tetrathiafulvalene derivatives 1–4.
![[1860-5397-16-86-1]](/bjoc/content/figures/1860-5397-16-86-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Results and Discussion
Synthesis
First, we tried to synthesize compounds 1 and 2 in one step from pristine TTF and 5, respectively, through palladium-catalyzed C–H arylation (Table 1). When the aryl bromides 6a,b were allowed to react with TTF under the conditions A, the products 1a,b were produced in 46 and 48% yields, respectively (Table 1, entries 1 and 2). Attempted isolations of products 1c and 1d failed, despite complete conversions of TTF, because the polarities of the mono-, di-, and triarylated TTFs were extremely close to that of the tetraarylated TTF 1c and the solubility of these compounds were low and almost beyond isolation for 1d (Table 1, entries 3 and 4). The palladium-catalyzed C–H arylation of 5 with 6a,b proceeded to give products 2a,b in 75 and 86% yields, respectively (Table 1, entries 5 and 6). On the other hand, it was difficult to produce 3 in the same manner because 2-bromothiophenes 7 bearing a 1,3-dithiole ring at the 5-position were unstable, for example, 7a decomposed at around 41–42 °C (Scheme 1a). Therefore, we achieved the synthesis of 3a by Pd-catalyzed thienylation of TTF using acetal-protected 8, followed by deprotection using PTSA·H2O and P(OEt)3-mediated cross coupling with 11 (Scheme 1b). The cross-conjugated molecule 4 was prepared in two procedures; one was the palladium-catalyzed C–H arylation of TTF with bromide 12 (Scheme 2a) and the other was the Vilsmeier–Haack reaction of 1a, followed by triethyl phosphite-mediated cross coupling with 11 (Scheme 2b).
Table 1: Synthesis of compounds 1 and 2.
![[Graphic 1]](/bjoc/content/inline/1860-5397-16-86-i3.svg?max-width=637&scale=1.0)
|
||||
| entry | TTF or 5 | 6 (equiv) | conditions | yield of 1 or 2 (%) |
| 1 | TTF | 6a (5) | A | 1a: 46 |
| 2 | TTF | 6b (5) | A | 1b: 48 |
| 3 | TTF | 6c (5) | A or B | 1c: 0 (mixture) |
| 4 | TTF | 6d (5) | A or B | 1d: 0 (mixture) |
| 5 | 5 | 6a (2.5) | Aa | 2a: 75 |
| 6 | 5 | 6b (2.5) | Aa | 2b: 86 |
aReaction time 24 h.
![[1860-5397-16-86-i1]](/bjoc/content/inline/1860-5397-16-86-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-16-86-i2]](/bjoc/content/inline/1860-5397-16-86-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Theoretical calculations
The DFT calculations of compounds 1a, 3a, and 4 have been carried out by using the B3LYP/6-31G(d,p) method [42]. Figure 2 shows an optimized geometry of 1a, and the 1,3-dithiole rings are labeled as A–E and A'–E'. This molecule adopted a nonplanar structure. The angle between the two 1,3-dithiole rings in the center (A–A') was 158.0°. The dihedral angles between A and B, A and B', A' and C, and A' and C' were 49.8°, 137.7°, 137.7°, and 49.9°, respectively. The HOMO, HOMO−1, and LUMO of 1a are shown in Figure 3. The HOMO of 1a was mainly located on the TTF moiety, whereas the HOMO−1 was located on the benzene ring and the outer 1,3-dithiole rings at the peripheral part of TTF. The LUMO of 1a spread over the whole molecule except the outer 1,3-dithiole rings. The orbital energy of the HOMO of 1a (−4.41 eV) was slightly higher than that of TTF (−4.57 eV). As such, the first oxidation of 1a might occur at a lower potential than TTF [43].
![[1860-5397-16-86-2]](/bjoc/content/figures/1860-5397-16-86-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: An optimized structure of 1a. a) Top view, b) side view, and c) labeling of the 1,3-dithiole rings.
Figure 2: An optimized structure of 1a. a) Top view, b) side view, and c) labeling of the 1,3-dithiole rings.
![[1860-5397-16-86-3]](/bjoc/content/figures/1860-5397-16-86-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Cyclic voltammetry analysis
The redox behavior of 1–4 was investigated by cyclic voltammetry. The compounds 1a and 1b exhibited four and three pairs of redox waves, respectively (around +0.03, +0.10, +0.17, and +0.42 V vs Fc/Fc+ for 1a and −0.05, +0.10, and +0.46 V vs Fc/Fc+ for 1b, Figure 4). The redox potentials of 1a,b are summarized in Table 2 together with the related compound TTF. The redox waves observed at +0.42 V for 1a and +0.46 V for 1b were likely related to the second redox of the central TTF moiety because they were close to the E2 of TTF (+0.37 V). The remaining redox processes observed at around +0.03, +0.10, and +0.17 V for 1a, and −0.05 and +0.10 V for 1b might have been derived from the first redox of the central TTF moiety and the redox of the four outer 1,3-dithiole rings. To elaborate the exact oxidation potentials and the number of electrons involved in each oxidation step, a digital simulation technique was applied [44]. As a result, the observed redox waves of 1a matched the simulated waves (Table 2). It was indicated that the redox wave at +0.10 V was due to an overlap of the sequential two stages of the one- and two-electron transfer waves at +0.07 and +0.12 V, while the other waves corresponded to one-electron transfer processes. The simulation results of 1a also showed that the redox wave simulated at +0.020 V might have been derived from the central TTF moiety because of the close ΔE values (+0.40 V for 1a and +0.46 V for TTF). The same discussion was applied to 1b. In addition, the potentials related to the outer 1,3-dithiole rings of 1a,b were influenced by the substituents, that is, 1b bearing electron-donating methyl groups had more negative redox potentials than 1a. As a consequence, the one-electron redox process of the TTF moiety and the multi-electron redox processes of the outer 1,3-dithiole rings might have overlapped in 1b.
![[1860-5397-16-86-4]](/bjoc/content/figures/1860-5397-16-86-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Cyclic voltammograms of 1a,b, 2a, and 4 in PhCN/CS2 1:1 (v/v) solution.
Figure 4: Cyclic voltammograms of 1a,b, 2a, and 4 in PhCN/CS2 1:1 (v/v) solution.
Table 2: Redox potentials of 1, 2a, 4, and related compoundsa.
| compound | observed or simulated value | E1 | E2 | E3 | E4 | E5 | E6 | E7 | E8 | E9 | E10 |
| 1a | observed | around +0.03b | +0.10 | +0.17 | +0.42 | ||||||
| simulated | +0.020 | +0.070 | +0.120 | +0.200 | +0.420 | ||||||
| 1b | observed | −0.05 | +0.10 | +0.46b | |||||||
| 2a | observed | −0.05 | +0.09 | +0.49 | |||||||
| 4 | observed | around −0.09 | +0.09 | +0.53b | |||||||
| simulated | −0.060 | −0.030 | +0.010 | +0.047 | +0.053 | +0.098 | +0.102 | +0.110 | +0.180 | +0.500 | |
| TTFc | observed | −0.09 | +0.37 | ||||||||
| 5c | observed | −0.01 | +0.42 | ||||||||
| 14c | observed | −0.07 | +0.09 | ||||||||
aIn PhCN/CS2 1:1 (v/v) containing 0.1 M n-Bu4NPF6. bAnodic peak. cIn PhCN containing 0.1 M n-Bu4NPF6. All potentials were measured against an Ag/Ag+ reference electrode and converted to vs Fc/Fc+.
Compound 2a exhibited three pairs of redox waves (−0.05, +0.09, and +0.49 V vs Fc/Fc+). The redox waves observed at −0.05 and +0.49 V were likely related to the TTF derivative (5) moiety, because they were close to the E1 and E2 of 5, respectively (Table 2). The comparison of the peak currents of each wave indicated that the redox wave observed at +0.09 V involved a two-electron transfer, while the redox waves observed at −0.05 and +0.49 V corresponded to one-electron transfer processes (see the differential pulse voltammetry (DPV) in Supporting Information File 1, Figure S2). These results supported the above-mentioned oxidation potentials and the number of electrons involved in each oxidation step of 1a,b.
Compound 4 exhibited three pairs of redox waves (around −0.09, +0.09, and +0.53 V vs Fc/Fc+). The redox potentials of 4 and the simulation results are also summarized in Table 2, together with those of the related compounds TTF and 14 (see Figure 5). The redox process observed at +0.53 V was likely related to the second redox of the central TTF moiety because this was the closest to the value of the E2 of TTF (+0.37 V) out of all potentials of the related compounds TTF and 14. The remaining redox processes observed at around −0.09 and +0.09 V might have been due to the first redox of the central TTF moiety, and the overall redox of the EBDT sites, respectively. The observed potentials of 4 were generally consistent with the simulated ones. The results of a digital simulation also showed that the redox wave observed at around −0.09 V and +0.09 V corresponded to three stages of one-electron transfer and six stages of one-electron transfer processes, respectively. In addition to the overlap of the first redox of the central TTF moiety and the redox of the EBDT sites, each redox potential of the succeeding eight-electron oxidations of the EBDT sites might have slightly shifted due to the non-equivalence of them. Also, the small ΔE value (0.16 V) of 14 caused the redox wave overlap. For these reasons, the first and second redox waves of 4 were broad compared to those of 1a and 1b.
![[1860-5397-16-86-5]](/bjoc/content/figures/1860-5397-16-86-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
The redox waves of 1a,b and 4 derived from the second redox of the central TTF moiety (+0.42 V for 1a, +0.46 V for 1b, and +0.53 V for 4) shifted to higher potentials than the second redox of TTF because of the instability of the hexacationic state of 1a,b, and the decacationic state of 4 compared to the dicationic states of TTF caused by on-site Coulomb repulsion between positive charges in the central TTF moiety and the outer 1,3-dithiole rings. The same discussion could be applied to compounds 2a. In addition, the observed peak currents of 1a and 4 in the high potential region at +0.4 to +0.5 V were smaller than those of the simulated waves. This phenomenon might be understood by considering that electron transfer of this redox reaction was slow enough to become the rate-determining step because the crowded structure by which the TTF core is participating in this redox process is surrounded by extended aromatic rings bearing 1,3-dithiol rings.
Conclusion
We have synthesized novel multistage TTF derivatives 1–4 bearing 6-aryl-1,4-dithiafulvene moieties by palladium-catalyzed direct C–H arylation. The DFT calculations revealed the nonplanar structure of the compounds. Cyclic voltammograms of 1a and 4 comprised four and three pairs of redox waves, respectively. As a result of the digital simulation of 1a, it was shown that the redox wave observed at +0.10 V involved two stages of one- and two-electron transfer(s), while the other redox waves corresponded to one-electron transfer. The digital simulation of 4 showed 10 stages of one-electron transfer in total. In addition, the first and second redox waves of 4 were broad compared to those of 1 owing to the following three reasons: a) overlap of the central TTF moiety and the redox of the EBDT sites, b) the succeeding eight-electron oxidations of the non-equivalent EBDT sites, and c) the small ΔE value (0.16 V) of the EBDT sites.
Supporting Information
| Supporting Information File 1: Synthetic procedures, theoretical chemical and electrochemical details, and copies of NMR spectra. | ||
| Format: PDF | Size: 6.2 MB | Download |
Funding
This work was partially supported by a Grant-in-Aid for Scientific Research (JP15H03798), from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). This work was also supported by a Grant-in-Aid for Research Promotion, Ehime University, to The Research Unit for Development of Organic Superconductors and to The Research Unit for Power Generation and Storage Materials.
References
-
Yamada, J.; Sugimoto, T., Eds. TTF Chemistry-Fundamental and Applications of Tetrathiafulvalene; Kodansha-Springer: Tokyo, 2004.
Return to citation in text: [1] [2] -
Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n
Return to citation in text: [1] -
Gorgues, A.; Hudhomme, P.; Sallé, M. Chem. Rev. 2004, 104, 5151–5184. doi:10.1021/cr0306485
Return to citation in text: [1] -
Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::aid-anie1372>3.0.co;2-i
Return to citation in text: [1] -
Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5114. doi:10.1021/cr030651o
Return to citation in text: [1] -
Hasegawa, M.; Iyoda, M. Tetrathiafulvalene: A Redox Unit for Functional Materials and a Building Block for Supramolecular Self-Assembly. In Organic Redox Systems; Nishinaga, T., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp 89–129. doi:10.1002/9781118858981.ch4
Return to citation in text: [1] -
Misaki, Y. Sci. Technol. Adv. Mater. 2009, 10, 024301. doi:10.1088/1468-6996/10/2/024301
Return to citation in text: [1] -
Inatomi, Y.; Hojo, N.; Yamamoto, T.; Shimada, M.; Watanabe, S. Examination of Multi-Electron Reaction Type pi-Conjugated Organic Compounds as Cathode Active Material for Rechargeable Power Supply. In ECS Meeting Abstracts, Volume MA2008-01, B1- Batteries General Session, Abstract #167, 213th ECS meeting, Phoenix, Arizona, May 18–22, 2008; The Electrochemical Society, 2008. doi:10.1149/ma2008-01/5/167
Return to citation in text: [1] -
Inatomi, Y.; Hojo, N.; Yamamoto, T.; Watanabe, S.-i.; Misaki, Y. ChemPlusChem 2012, 77, 973–976. doi:10.1002/cplu.201200197
Return to citation in text: [1] -
Kato, M.; Ogi, D.; Yao, M.; Misaki, Y. Chem. Lett. 2013, 42, 1556–1558. doi:10.1246/cl.130841
Return to citation in text: [1] -
Kato, M.; Senoo, K.-i.; Yao, M.; Misaki, Y. J. Mater. Chem. A 2014, 2, 6747–6754. doi:10.1039/c3ta14920j
Return to citation in text: [1] -
Iwamoto, S.; Inatomi, Y.; Ogi, D.; Shibayama, S.; Murakami, Y.; Kato, M.; Takahashi, K.; Tanaka, K.; Hojo, N.; Misaki, Y. Beilstein J. Org. Chem. 2015, 11, 1136–1147. doi:10.3762/bjoc.11.128
Return to citation in text: [1] -
Yamauchi, T.; Shibata, Y.; Aki, T.; Yoshimura, A.; Yao, M.; Misaki, Y. Chem. Lett. 2018, 47, 1176–1179. doi:10.1246/cl.180496
Return to citation in text: [1] -
Ogi, D.; Fujita, Y.; Kato, M.; Yamauchi, T.; Shirahata, T.; Yao, M.; Misaki, Y. Eur. J. Org. Chem. 2019, 2725–2728. doi:10.1002/ejoc.201801877
Return to citation in text: [1] -
Yamauchi, T.; Kato, M.; Shirahata, T.; Yao, M.; Misaki, Y. Chem. Lett. 2019, 48, 1507–1510. doi:10.1246/cl.190703
Return to citation in text: [1] -
Bryce, M. R. J. Mater. Chem. 2000, 10, 589–598. doi:10.1039/a908385e
Return to citation in text: [1] -
Coffen, D. L.; Chambers, J. Q.; Williams, D. R.; Garrett, P. E.; Canfield, N. D. J. Am. Chem. Soc. 1971, 93, 2258–2268. doi:10.1021/ja00738a028
Return to citation in text: [1] -
Ueno, Y.; Okawara, M. Chem. Lett. 1974, 3, 1135–1138. doi:10.1246/cl.1974.1135
Return to citation in text: [1] -
Ueno, Y.; Nakayama, A.; Okawara, M. J. Am. Chem. Soc. 1976, 98, 7440–7441. doi:10.1021/ja00439a064
Return to citation in text: [1] -
Yoneda, S.; Kawase, T.; Inaba, M.; Yoshida, Z.-i. J. Org. Chem. 1978, 43, 595–598. doi:10.1021/jo00398a015
Return to citation in text: [1] -
Le Coustumer, G.; Mollier, Y. J. Chem. Soc., Chem. Commun. 1980, 38–39. doi:10.1039/c39800000038
Return to citation in text: [1] -
Babeau, A.; Tinh, N. H.; Gasparoux, H.; Polycarpe, C.; Torreilles, E.; Giral, L. Mol. Cryst. Liq. Cryst. 1982, 72, 171–176. doi:10.1080/01406568208084054
Return to citation in text: [1] -
Starodub, V. A.; Baumer, V. N.; Guella, I. M.; Golovkina, I. F.; Alyoshin, V. G.; Nemoshkalenko, V. V.; Senkiewicz, A. I. Synth. Met. 1983, 5, 101–111. doi:10.1016/0379-6779(83)90124-8
Return to citation in text: [1] -
Tsujimoto, K.; Okeda, Y.; Ohashi, M. J. Chem. Soc., Chem. Commun. 1985, 1803–1804. doi:10.1039/c39850001803
Return to citation in text: [1] -
Charlton, A.; Underhill, A. E.; Williams, G.; Kalaji, M.; Murphy, P. J.; Hibbs, D. E.; Hursthouse, M. B.; Malik, K. M. A. Chem. Commun. 1996, 2423–2424. doi:10.1039/cc9960002423
Return to citation in text: [1] -
Charlton, A.; Underhill, A. E.; Williams, G.; Kalaji, M.; Murphy, P. J.; Malik, K. M. A.; Hursthouse, M. B. J. Org. Chem. 1997, 62, 3098–3102. doi:10.1021/jo962301q
Return to citation in text: [1] -
Gompper, R.; Hock, J. Synth. Met. 1997, 84, 339–340. doi:10.1016/s0379-6779(97)80771-0
Return to citation in text: [1] -
Zotti, G.; Zecchin, S.; Schiavon, G.; Berlin, A.; Huchet, L.; Roncali, J. J. Electroanal. Chem. 2001, 504, 64–70. doi:10.1016/s0022-0728(01)00429-6
Return to citation in text: [1] -
Kakinuma, T.; Kojima, H.; Kawamoto, T.; Mori, T. J. Mater. Chem. C 2013, 1, 2900–2905. doi:10.1039/c3tc30089g
Return to citation in text: [1] -
Mitamura, Y.; Yorimitsu, H.; Oshima, K.; Osuka, A. Chem. Sci. 2011, 2, 2017–2021. doi:10.1039/c1sc00372k
Return to citation in text: [1] -
Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573
Return to citation in text: [1] -
Sugimoto, T.; Awaji, H.; Misaki, Y.; Yoshida, Z.-i.; Kai, Y.; Nakagawa, H.; Kasai, N. J. Am. Chem. Soc. 1985, 107, 5792–5793. doi:10.1021/ja00306a030
Return to citation in text: [1] -
Sugimoto, T.; Misaki, Y.; Arai, Y.; Yamamoto, Y.; Yoshida, Z.-i.; Kai, Y.; Kasai, N. J. Am. Chem. Soc. 1988, 110, 628–629. doi:10.1021/ja00210a069
Return to citation in text: [1] -
Misaki, Y.; Matsumura, Y.; Sugimoto, T.; Yoshida, Z.-i. Tetrahedron Lett. 1989, 30, 5289–5292. doi:10.1016/s0040-4039(01)93767-0
Return to citation in text: [1] -
Amaresh, R. R.; Liu, D.; Konovalova, T.; Lakshmikantham, M. V.; Cava, M. P.; Kispert, L. D. J. Org. Chem. 2001, 66, 7757–7764. doi:10.1021/jo010663e
Return to citation in text: [1] -
Rajagopal, D.; Lakshmikantham, M. V.; Cava, M. P. Org. Lett. 2002, 4, 2581–2583. doi:10.1021/ol026227b
Return to citation in text: [1] -
Coffin, M. A.; Bryce, M. R.; Batsanov, A. S.; Howard, J. A. K. J. Chem. Soc., Chem. Commun. 1993, 552–554. doi:10.1039/c39930000552
Return to citation in text: [1] -
Bryce, M. R.; Coffin, M. A.; Skabara, P. J.; Moore, A. J.; Batsanov, A. S.; Howard, J. A. K. Chem. – Eur. J. 2000, 6, 1955–1962. doi:10.1002/1521-3765(20000602)6:11<1955::aid-chem1955>3.0.co;2-b
Return to citation in text: [1] -
Hasegawa, M.; Fujioka, A.; Kubo, T.; Honda, T.; Miyamoto, H.; Misaki, Y. Chem. Lett. 2008, 37, 474–475. doi:10.1246/cl.2008.474
Return to citation in text: [1] -
Horiuchi, H.; Misaki, Y. Chem. Lett. 2010, 39, 989–991. doi:10.1246/cl.2010.989
Return to citation in text: [1] -
Nishiwaki, M.; Tezuka, M.; Shirahata, T.; Misaki, Y. Chem. Lett. 2011, 40, 467–469. doi:10.1246/cl.2011.467
Return to citation in text: [1] -
Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, 2019.
Return to citation in text: [1] -
The optimized geometries and energy levels of the LUMO, HOMO, and HOMO−1 of 3a and 4 are shown in Supporting Information File 1, Figure S1.
Return to citation in text: [1] -
DigiElch 7 Prof software; Elchsoft: Kleinromstedt, Germany.
Return to citation in text: [1]
| 1. | Yamada, J.; Sugimoto, T., Eds. TTF Chemistry-Fundamental and Applications of Tetrathiafulvalene; Kodansha-Springer: Tokyo, 2004. |
| 2. | Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n |
| 3. | Gorgues, A.; Hudhomme, P.; Sallé, M. Chem. Rev. 2004, 104, 5151–5184. doi:10.1021/cr0306485 |
| 4. | Segura, J. L.; Martín, N. Angew. Chem., Int. Ed. 2001, 40, 1372–1409. doi:10.1002/1521-3773(20010417)40:8<1372::aid-anie1372>3.0.co;2-i |
| 5. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5114. doi:10.1021/cr030651o |
| 6. | Hasegawa, M.; Iyoda, M. Tetrathiafulvalene: A Redox Unit for Functional Materials and a Building Block for Supramolecular Self-Assembly. In Organic Redox Systems; Nishinaga, T., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp 89–129. doi:10.1002/9781118858981.ch4 |
| 7. | Misaki, Y. Sci. Technol. Adv. Mater. 2009, 10, 024301. doi:10.1088/1468-6996/10/2/024301 |
| 8. | Inatomi, Y.; Hojo, N.; Yamamoto, T.; Shimada, M.; Watanabe, S. Examination of Multi-Electron Reaction Type pi-Conjugated Organic Compounds as Cathode Active Material for Rechargeable Power Supply. In ECS Meeting Abstracts, Volume MA2008-01, B1- Batteries General Session, Abstract #167, 213th ECS meeting, Phoenix, Arizona, May 18–22, 2008; The Electrochemical Society, 2008. doi:10.1149/ma2008-01/5/167 |
| 9. | Inatomi, Y.; Hojo, N.; Yamamoto, T.; Watanabe, S.-i.; Misaki, Y. ChemPlusChem 2012, 77, 973–976. doi:10.1002/cplu.201200197 |
| 10. | Kato, M.; Ogi, D.; Yao, M.; Misaki, Y. Chem. Lett. 2013, 42, 1556–1558. doi:10.1246/cl.130841 |
| 11. | Kato, M.; Senoo, K.-i.; Yao, M.; Misaki, Y. J. Mater. Chem. A 2014, 2, 6747–6754. doi:10.1039/c3ta14920j |
| 12. | Iwamoto, S.; Inatomi, Y.; Ogi, D.; Shibayama, S.; Murakami, Y.; Kato, M.; Takahashi, K.; Tanaka, K.; Hojo, N.; Misaki, Y. Beilstein J. Org. Chem. 2015, 11, 1136–1147. doi:10.3762/bjoc.11.128 |
| 13. | Yamauchi, T.; Shibata, Y.; Aki, T.; Yoshimura, A.; Yao, M.; Misaki, Y. Chem. Lett. 2018, 47, 1176–1179. doi:10.1246/cl.180496 |
| 14. | Ogi, D.; Fujita, Y.; Kato, M.; Yamauchi, T.; Shirahata, T.; Yao, M.; Misaki, Y. Eur. J. Org. Chem. 2019, 2725–2728. doi:10.1002/ejoc.201801877 |
| 15. | Yamauchi, T.; Kato, M.; Shirahata, T.; Yao, M.; Misaki, Y. Chem. Lett. 2019, 48, 1507–1510. doi:10.1246/cl.190703 |
| 16. | Bryce, M. R. J. Mater. Chem. 2000, 10, 589–598. doi:10.1039/a908385e |
| 1. | Yamada, J.; Sugimoto, T., Eds. TTF Chemistry-Fundamental and Applications of Tetrathiafulvalene; Kodansha-Springer: Tokyo, 2004. |
| 31. | Gholami, M.; Tykwinski, R. R. Chem. Rev. 2006, 106, 4997–5027. doi:10.1021/cr0505573 |
| 32. | Sugimoto, T.; Awaji, H.; Misaki, Y.; Yoshida, Z.-i.; Kai, Y.; Nakagawa, H.; Kasai, N. J. Am. Chem. Soc. 1985, 107, 5792–5793. doi:10.1021/ja00306a030 |
| 33. | Sugimoto, T.; Misaki, Y.; Arai, Y.; Yamamoto, Y.; Yoshida, Z.-i.; Kai, Y.; Kasai, N. J. Am. Chem. Soc. 1988, 110, 628–629. doi:10.1021/ja00210a069 |
| 34. | Misaki, Y.; Matsumura, Y.; Sugimoto, T.; Yoshida, Z.-i. Tetrahedron Lett. 1989, 30, 5289–5292. doi:10.1016/s0040-4039(01)93767-0 |
| 35. | Amaresh, R. R.; Liu, D.; Konovalova, T.; Lakshmikantham, M. V.; Cava, M. P.; Kispert, L. D. J. Org. Chem. 2001, 66, 7757–7764. doi:10.1021/jo010663e |
| 36. | Rajagopal, D.; Lakshmikantham, M. V.; Cava, M. P. Org. Lett. 2002, 4, 2581–2583. doi:10.1021/ol026227b |
| 37. | Coffin, M. A.; Bryce, M. R.; Batsanov, A. S.; Howard, J. A. K. J. Chem. Soc., Chem. Commun. 1993, 552–554. doi:10.1039/c39930000552 |
| 38. | Bryce, M. R.; Coffin, M. A.; Skabara, P. J.; Moore, A. J.; Batsanov, A. S.; Howard, J. A. K. Chem. – Eur. J. 2000, 6, 1955–1962. doi:10.1002/1521-3765(20000602)6:11<1955::aid-chem1955>3.0.co;2-b |
| 39. | Hasegawa, M.; Fujioka, A.; Kubo, T.; Honda, T.; Miyamoto, H.; Misaki, Y. Chem. Lett. 2008, 37, 474–475. doi:10.1246/cl.2008.474 |
| 40. | Horiuchi, H.; Misaki, Y. Chem. Lett. 2010, 39, 989–991. doi:10.1246/cl.2010.989 |
| 41. | Nishiwaki, M.; Tezuka, M.; Shirahata, T.; Misaki, Y. Chem. Lett. 2011, 40, 467–469. doi:10.1246/cl.2011.467 |
| 30. | Mitamura, Y.; Yorimitsu, H.; Oshima, K.; Osuka, A. Chem. Sci. 2011, 2, 2017–2021. doi:10.1039/c1sc00372k |
| 25. | Charlton, A.; Underhill, A. E.; Williams, G.; Kalaji, M.; Murphy, P. J.; Hibbs, D. E.; Hursthouse, M. B.; Malik, K. M. A. Chem. Commun. 1996, 2423–2424. doi:10.1039/cc9960002423 |
| 26. | Charlton, A.; Underhill, A. E.; Williams, G.; Kalaji, M.; Murphy, P. J.; Malik, K. M. A.; Hursthouse, M. B. J. Org. Chem. 1997, 62, 3098–3102. doi:10.1021/jo962301q |
| 27. | Gompper, R.; Hock, J. Synth. Met. 1997, 84, 339–340. doi:10.1016/s0379-6779(97)80771-0 |
| 28. | Zotti, G.; Zecchin, S.; Schiavon, G.; Berlin, A.; Huchet, L.; Roncali, J. J. Electroanal. Chem. 2001, 504, 64–70. doi:10.1016/s0022-0728(01)00429-6 |
| 29. | Kakinuma, T.; Kojima, H.; Kawamoto, T.; Mori, T. J. Mater. Chem. C 2013, 1, 2900–2905. doi:10.1039/c3tc30089g |
| 17. | Coffen, D. L.; Chambers, J. Q.; Williams, D. R.; Garrett, P. E.; Canfield, N. D. J. Am. Chem. Soc. 1971, 93, 2258–2268. doi:10.1021/ja00738a028 |
| 18. | Ueno, Y.; Okawara, M. Chem. Lett. 1974, 3, 1135–1138. doi:10.1246/cl.1974.1135 |
| 19. | Ueno, Y.; Nakayama, A.; Okawara, M. J. Am. Chem. Soc. 1976, 98, 7440–7441. doi:10.1021/ja00439a064 |
| 20. | Yoneda, S.; Kawase, T.; Inaba, M.; Yoshida, Z.-i. J. Org. Chem. 1978, 43, 595–598. doi:10.1021/jo00398a015 |
| 21. | Le Coustumer, G.; Mollier, Y. J. Chem. Soc., Chem. Commun. 1980, 38–39. doi:10.1039/c39800000038 |
| 22. | Babeau, A.; Tinh, N. H.; Gasparoux, H.; Polycarpe, C.; Torreilles, E.; Giral, L. Mol. Cryst. Liq. Cryst. 1982, 72, 171–176. doi:10.1080/01406568208084054 |
| 23. | Starodub, V. A.; Baumer, V. N.; Guella, I. M.; Golovkina, I. F.; Alyoshin, V. G.; Nemoshkalenko, V. V.; Senkiewicz, A. I. Synth. Met. 1983, 5, 101–111. doi:10.1016/0379-6779(83)90124-8 |
| 24. | Tsujimoto, K.; Okeda, Y.; Ohashi, M. J. Chem. Soc., Chem. Commun. 1985, 1803–1804. doi:10.1039/c39850001803 |
| 43. | The optimized geometries and energy levels of the LUMO, HOMO, and HOMO−1 of 3a and 4 are shown in Supporting Information File 1, Figure S1. |
© 2020 Yoshimura et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)