Abstract
The construction of redox-active supramolecular assemblies based on star-shaped and radially expanded tetrathiafulvalene (TTF) oligomers with divergent and extended conjugation is summarized. Star-shaped TTF oligomers easily self-aggregate with a nanophase separation to produce supramolecular structures, and their TTF units stack face-to-face to form columnar structures using the fastener effect. Based on redox-active self-organizing supramolecular structures, conducting nanoobjects are constructed by doping of TTF oligomers with oxidants after the formation of such nanostructures. Although radical cations derived from TTF oligomers strongly interact in solution to produce a mixed-valence dimer and π-dimer, it seems to be difficult to produce nanoobjects of radical cations different from those of neutral TTF oligomers. In some cases, however, radical cations form nanostructured fibers and rods by controlling the supramolecular assembly, oxidation states, and counter anions employed.
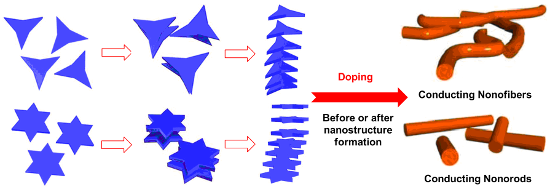
Graphical Abstract
Introduction
Tetrathiafulvalene (TTF) chemistry first attracted enthusiastic attention of chemists and physicists on high electrical conductivity and superconductivity with high Tc temperature. Recently, however, TTF and its derivatives are frequently employed as a redox-active moiety for organic electronic devices such as field-effect-transistors (FET), dye-sensitized solar cells (DSC), positive electrode materials for rechargeable batteries, and electrochromic (EC) materials [1].
TTF derivatives are versatile building blocks to form aggregates in the solid state with interesting conducting and magnetic behavior [2]. Although these properties are mainly originated from specific interactions between molecules having one or more unpaired electrons [3,4], neutral TTF and its derivatives also easily form stacked columnar structures with face-to-face π···π stacking and side-by-side S···S interactions in the crystalline state. Furthermore, weak intermolecular interactions (hydrogen bonding, metal coordination, CT interaction, π···π stacking, van der Waals interaction, etc.) play an important role in the formation of the three-dimensional (3D) crystal structures [5]. For the construction of nanostructured objects, π···π, S···S, and other weak intermolecular interactions first accelerate self-aggregation of molecules in solution [6-8] and then produce the functional one-dimensional (1D) or two-dimensional (2D) supramolecular structures, which are very important in advanced nanosciences [9-11]. For the formation of the nanostructured objects such as fibers, rods, tubes, and particles, amphiphilic TTFs having a rigid core and long alkyl chains are one of the best molecular systems. The self-assembly of TTF derivatives in solid and on surface gives rise to a long-distance dynamic ordering as compared with single crystals.
Among the recent researches on TTF and its derivatives, radially expanded or star-shaped multi-TTFs with C3 and C6 symmetries have attracted considerable attention in the field of materials science because of their divergent and extended π-conjugation. Various C3-symmetric compounds incorporating three conjugated TTF units were designed and synthesized to realize TTF-based conducting organic magnets using ferromagnetic interaction between the two TTF radical cations [12-21]. On the other hand, compounds with a hexagonal molecular symmetry were used as core structures for constructing conductive fibers and functional dyes [22,23]. Furthermore, various multifunctional TTF-based supramolecular architectures have been designed and synthesized to realize molecular sensors, redox switches, multi-input systems for logic gates, electrochemically-driven conformational controls, molecular clips and tweezers, and redox-controlled gelation processes. TTF-based supramolecular chemistry in solution was thoroughly outlined in recent reviews of Jeppesen, Nielsen, and Becher (2004) [24], Iyoda, Hasegawa, and Miyake (2004) [25,26], Sallé and Zhang (2009) [27], and Martin (2009, 2012) [28,29]. However, limited examples of redox-active nanostructures in the solid state were summarized so far. Therefore, this review focuses on the conducting nanostructures of TTF derivatives in the solid state, together with association behavior in solution.
Review
Redox-active radially expanded TTF oligomers in solution and the solid state
TTF oligomers with radially expanded structures can be expected to demonstrate multifunctional properties because a central core and TTF branches exhibit individual and/or cooperative functionalities [25]. For example, dendrimers composed of central benzenoid cores and TTF branches are cited as representative examples [30-33]. Other functional units such as fullerenes [34,35], cyclodextrins [36], porphyrin [37], and phthalocyanine [38-40] can also be introduced into the core of radial oligo-TTFs. As shown in Figure 1, TTF-annelated porphyrin 1 was synthesized by Becher and co-workers in 2001 [37]. Reflecting its strong π-donor ability, 1 was oxidized spontaneously in solution to afford 1•+ under ambient conditions. Multifunctional TTF-crown ether-substituted phthalocyanine (Pc) 2a and its copper(II) complex 2b were reported by Amabilino, Rowan, Nolte, and co-workers in 2005 [40]. The giant molecule 2a self-aggregated in chloroform–dioxane to form a gel. TEM images of the xerogel exhibited helical molecular tapes nanometer wide and micrometer long. A cyclic voltammetry (CV) study on 2b showed the redox properties expected for Pc and TTF, and doping of 2b in CH2Cl2 with I2 produced a radical cation species.
![[1860-5397-11-175-1]](/bjoc/content/figures/1860-5397-11-175-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Radially expanded TTF oligomers 1 and 2a,b.
Figure 1: Radially expanded TTF oligomers 1 and 2a,b.
Among radially expanded TTFs, Jeppesen, Becher, Nielsen, Sessler, and co-workers reported TTF-calix[4]pyrrole 3 as a valuable supramolecular receptor, and 3 easily incorporated 1,3,5-trinitrobenzene (TNB) in the cavity to form 4 (3:TNB = 1:2) (Figure 2) [41,42]. Furthermore, in the presence of halide ions, 3 formed the C60 complex 5, in which C60 was bound within the bowl-like cup of the TTF-calix[4]pyrrole core in a ball-and-socket binding mode [43].
![[1860-5397-11-175-2]](/bjoc/content/figures/1860-5397-11-175-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: TTF-calix[4]pyrrole 3 and its TNT and C60 complexes 4 and 5.
Figure 2: TTF-calix[4]pyrrole 3 and its TNT and C60 complexes 4 and 5.
Recently, the C3-symmetric compounds 6a,b incorporating three TTF residues were reported by Amabilino, Avarvari, and co-workers (Figure 3) [21]. The three TTF units with chiral citronellyl and dihydrocitronellyl chains led to helical one-dimensional stacks in solution to produce fibers that have morphologies depending on the nature of the chiral alkyl group, although an achiral counterpart showed no helicity. C3-symmetric truxene-TTFs 7a–c were synthesized by Ortí, Martín, and co-workers (Figure 3) [44].
![[1860-5397-11-175-3]](/bjoc/content/figures/1860-5397-11-175-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: C3-symmetric TTF derivatives 6a,b and 7a–c.
Figure 3: C3-symmetric TTF derivatives 6a,b and 7a–c.
The pioneering studies on the synthesis of tetrakis(1,3-dithiol-2-ylidene)cyclobutane (8) and related [5] and [6]radialenes 9 and 10a,b were reported by Yoshida and co-workers in the 1980’s (Figure 4). These π-expanded TTFs 8–10a,b exhibited unique X-ray structures and multi-redox behavior [45-47].
![[1860-5397-11-175-4]](/bjoc/content/figures/1860-5397-11-175-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Radially expanded TTF derivatives 8, 9, and 10a,b.
Figure 4: Radially expanded TTF derivatives 8, 9, and 10a,b.
Conducting supramolecular assembly of oligo-TTFs
The electric conductivities of doped nanofibers and nanorods derived from TTF and its derivatives are measured by mounting them on Au electrodes with a μm-sized spacing. On the other hand, the conductivities of the corresponding neutral nanoobjects are determined by pulse-radiolysis or flash-photolysis time-resolved microwave conductivity techniques [48,49]. Current-sensing atomic force microscopy (CS-AFM) and combination of scanning tunneling microscopy and spectroscopy (STM/STS) are also employed for determining the conductivities of nanoobjects [50,51]. The electrical conductivity of nanostructures mainly depends on the alignment of stacked TTFs or their radical salts. The first fibrous material was fabricated by using arborol-TTF 11 in 1994 by Joergensen, Bechgaard, and co-workers (Figure 5) [52]. Although 11 showed no conductivity, Bryce and co-workers synthesized arborol-functionalized TTF derivative 12 in 2003, whose doped film exhibited a moderate level of conductivity (σrt ≈ 10−4 S cm−1) [53]. In 2005, several groups reported the formation of nanofibers using amphiphilic TTFs (13 and 14) (Figure 5) [54-56] After that, many research efforts have been focused on the construction of conducting nanoobjects possibly employed as nanosized electric wires, wirings, molecular switches, and devices. Some neutral nanoobjects derived from TTFs show electroconductivity owing to the fastener effect [57]; however, the oxidation of face-to-face stacked TTFs easily generates highly conducting states with unfilled band structure. Thus, the doping of neutral nanoobjects with iodine is generally used for preparing conducting nanostructures. Chemically oxidized TTFs in solution are also available for preparing conducting nanofibers.
![[1860-5397-11-175-5]](/bjoc/content/figures/1860-5397-11-175-5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Amphiphilic TTFs 11–14 and 15a,b.
Figure 5: Amphiphilic TTFs 11–14 and 15a,b.
The fastener effect [57], which enhances the face-to-face interaction between the two TTFs, can be used to construct conducting nanostructured fibers in the neutral state, and doping of the fibers with iodine affords black conducting fibers. For example, the bi-TTF derivative 15a with long alkylthio chains as substituents was synthesized (Figure 5) [58]. Bi-TTF 15a formed reddish orange rods which exhibited a bulk conductivity of σrt = 1.0 × 10−6 S cm−1 without doping. The p-type semiconductivity was detected by CS-AFM. Furthermore, the doping of 15a with iodine and bromine vapors afforded black conducting complexes (σrt = 1.1 × 10−4 and 1.5 × 10−4 S cm−1, respectively).
For conjugated TTF dimers linked by π-systems or chalcogen atoms, intramolecular through-bond and/or through-space interactions can be expected between two TTF parts. The intramolecular through-bond interaction between the two TTF parts linked in a head-to-tail manner is calculated to be weak in the ground state [25]. Thus, the conjugation of the two neutral TTF parts in 16–19 is weak (Figure 6) [59]. In the cyclic voltammetry (CV) measurements, tetraethylthio-bi-TTF 16 showed two one-electron and one two-electron redox waves (Table 1), while other TTF dimers of 17–19 exhibited only two two-electron reversible redox waves corresponding to TTF/TTF•+ and TTF•+/TTF2+ at a normal scan rate (100 mV s-1). As shown in Table 1, however, steady-state electronic spectra of 16•+, 17•+, and 18•+ show intramolecular interaction between TTF and TTF•+, and the absorption maxima were observed at ca. 450 and 750 nm, together with broad absorption of intramolecular CT interactions between two TTF units at 1400, 1300, 1200 nm, respectively. The magnitude of these broad absorption bands is clearly affected by the distance between two TTFs, and TTF dimer 19•+ linked with a longer spacer exhibited no intramolecular CT absorption band. Moreover, the longest absorption maxima of the dications 162+, 172+, and 182+ exhibit a bathochromic shift of 44, 30, and 14 nm, respectively, from the corresponding absorption maximum of 16•+, 17•+, and 18•+ due to the head-to-tail orientation of two TTF•+ (Davydov red shift) [25]. It is worth noting that the redox behavior of TTF dimers in CV measurements are sensitive to the concentration and the solvent used, and pristine bi-TTF 15b showed two reversible two-electron redox waves at −0.03 and 0.38 V vs Fc/Fc+ in benzonitrile under normal conditions [25,60].
![[1860-5397-11-175-6]](/bjoc/content/figures/1860-5397-11-175-6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: TTF dimers linked by σ-bond (16) and conjugated π-systems (17–19).
Figure 6: TTF dimers linked by σ-bond (16) and conjugated π-systems (17–19).
Table 1: Redox potentials of 16–19 and absorption maxima of monocations 16•+, 17•+, 18•+ and 19•+, and dications 162+, 172+, 182+ and 192+ [25,59].
| Compound | Redox potentialsa vs Fc/Fc+ | Absorption maximab | |||
|---|---|---|---|---|---|
| E1/21 (V) | E1/22 (V) | E1/23 (V) | Monocation (nm) | Dication (nm) | |
| 16 | 0.06 | 0.17 | 0.44 | 772, 1400 br | 816, 1098 sh |
| 17 | 0.11 | 0.42 | 778, 1300 br | 808 | |
| 18 | 0.12 | 0.42 | 790, 1200 br | 804 | |
| 19 | 0.08 | 0.38 | 790 | 796 | |
aPotentials were measured by cyclic voltammetry (CV) in benzonitrile against a Ag/Ag+ electrode and adjusted to the Fc/Fc+ potential. bMeasured in CH2Cl2/CH3CN (4:1) using Fe(ClO4)3 as the oxidation reagent.
Conducting nanostructure formation from star-shaped oligo-TTFs
Although pristine TTF does not self-associate in solution due to the low association constant for dimerization, the mixed-valence (MV) dyad (TTF/TTF)•+ and the dicationic dyad (TTF•+)2, so-called π-dimer, are formed in concentrated solution or at low temperature [61]. On the other hand, the synergy of either the fastener effect or π-expansion allows star-shaped C3-symmetric oligo-TTFs 22 and 23 to self-associate both in solution and in the solid state even in neutral state [18]. Compounds 22 and 23 were synthesized in good yields by Sonogashira coupling reaction of 1,3,5-triethynylbenzene with 20 and 1,3,5-triiodobenzene with 21, respectively (Scheme 1). X-ray analysis of 22 revealed the columnar structure, in which the three TTF units stack in face-to-face manner to form single crystals (Figure 7).
![[1860-5397-11-175-i1]](/bjoc/content/inline/1860-5397-11-175-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthesis of star-shaped TTF trimers 22 and 23.
Scheme 1: Synthesis of star-shaped TTF trimers 22 and 23.
![[1860-5397-11-175-7]](/bjoc/content/figures/1860-5397-11-175-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Projections of the molecular array of 22 in crystal structure (a) along with the c axis and (b) from side view.
Figure 7: Projections of the molecular array of 22 in crystal structure (a) along with the c axis and (b) fro...
In the case of 23 with butyl chains, this molecule dimerized in CDCl3 solution (K2 = 1.58 ± 0.30 M−1 at 293 K). The chemical shift of the central benzene ring clearly shifted higher field with an increase of concentration or lowering temperature. The observed shift is attributed to the shielding effect from the neighboring molecule that settles in face-to-face mode. The thermodynamic parameters were estimated to be ΔH = −9.43 kJ mol−1 and ΔS = −28.3 J mol−1 by the van’t Hoff plot (Table 2 and Supporting Information File 1). The self-association behavior is significantly affected by the solvent. While no association was observed in benzene-d6 solution in the concentration ranges of 0.7–21 mM even at low temperatures, a larger K2 value was estimated in CDCl3–CD3CN solution (3:7 v/v, K2 = 5.01 ± 0.98 M−1 at 293 K). Moreover, only a small concentration dependence of the chemical shift, which could not be used for determination of the K2 value, was observed in acetone–CS2 solution owing to very weak self-association. These results clearly suggest that the association behavior is driven by intermolecular π–π, S···S, and/or S···H interactions in solution. Note that these K2 values of 23 in the neutral state are similar to that of the mixed valence dimer (TTF•+ + TTF) (K2 = 6.0 M−1) and much larger than that of the π-dimer (TTF•+ + TTF•+) (K2 = 0.6 M−1) described in the literature [61].
Table 2: Association (dimerization) constants and thermodynamic parameters of 23 in various solventsa.
| Solvent | K2 (M−1)b | ΔG (kJ mol−1)b | ΔH (kJ mol−1) | ΔS (J mol−1) |
|---|---|---|---|---|
| CDCl3 | 1.58 | −1.13 | −9.41 | −28.2 |
| CDCl3–CD3CN (3:7) | 5.01 | −3.64 | −16.6 | −43.1 |
| benzene-d6 | –c | –c | –c | –c |
aParameters were estimated from titration experiments using 1H NMR with the assumption of the dimerization process of 23. bAt 298 K. cNo association was observed.
Strong self-association of 23 was observed in the oxidation state. CV analysis of 23 in a dilute CH2Cl2 solution (1.9 × 10−5 M) showed two three-electron redox waves at 0.05 and 0.40 V vs Fc/Fc+ corresponding to the formation of 233+ and 236+, whereas a similar CV analysis of 23 in a concentrated CH2Cl2 solution (1.2 × 10−3 M) displayed three reversible waves at −0.04, 0.14, and 0.47 V vs Fc/Fc+ corresponding to the formation of (23)23+, (23)26+, and (23)212+ (Figure S2, Supporting Information File 1). Interestingly, the three cationic species 23•+, 232+ and 233+ prepared by chemical oxidation with Fe(ClO4)3 in CH2Cl2/CH3CN (4:1) showed a strong self-association, and electronic spectra of 23•+ and 232+ exhibited marked intermolecular charge resonance (CR) bands at λmax 2000 (br, ε 1500) and 2000 (br, ε 1700) owing to the face-to-face mixed valence interaction (Figure 8), and 233+ exhibited a typical Davydov blue shift (λmax 738 nm, ε 27000) as compared with 19 (λmax 796 nm, Table 1) [25]. To determine the conducting behavior of 22 and 23, a pellet of 22 was treated with iodine to produce a semiconducting black solid (σrt = 3.6 × 10−4 S cm−1), whereas a similar doping of 23 with iodine resulted in the formation of the conducting liquid.
![[1860-5397-11-175-8]](/bjoc/content/figures/1860-5397-11-175-8.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 8: UV–vis/NIR spectra of 23, 23•+, 233+, and 236+.
Figure 8: UV–vis/NIR spectra of 23, 23•+, 233+, and 236+.
Radially expanded TTF oligomers with a large central π-surface can be expected to show effective intra- and intermolecular delocalization of electrons in the neutral and mixed-valence states. Furthermore, the supramolecular self-assembly of these large molecules having nanophase separation is a promising way of realizing molecular switches and devices [62-65]. With this in mind, hexadehydrotris(TTF)[12]annulenes 28 and 29 and dodecadehydrotris(TTF)[18]annulenes 30 and 31 were synthesized using palladium-mediated coupling reactions (Scheme 2) [20,25,26,66-68]. Tris(TTF)[12]annulenes 28 and 29 were pepared by Sonogashira coupling of 26 with 24 and 27 with 25 in 25 and 36% yields, respectively. For the synthesis of 30 and 31, cyclotrimerization of 24 and 25 with a stoichiometric amount of PdCl2(PPh3)2 and CuI in triethylamine–THF was employed to afford 30 and 31 in 32 and 29% yields, respectively. Although tris(TTF)[18]annulenes are stable at room temperature in air, tris(TTF)[12]annulenes 28 and 29 gradually decomposed under ambient conditions due to the instability of central 4n π-electron system.
![[1860-5397-11-175-i2]](/bjoc/content/inline/1860-5397-11-175-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tris(TTF)[12]annulenes 28 and 29 and tris(TTF)[18]annulenes 30 and 31, together with hexyl-substituted tris(TTF)[18]annulene 32.
Scheme 2: Synthesis of tris(TTF)[12]annulenes 28 and 29 and tris(TTF)[18]annulenes 30 and 31, together with h...
In order to investigate the effect of fused two TTF units on the cyclic conjugation and the interaction of the two TTF units in the neutral and cationic states, TTF-fused annulenes 33 [69] and radiannulenes 34 and 35 [70] were synthesized using a Sonogashira coupling in moderate yields (Figure 9).
![[1860-5397-11-175-9]](/bjoc/content/figures/1860-5397-11-175-9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 9: TTF-fused annulene 33 and radiannulenes 34 and 35.
Figure 9: TTF-fused annulene 33 and radiannulenes 34 and 35.
The thermodynamic study on the self-aggregation of tris(TTF)annulenes indicates that the aggregation of 28, 30, and 31 is an enthalpically driven process that is entropically disfavored (Table 3) [68], although the aggregation of planar macrocyclic belts is both enthalpically and entropically driven [71]. The TTF[18]annulene 30 has smaller ΔH and ΔS values than the TTF[12]annulene 28, suggesting a higher stacking ability and a larger ring size for 30. Alkyl-substituted TTF[18]annulene 32 was reported to show almost no aggregation behavior in solution [72]. However, the slightly more amphiphilic 31 exhibits self-aggregation in benzene, toluene, and cyclohexane owing to a slightly larger nanophase separation in 31. It is worth noting that the self-aggregation of TTF-annulenes results in the appearance of solvatochromism and thermochromism [68]. As shown in Figure 10, 30 exhibits a supramolecular thermochromism in toluene, and the color at −10 °C is reddish purple, whereas the color at −70 °C is purple. On the other hand, as shown in Figure 11, a solution of 33 exhibits deep green in CS2 but purple in CH2Cl2 [69].
Table 3: Self-aggregation data in toluene-d8.a
| Comp. | ΔG (kJ mol−1) at 303 K | ΔH (kJ mol−1) | ΔS (J mol−1·K−1) |
|---|---|---|---|
| 28 | −11.8 | −32.0 | −66.3 |
| 30 | −14.5 | –37.8 | −77.0 |
| 31 | −10.1 | −21.5 | −37.1 |
aDetermined with concentration/temperature-dependent 1H NMR assuming an infinite association model [68].
![[1860-5397-11-175-10]](/bjoc/content/figures/1860-5397-11-175-10.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 10: Colors of 30 solutions a–d in toluene (0.025 mM) at various temperatures. (a) λmax: 511 nm, (b) λmax: 512 nm, (c) λmax: 517 nm, (d) λmax: 520 nm. Reprinted with permission from [68]. Copyright 2012 Chemical Society of Japan.
Figure 10: Colors of 30 solutions a–d in toluene (0.025 mM) at various temperatures. (a) λmax: 511 nm, (b) λmax...
![[1860-5397-11-175-11]](/bjoc/content/figures/1860-5397-11-175-11.png?scale=1.84&max-width=1024&background=FFFFFF)
Figure 11: Solutions of 33. (a) In CS2, λmax: 608 nm. (b) In CH2Cl2, λmax: 577 nm. Reprinted with permission from [69]. Copyright 2004 Royal Society of Chemistry.
Figure 11: Solutions of 33. (a) In CS2, λmax: 608 nm. (b) In CH2Cl2, λmax: 577 nm. Reprinted with permission f...
CV analysis of 28–34 in solution showed different behaviors (Table 4). The [12]annulenes 28, 29, and 33 exhibited two reversible one-electron reductions due to the smooth reduction of the 12π electron system to a 14π electron system, whereas the [18]annulenes 30 and 31 showed an irreversible reduction wave, due to the unfavorable reduction of the aromatic 18π electron system. On the other hand, all the molecules exhibited reversible oxidation waves in CH2Cl2 based on the high HOMO levels of TTF units. Another important feature in the CV data of 28–32 is broadening or splitting of the first oxidation wave, indicating intra- and/or intermolecular interactions between TTF units [68]. Interestingly, the first oxidation potential of 28 and 29 splited at the slow scanning rate of 3 mV s−1 owing to the intermolecular mixed-valence interaction between the TTF•+ and TTF moieties under diffusion-controlled conditions. In the case of 31, the first oxidation potential (E1/2 = 0.14 V vs Fc/Fc+) is lower than that of 32 with alkyl groups (E1/2 = 0.20 V). Since the first oxidation potential of 31 in a dilute solution broadened but did not split (E1/2ox1 in CH2Cl2: 0.23 (3e) V), the potential of 31 at 0.14 V (Table 4) reflected the strong intermolecular interaction between the TTF•+ and TTF moieties in 31•+. By comparison with the known UV–vis/NIR spectra of mixed valence dimers [18,73], the association constant Ka of 31•+ measured in CH2Cl2–MeCN 4:1 assuming an infinite association model [74] is large (Ka = 3.12 ± 0.48 × 105 M−1 at 298 K) owing to 18 sulfur atoms in 31. Therefore, the oxidation of 31 solution (0.1 mM) in CH2Cl2 first forms (31)22+ owing to the intermolecular mix-valence interaction between the TTF•+ and TTF moieties, and the further oxidation forms 313+ [68]. In summary, the oxidation of 28–31 showed multistep processes owing to intra- and/or intermolecular interactions between TTF units. In the case of [18]annulene 31, the first oxidation potential splited in two with the strong intermolecular interaction in 31•+. TTF-functionalized radiaannulenes (RAs) 34 and 35 also exhibit multiple redox states [70]. CV analysis of 34 shows the two reversible one-electron reductions as the reduction of the RA core, whereas the three reversible oxidations at 0.20, 0.29, and 0.61 V correspond to the formation of 34•+, 342+, and 344+. Therefore, the redox behavior of 34 is similar to those of 28, 29, and 31.
Table 4: Redox potentials of 28–34 measured by CVa.
| Compound | E1/2red2 (V) | E1/2red1 (V) | E1/2ox1 (V) | E1/2ox2 (V) |
|---|---|---|---|---|
| 28b | −1.52 (1e) | −1.16 (1e) | 0.38 (3e) [0.29, 0.44]c | 0.66 (3e) |
| 29b | −1.78 (1e) | −1.41 (1e) | 0.21 (3e) [0.12, 0.26]c | 0.49 (3e) |
| 30b | –d | –1.35e | 0.43 (3e)f | 0.70 (3e) |
| 31b | –d | –1.48e | 0.14 (1e), 0.29 (2e) | 0.53 (3e) |
| 32g | –d | –1.40e | 0.20 (3e) | 0.64 (3e) |
| 33b,i | −1.87 (1e) | –1.50 (1e) | 0.19 (2e)f,h | 0.46 (2e)h |
| 34i | −1.52 (1e) | −1.16 (1e) | 0.20 (1e), 0.29 (1e) | 0.61 (2e) |
aConditions: 0.1 M Bu4NClO4, 100 mV s–1, Pt as a working electrode, Ag/Ag+ as a reference electrode, Pt wire as a counter electrode. Potentials were referenced to Fc/Fc+. Solvent: THF for reduction, and CH2Cl2 for oxidation. bConcentration: 0.1 mM. cMeasured at 3 mV s–1. dNot observed. eIrreversible process. fBroad redox wave. gAccording to [72]. hSolvent: benzonitrile. iAccording to [70].
The [18]annulenes 30 and 31 formed a fibrous structure in H2O–THF 1:1, and 31 required longer time for fiber formation than 30 owing to weaker association constant in solution (Ka in toluene-d8 at 303 K = 634 M−1 (30), 101 M−1 (31)) [67,68]. Both 30 and 31 fibers showed roughly the same behavior for doping with iodine, and the color of fibers quickly changed from bluish purple to dark brown due to the partial oxidation of 30 and 31 as shown Figure 12 (the maximum conductivities: 30 σrt 2.0 × 10−2 S cm−1, 31 σrt 2.6 × 10−3 S cm−1). The color of the doped fibers gradually returned to the original bluish purple under vacuum, but the speed of the iodine desorption for fiber 31 was very slow. The conductivity of the doped pellet prepared from fiber 30 is estimated to be ca. 1000 times higher than that of the neutral fiber (before doping: σrt 3 × 10−6 S cm−1, after doping: σrt 3 × 10−3 S cm−1) [68].
![[1860-5397-11-175-12]](/bjoc/content/figures/1860-5397-11-175-12.png?scale=1.72&max-width=1024&background=FFFFFF)
Figure 12: Optical micrographs (1000× magnified) of fibers, prepared from 30 in THF–H2O 1:1, on a glass plate at 23 °C. (a) Before iodine doping. (b) After iodine doping (3 min). Reprinted with permission from [26]. Copyright 2010 Royal Society of Chemistry.
Figure 12: Optical micrographs (1000× magnified) of fibers, prepared from 30 in THF–H2O 1:1, on a glass plate ...
Star-shaped pyrrole-fused TTF oligomers 38–43 were synthesized by nucleophilic aromatic substitution (SNAr) reactions of fluorinated benzenes with the pyrrolyl sodium salts derived from 36 and 37 in moderate yields (Scheme 3) [23]. X-ray analysis of 38 revealed that the three TTF units are bent simply to fill an empty space and stacked to form a columnar structure. The torsion angle between the mean planes of the pyrrole and central benzene is 7–32°, indicating the conformational flexibility of the pyrrole–benzene linkage. The calculated torsion angles between the pyrroles and central benzenes of 38, 40, and 42 are 34, 45, and 59°, respectively, and the non-planar structures of 38, 40, and 42 are in good agreement with the high-field shift of α-protons of pyrroles in the 1H NMR spectra: δ 6.89 (38), 6.41 (40), 5.93 ppm (42). Star-shaped TTF 10-mer 44 was also synthesized by SNAr reaction of the sodium salt of 36 with decafluorobiphenyl (44%) [75] (Figure 13).
![[1860-5397-11-175-i3]](/bjoc/content/inline/1860-5397-11-175-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Star-shaped TTF oligomers 38–43.
Scheme 3: Star-shaped TTF oligomers 38–43.
![[1860-5397-11-175-13]](/bjoc/content/figures/1860-5397-11-175-13.svg?scale=2.0&max-width=1024&background=FFFFFF)
In the CV measurements (Figure 14), tetrasubstituted 40 shows typical two reversible oxidation waves at E1/2ox1 = 0.044 and E1/2ox2 = 0.35 V (vs Fc/Fc+). However, trisubstituted 38 and hexasubstituted 42 exhibit split and broad first peaks, respectively, at −0.086 and 0.020 V (38) and 0.097 V (42), followed by second peaks at 0.45 V (38) and 0.37 V (42). The CV data of tetrasubstituted 40 suggests no intramolecular charge delocalization between the adjacent TTF units. The splitting and broadening of the first oxidation waves in 38 and 42 are considered to be caused by intermolecular interactions between the neutral and cationic TTF units.
![[1860-5397-11-175-14]](/bjoc/content/figures/1860-5397-11-175-14.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 14: Cyclic voltammograms of 38, 40, and 42 (0.1 mM) in benzonitrile with 0.1 M n-Bu4PF6 as a supporting electrolyte, Ag/AgNO3 as a reference electrode, glassy carbon as a working electrode, Pt wire as a counter electrode, and a scan rate of 100 mV s−1. Values are half-wave potentials. Adapted with permission from [23]. Copyright 2011 American Chemical Society.
Figure 14: Cyclic voltammograms of 38, 40, and 42 (0.1 mM) in benzonitrile with 0.1 M n-Bu4PF6 as a supporting...
As shown in Figure 15, the stepwise chemical oxidation of 38, 40, and 42 with Fe(ClO4)3 in CH2Cl2–CH3CN 2:1 exhibits the typical changes in the absorption spectra. The addition of Fe(ClO4)3 up to 1 equiv with respect to each of the TTF units causes new absorption maxima at longer wavelength region (blue to green spectra). For the oxidation of 40, the changes show several isosbestic points, indicating that each TTF unit is oxidized from the neutral to the radical cation (TTF•+) in a stepwise manner (Figure 15b). On the other hand, for 38 and 42, there are no isosbestic points (Figure 15a,c). For 38, a new broad peak around 1850 nm (intermolecular CR absorption) appears in the presence of 1.5 equiv of Fe(ClO4)3, which is attributed to the formation of an intermolecular face-to-face mixed valence complex. These results are consistent with the peak splitting of the CV. Furthermore, CV analysis of 44 exhibited two reversible ten-electron redox waves corresponding to the formation of 4410+ and 4420+.
![[1860-5397-11-175-15]](/bjoc/content/figures/1860-5397-11-175-15.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 15: Stepwise oxidation of (a) 38 (0.02 mM), (b) 40 (0.05 mM), and (c) 42 (0.03 mM) with incremental addition of Fe(ClO4)3 in a mixture of CH2Cl2–CH3CN (2:1, v/v) at 25 °C. The blue line indicates the neutral absorption spectra, the green line the multiple TTF radical cations 383(•+), 404(•+), and 426(•+), and the red line the TTF dications 386+, 408+, and 4212+. Adapted with permission from [23]. Copyright 2011 American Chemical Society.
Figure 15: Stepwise oxidation of (a) 38 (0.02 mM), (b) 40 (0.05 mM), and (c) 42 (0.03 mM) with incremental add...
Trisubstituted 38 showed polymorphism and formed single crystals from CH2Cl2, whereas it produced a yellow fibrous material from CH2Cl2–hexane 1:4. X-ray diffractometry (XRD) exhibited that fiber 38 possesses a hexagonal columnar structure different from single crystals. Furthermore, the spin-coated film of 38 has an amorphous structure. Interestingly, doping of single crystals, hexagonal fiber, and amorphous film of 38 with iodine vapor produced black CT-complexes having different assembled structure. After doping, electric conductivity of single crystals was σrt = 1.8 × 10−2 S cm−1 and the fiber was 1.9 × 10−2 S cm−1, whereas the amorphous film was 2.5 × 10−3 S cm−1. The difference in the conductivity reflects the molecular level alignments. Other star-shaped oligomers 39–44 also formed nanostructures fibers, particles and film, and doping with iodine produced black complexes which exhibited electric conductivities of σrt = 2.7 × 10−3–2.4 × 10−2 S cm−1 in spite of the non-planarity of the molecular frame of 39–44.
The pyridazine-3,6-diol-annulated TTF derivative 45 produced trimer 46 via hydrogen bonds in a THF–H2O solution (Scheme 4), in which micrometer-sized fibrous material was gradually formed [76]. The compressed pellet of the 46 fibers showed an electrical conductivity of σrt = 2.3 × 10−4 S cm−1 after doping with iodine vapor. The addition of ethylene diamine triggered the reorganization of the supramolecular structure 46, and fine nanoscopic fibers composed of 45 and ethylene diamine (1:1) were produced from the CHCl3 solution. A compressed pellet of the fibers of 45·H2NCH2CH2NH2 exhibited an electrical conductivity in the range of σrt = 1.5–10.0 × 10−5 S cm−1 after iodine doping.
![[1860-5397-11-175-i4]](/bjoc/content/inline/1860-5397-11-175-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Pyridazine-3,6-diol-TTF 45 and its trimer 46.
Scheme 4: Pyridazine-3,6-diol-TTF 45 and its trimer 46.
Recently, conducting nanofibers derived from the self-assembly of TTF-appended dipeptides were reported [77]. Conductivity measurements performed on the nanofibers of TTF-appended dipeptides indicate a remarkable enhancement in the conductivity after doping with TCNQ (σrt = 1 × 10−5 S cm−1).
Conducting nanostructures prepared from cation radicals
Molecular conductors derived from CT complexes and radical salts of TTFs are widely known [1], and mixed-valence (TTF2)n+ (0 < n < 1) was reported to form self-accembled conducting nanofibers (σrt = ~10−2 S cm−1) [78-82]. However, there is only a limited number of examples of nanofibers and nanorods prepared from CT complexes and radical salts of star-shaped and radially expanded TTF oligomers. One typical example is the conducting CT complex 472+·(TCNQF4•−)2 of amphiphilic TTF 47 and TCNQF4 (Figure 16) [83,84]. The fiber structure with typical dimensions of 2.5 nm (height) × 50 nm (width) × 1 μm (length) was constructed on a mica substrate by using the Langmuir–Blodgett (LB) technique, and the conductivity of the film composed the 472+·(TCNQF4•−)2 fiber was found to be on the order of σrt = 10−3 S cm−1.
![[1860-5397-11-175-16]](/bjoc/content/figures/1860-5397-11-175-16.svg?scale=2.0&max-width=1024&background=FFFFFF)
The stacking behavior of TTF in solution and in the solid state was employed as a driving force to construct higher aggregates by using the star-shaped hexakis(tetrathiafulvalenylethynyl)benzene 48 (Figure 17a). The TTF-hexamer 48 was synthesized by Sonogashira coupling of 21 with hexaiodobenzene (52%) [22]. As expected, 48 strongly self-aggregates in CHCl3 (Ka = 2.1 × 104 M–1, 23 °C) and in other common organic solvents. To construct nanoobjects, a CHCl3 solution of 48 was diluted with hexane to afford dark blue fibers with a slim and curled fiber structure (40–90 nm wide, 30–100 nm thick and more than 10 μm long) (Figure 17b). On the other hand, a dark blue film was formed by casting a solution of 48 on a glass surface (Figure 17c). XRD studies on the fiber and the film of 48 revealed that the fiber has a hexagonal alignment, whereas the film has a lamellar structure with lateral order and π···π stacking. It is worth noting that the film of 48 prepared by casting a 0.1 wt % solution of 48 in CHCl3 exhibited a low carrier mobility of μ = 3 × 10−6 cm2 V−1 s−1, indicating a lamellar structure vertical to the substrate surface.
![[1860-5397-11-175-17]](/bjoc/content/figures/1860-5397-11-175-17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 17: (a) Star-shaped TTF hexamer 48. (b) Optical image of 48 fiber with a hexagonal structure. (c) Optical image of 48 film with a lamellar structure. Adapted with permission from [22]. Copyright 2007 American Chemical Society.
Figure 17: (a) Star-shaped TTF hexamer 48. (b) Optical image of 48 fiber with a hexagonal structure. (c) Optic...
Oxidation of 48 with 1 and 3 equiv of Fe(ClO4)3 produced the analytically pure monocation 48•+ClO4− and trication 483+(ClO4−)3, respectively. The cationic species 48•+ClO4− and 483+(ClO4−)3 strongly self-aggregate in CHCl3 (Ka = 2.3–2.5 × 106 M–1) and rather weakly aggregate in THF. Interestingly, in THF, 48•+ClO4− and 483+(ClO4−)3 exhibited the formation of stacked cylindrical structures with a radius of 11 Å and a height of 14–16 Å by small-angle X-ray scattering (SAXS). ESR spectra of 48•+ and 483+ in CHCl3 at 23 °C showed 100% of spin for 48•+ and 33% of spin for 483+. Therefore, the spin–spin interaction in 48•+ is weak, whereas the spin–spin interaction in 483+ is strong.
The monocation 48•+ClO4− easily formed a hexagonal fiber from CHCl3–hexane solution, probably owing to the strong aggregation properties and molecular shape of the stacked 48•+ClO4−, whereas trication 483+(ClO4−)3 produced a nanoparticle having a low internal regularity, presumably owing to the strong intermolecular TTF•+–TTF•+ interaction of 483+. A cast film of 48•+ClO4− shows a lamellar structure vertical to the substrate in a similar manner to the neutral 48 (Figure 17c). Interestingly, the structural difference between nanofiber and film of 48•+ClO4− leads to the different electric conductivities of wires (σrt = 1.1 × 10−3 S cm−1) and film (σrt = 3.1 × 10-5 S cm−1) depending on their stacking structures.
Cation radicals of pyrrole-fused TTF trimer 38 also formed conducting nanostructures when a CH2Cl2 solution of 381.5(•+) was mixed with an excess amount of hexane. The XRD pattern of the fiber 381.5(•+) is composed of a lamellar structure different from the neutral 38 fiber and an exhibited electric conductivity of σrt = 2.9 × 10−4 S cm−1. The lower conductivity of the fiber 381.5(•+) as compared to the doped 38 fiber (σrt = 1.9 × 10−2 S cm−1) may be due to the difference in their internal structures.
Conclusion
The construction of nanoobjects based on the self-assembly of TTFs were rapidly advanced, and a large number of functional properties such as electronic, magnetic, and optical properties were recently reported. Based on these developments of nanoscience, the construction of conducting nanoobjects has also been investigated to realize electrochemically-driven conformational control, redox-controlled gelation processes, redox switches, and molecular sensors. Furthermore, semiconductive fibers and rods of TTFs can be utilized for nanosized electric wires and wirings in nanoelectronics. The next key innovation in TTF-based nanoobjects is the fulfillment of nanofiber and nanorod with metallic conductivity and superconductivity. To achieve a high electric conductivity, further knowledge is necessary to fabricate a closely stacked ionic state with unfilled bands. If these innovative systems can be implemented, conducting nanoobjects find functions in a variety of mass use devices.
Supporting Information
| Supporting Information File 1: Determination of association constants (K2) of 23 by NMR and cyclic voltammetry analysis of 23. | ||
| Format: PDF | Size: 408.8 KB | Download |
Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research from JSPS and by Strategic Japanese-German Cooperative Program of JST (Japan Science and Technology Corporation). We thank Prof. Tohru Nishinaga (Tokyo Metropolitan University) and Prof. Masayoshi Takase (Ehime University) for their helpful assistance.
References
-
Batail, P. Chem. Rev. 2004, 104, 4887–4890. doi:10.1021/cr040697x
Return to citation in text: [1] [2] -
Yamada, Y.; Sugimoto, T. TTF Chemistry. Fundamentals and Applications of Tetrathiafulvalene; KODANSHA–Springer: New York, NY, U.S.A., 2004.
Return to citation in text: [1] -
Ishiguro, T.; Yamaji, K.; Saito, G. Organic Superconductors, 2nd ed.; Springer Series in Solid-State Sciences, Vol. 88; Springer: Berlin, Germany, 1998. doi:10.1007/978-3-642-58262-2
Return to citation in text: [1] -
Saito, G.; Yoshida, Y. Bull. Chem. Soc. Jpn. 2007, 80, 1–137. doi:10.1246/bcsj.80.1
Return to citation in text: [1] -
Desiraju, G. R. Angew. Chem., Int. Ed. Engl. 1995, 34, 2311–2327. doi:10.1002/anie.199523111
Return to citation in text: [1] -
Lehn, J.-M. Science 2002, 295, 2400–2403. doi:10.1126/science.1071063
Return to citation in text: [1] -
Hoeben, F. J. M.; Jonkeijm, P.; Meijer, E. W.; Schenning, A. P. H. J. Chem. Rev. 2005, 105, 1491–1546. doi:10.1021/cr030070z
Return to citation in text: [1] -
Whitesides, G. M.; Grzybowski, B. Science 2002, 295, 2418–2421. doi:10.1126/science.1070821
Return to citation in text: [1] -
Nayak, S.; Lyon, L. A. Angew. Chem., Int. Ed. 2005, 44, 7686–7708. doi:10.1002/anie.200501321
Return to citation in text: [1] -
Gomar-Nadal, E.; Puigmartí-Luis, J.; Amabilino, D. B. Chem. Soc. Rev. 2008, 37, 490–504. doi:10.1039/B703825A
Return to citation in text: [1] -
Li, C.; Bai, H.; Shi, G. Chem. Soc. Rev. 2009, 38, 2397–2409. doi:10.1039/b816681c
Return to citation in text: [1] -
Bryce, M. R.; Marshallsay, G. J.; Moore, A. J. J. Org. Chem. 1992, 57, 4859–4862. doi:10.1021/jo00044a020
Return to citation in text: [1] -
Formigué, M.; Johannsen, I.; Boubekeur, K.; Nelson, C.; Batail, P. J. Am. Chem. Soc. 1993, 115, 3752–3759. doi:10.1021/ja00062a047
Return to citation in text: [1] -
Iyoda, M.; Fukuda, M.; Yoshida, M.; Sasaki, S. Synth. Met. 1995, 70, 1171–1172. doi:10.1016/0379-6779(94)02806-A
Return to citation in text: [1] -
Iyoda, M.; Fukuda, M.; Yoshida, M.; Sasaki, S. Chem. Lett. 1994, 23, 2369–2372. doi:10.1246/cl.1994.2369
Return to citation in text: [1] -
González, A.; Segura, J. K.; Martín, N. Tetrahedron Lett. 2000, 41, 3083–3086. doi:10.1016/S0040-4039(00)00344-0
Return to citation in text: [1] -
Kanibolotsky, A.; Roquet, S.; Cariou, M.; Leriche, P.; Turrin, C.-O.; de Bettingnies, R.; Caminade, A.-M.; Majoral, J.-P.; Khodorkovsky, V.; Gorgues, A. Org. Lett. 2004, 6, 2109–2112. doi:10.1021/ol049648x
Return to citation in text: [1] -
Hasegawa, M.; Takano, J.-i.; Enozawa, H.; Kuwatani, Y.; Iyoda, M. Tetrahedron Lett. 2004, 45, 4109–4112. doi:10.1016/j.tetlet.2004.03.150
Return to citation in text: [1] [2] [3] -
Jia, H.-P.; Liu, S.-X.; Sanguine, L.; Levillain, E.; Decurtins, S. J. Org. Chem. 2009, 74, 5727–5729. doi:10.1021/jo901054b
Return to citation in text: [1] -
Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Heterocycles 2010, 80, 909–915. doi:10.3987/COM-09-S(S)122
Return to citation in text: [1] [2] -
Pop, F.; Melan, C.; Danila, I.; Linares, M.; Beljonne, D.; Amabilino, D. B.; Avarvari, N. Chem. – Eur. J. 2014, 20, 17443–17453. doi:10.1002/chem.201404753
Return to citation in text: [1] [2] -
Hasegawa, M.; Enozawa, H.; Kawabata, Y.; Iyoda, M. J. Am. Chem. Soc. 2007, 129, 3072–3073. doi:10.1021/ja069025+
Return to citation in text: [1] [2] [3] -
Takase, M.; Yoshida, N.; Nishinaga, T.; Iyoda, M. Org. Lett. 2011, 13, 3896–3899. doi:10.1021/ol2014279
Return to citation in text: [1] [2] [3] [4] -
Jeppesen, J. O.; Nielsen, M. B.; Becher, J. Chem. Rev. 2004, 104, 5115–5131. doi:10.1021/cr030630u
Return to citation in text: [1] -
Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Hasegawa, M.; Iyoda, M. Chem. Soc. Rev. 2010, 39, 2420–2427. doi:10.1039/b909347h
Return to citation in text: [1] [2] [3] -
Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n
Return to citation in text: [1] -
Pérez, E. M.; Illescas, B. M.; Herranz, M. Á.; Martín, N. New J. Chem. 2009, 33, 228–234. doi:10.1039/B816272G
Return to citation in text: [1] -
Brunetti, F. G.; López, J. L.; Atienza, C.; Martín, N. J. Mater. Chem. 2012, 22, 4188–4205. doi:10.1039/c2jm15710a
Return to citation in text: [1] -
Christensen, C. A.; Bryce, M. R.; Batsanov, A. S.; Becher, J. Chem. Commun. 2000, 331–332. doi:10.1039/A909882H
Return to citation in text: [1] -
Christensen, C. A.; Bryce, M. R.; Becher, J. Synthesis 2000, 1695–1704. doi:10.1055/s-2000-8203
Return to citation in text: [1] -
Zou, L.; Xu, W.; Shao, X.; Zhang, D.; Wang, Q.; Zhu, D. Org. Biomol. Chem. 2003, 1, 2157–2159. doi:10.1039/b301587d
Return to citation in text: [1] -
Le Derf, F.; Levillain, E.; Trippé, G.; Gorgues, A.; Sallé, M.; Sebastían, R.-M.; Caminade, A.-M.; Majoral, J.-P. Angew. Chem., Int. Ed. 2001, 40, 224–227. doi:10.1002/1521-3773(20010105)40:1<224::AID-ANIE224>3.0.CO;2-O
Return to citation in text: [1] -
Kreher, D.; Cariou, M.; Liu, S.-G.; Levillain, E.; Veciana, J.; Rovira, C.; Gorgues, A.; Hudhomme, P. J. Mater. Chem. 2002, 12, 2137–2159. doi:10.1039/b201695h
Return to citation in text: [1] -
Mas-Torrent, M.; Rodrígues-Mias, R. A.; Solà, M.; Molins, M. A.; Pons, M.; Vidal-Gancedo, J.; Veciana, J.; Rovira, C. J. Org. Chem. 2002, 67, 566–575. doi:10.1021/jo010748f
Return to citation in text: [1] -
Bras, Y. L.; Sallé, M.; Leriche, P.; Mingotaud, C.; Richomme, P.; Møller, J. J. Mater. Chem. 1997, 7, 2393–2396. doi:10.1039/a704005i
Return to citation in text: [1] -
Becher, J.; Brimert, T.; Jeppesen, J. O.; Pedersen, J. Z.; Zubarev, R.; Bjørnholm, T.; Reitzel, N.; Jensen, T. R.; Kjaer, K.; Levillain, E. Angew. Chem., Int. Ed. 2001, 40, 2497–2500. doi:10.1002/1521-3773(20010702)40:13<2497::AID-ANIE2497>3.0.CO;2-F
Return to citation in text: [1] [2] -
Cook, M. J.; Cooke, G.; Jafari-Fini, A. Chem. Commun. 1996, 1925. doi:10.1039/CC9960001925
Return to citation in text: [1] -
Farren, C.; Christensen, C. A.; FitzGerald, S.; Bryce, M. R.; Beeby, A. J. Org. Chem. 2002, 67, 9130. doi:10.1021/jo020340y
Return to citation in text: [1] -
Sly, J.; Kasák, P.; Gomar-Nadal, E.; Rovira, C.; Górriz, L.; Thordardarson, P.; Amabilino, D. B.; Rowan, A. E.; Nolte, R. J. M. Chem. Commun. 2005, 1255–1257. doi:10.1039/b416034g
Return to citation in text: [1] [2] -
Nielsen, K. A.; Cho, W.-S.; Jeppesen, J. O.; Lynch, V. M.; Becher, J.; Sessler, J. L. J. Am. Chem. Soc. 2004, 126, 16296–16297. doi:10.1021/ja044664a
Return to citation in text: [1] -
Nielsen, K. A.; Bähring, S.; Jeppesen, J. O. Chem. – Eur. J. 2011, 17, 11001–11007. doi:10.1002/chem.201101266
Return to citation in text: [1] -
Davis, C. M.; Lim, J. M.; Larsen, K. R.; Kim, D. S.; Sung, Y. M.; Lyons, D. M.; Lynch, V. M.; Nielsen, K. A.; Jeppesen, J. O.; Kim, D.; Park, J. S.; Sessler, J. L. J. Am. Chem. Soc. 2014, 136, 10410–10417. doi:10.1021/ja504077f
Return to citation in text: [1] -
Pérez, E. M.; Sierra, M.; Sánchez, L.; Torres, M. R.; Viruela, R.; Viruela, P. M.; Ortí, E.; Martín, N. Angew. Chem., Int. Ed. 2007, 46, 1847–1851. doi:10.1002/anie.200604327
Return to citation in text: [1] -
Sugimoto, T.; Awaji, H.; Misaki, Y.; Yoshida, Z.; Kai, Y.; Nakagawa, H.; Kasai, N. J. Am. Chem. Soc. 1985, 107, 5792–5793. doi:10.1021/ja00306a030
Return to citation in text: [1] -
Sugimoto, T.; Misaki, Y.; Arai, Y.; Yamamoto, Y.; Yoshida, Z.; Kai, Y.; Kasai, N. J. Am. Chem. Soc. 1988, 110, 628–629. doi:10.1021/ja00210a069
Return to citation in text: [1] -
Sugimoto, T.; Misaki, Y.; Kajita, T.; Yoshida, Z.; Kai, Y.; Kasai, N. J. Am. Chem. Soc. 1987, 109, 4106–4107. doi:10.1021/ja00247a042
Return to citation in text: [1] -
Warman, J. M.; de Haas, M. P.; Dicker, G.; Grozema, F. C.; Piris, J.; Debije, M. G. Chem. Mater. 2004, 16, 4600–4609. doi:10.1021/cm049577w
Return to citation in text: [1] -
Kroeze, J. E.; Savenije, T. J.; Vermeulen, M. J. W.; Warman, J. M. J. Phys. Chem. B 2003, 107, 7696–7705. doi:10.1021/jp0217738
Return to citation in text: [1] -
Kostecki, R.; Schnyder, B.; Alliata, D.; Song, X.; Kinoshita, K.; Kötz, R. Thin Solid Films 2001, 396, 36–43. doi:10.1016/S0040-6090(01)01185-3
Return to citation in text: [1] -
Müllen, K.; Rabe, J. P. Acc. Chem. Res. 2008, 41, 511–520. doi:10.1021/ar7001446
Return to citation in text: [1] -
Joergensen, M.; Bechgaard, K.; Bjoernholm, T.; Sommer-Larsen, P.; Hansen, L. G.; Schaumburg, K. J. Org. Chem. 1994, 59, 5877–5882. doi:10.1021/jo00099a012
Return to citation in text: [1] -
Le Gall, T.; Pearson, C.; Bryce, M. R.; Petty, M. C.; Dahlgaard, H.; Becher, J. Eur. J. Org. Chem. 2003, 3562–3568. doi:10.1002/ejoc.200300286
Return to citation in text: [1] -
Kitamura, T.; Nakaso, S.; Mizoshita, N.; Tochigi, Y.; Shimomura, T.; Moriyama, M.; Ito, K.; Kato, T. J. Am. Chem. Soc. 2005, 127, 14769–14775. doi:10.1021/ja053496z
Return to citation in text: [1] -
Kitahara, T.; Shirakawa, M.; Kawano, S.-i.; Beginn, U.; Fujita, N.; Shinkai, S. J. Am. Chem. Soc. 2005, 127, 14980–14981. doi:10.1021/ja0552038
Return to citation in text: [1] -
Wang, C.; Zhang, D.; Zhu, D. J. Am. Chem. Soc. 2005, 127, 16372–16373. doi:10.1021/ja055800u
Return to citation in text: [1] -
Inokuchi, H.; Saito, G.; Seki, K.; Wu, P.; Tanf, T. B.; Mori, T.; Imaeda, K.; Enoki, T.; Higuchi, Y.; Inaka, K.; Yasuoka, N. Chem. Lett. 1986, 15, 1263–1266. doi:10.1246/cl.1986.1263
Return to citation in text: [1] [2] -
Honna, Y.; Isomura, E.; Enozawa, H.; Hasegawa, M.; Takase, M.; Nishinaga, T.; Iyoda, M. Tetrahedron Lett. 2010, 51, 679–682. doi:10.1016/j.tetlet.2009.11.106
Return to citation in text: [1] -
Iyoda, M.; Hasegawa, M.; Takano, J.-i.; Hara, K.; Kuwatani, Y. Chem. Lett. 2002, 32, 590–591. doi:10.1246/cl.2002.590
Return to citation in text: [1] [2] -
Iyoda, M.; Kuwatani, Y.; Ueno, U.; Oda, M. Chem. Commun. 1992, 158–159. doi:10.1039/c39920000158
Return to citation in text: [1] -
Rosokha, S. V.; Kochi, J. K. J. Am. Chem. Soc. 2007, 129, 828–838. doi:10.1021/ja064166x
Return to citation in text: [1] [2] -
Benniston, A. C. Chem. Soc. Rev. 2004, 33, 573–578. doi:10.1039/B309963F
Return to citation in text: [1] -
Wouters, D.; Schubert, U. S. Angew. Chem., Int. Ed. 2004, 43, 2480–2495. doi:10.1002/anie.200300609
Return to citation in text: [1] -
Wassel, R. A.; Gorman, C. B. Angew. Chem., Int. Ed. 2004, 43, 5120–5123. doi:10.1002/anie.200301735
Return to citation in text: [1] -
Palermo, V.; Samorì, P. Angew. Chem., Int. Ed. 2007, 46, 4428–4432. doi:10.1002/anie.200700416
Return to citation in text: [1] -
Enozawa, H.; Hasegawa, M.; Takamatsu, D.; Fukui, K.-i.; Iyoda, M. Org. Lett. 2006, 8, 1917–1920. doi:10.1021/ol0605530
Return to citation in text: [1] -
Enozawa, H.; Hasegawa, M.; Isomura, E.; Nishinaga, T.; Kato, T.; Yamato, M.; Kimura, T.; Iyoda, M. ChemPhysChem 2009, 10, 2607–2611. doi:10.1002/cphc.200900545
Return to citation in text: [1] [2] -
Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] -
Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Chem. Commun. 2004, 2042–2043. doi:10.1039/b407200f
Return to citation in text: [1] [2] [3] -
Lincke, K.; Frellsen, A. F.; Parker, C. R.; Bond, A. D.; Hammerich, O.; Nielsen, M. B. Angew. Chem., Int. Ed. 2012, 51, 6099–6102. doi:10.1002/anie.201202324
Return to citation in text: [1] [2] [3] -
Hanai, Y.; Rahman, M. J.; Yamakawa, J.; Takase, M.; Nishinaga, T.; Hasegawa, M.; Kamada, K.; Iyoda, M. Chem. – Asian J. 2011, 6, 2940–2945.
Return to citation in text: [1] -
Andersson, A. S.; Kilså, K.; Hassenkam, T.; Gisselbrecht, J.-P.; Boudon, C.; Gross, M.; Nielsen, M. B.; Diederich, F. Chem. – Eur. J. 2006, 12, 8451–8459. doi:10.1002/chem.200600986
Return to citation in text: [1] [2] -
Hasegawa, M.; Daigoku, K.; Hashimoto, K.; Nishikawa, H.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 51–60. doi:10.1246/bcsj.20110224
Return to citation in text: [1] -
Martin, R. B. Chem. Rev. 1996, 96, 3043–3064. doi:10.1021/cr960037v
Return to citation in text: [1] -
Takase, M.; Yoshida, N.; Narita, T.; Fujio, F.; Nishinaga, T.; Iyoda, M. RSC Adv. 2012, 2, 3221–3224. doi:10.1039/c2ra00035k
Return to citation in text: [1] -
Inoue, R.; Hasegawa, M.; Mazaki, Y. Chem. Lett. 2015, 44, 448–450. doi:10.1246/cl.141165
Return to citation in text: [1] -
Nalluri, S. K. M.; Shivarova, N.; Kanibolotsky, A. L.; Zelzer, M.; Gupta, S.; Frederix, P. W. J. M.; Skabara, P. J.; Gleskova, H.; Ulijn, R. V. Langmuir 2014, 30, 12429–12437. doi:10.1021/la503459y
Return to citation in text: [1] -
Tatewaki, Y.; Hatanaka, T.; Tsunashima, R.; Nakamura, T.; Kimura, M.; Shirai, H. Chem. – Asian J. 2009, 4, 1474–1479. doi:10.1002/asia.200900044
Return to citation in text: [1] -
Tanaka, K.; Kunita, T.; Ishiguro, F.; Naka, K.; Chujo, Y. Langmuir 2009, 25, 6929–6933. doi:10.1021/la900219b
Return to citation in text: [1] -
Ahn, S.; Kim, Y.; Beak, S.; Ishimoto, S.; Enozawa, H.; Isomura, E.; Hasegawa, M.; Iyoda, M.; Park, Y. J. Mater. Chem. 2010, 20, 10817–10823. doi:10.1039/c0jm02628j
Return to citation in text: [1] -
Tanaka, K.; Matsumoto, T.; Ishiguro, F.; Chujo, Y. J. Mater. Chem. 2011, 21, 9603–9607. doi:10.1039/c1jm11161b
Return to citation in text: [1] -
Jain, A.; Rao, K. V.; Mogera, U.; Sagade, A. A.; George, S. J. Chem. – Eur. J. 2011, 17, 12355–12361. doi:10.1002/chem.201101813
Return to citation in text: [1] -
Akutagawa, T.; Ohta, T.; Hasegawa, T.; Nakamura, T.; Christensen, C. A.; Becher, J. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5028–5033. doi:10.1073/pnas.082644299
Return to citation in text: [1] -
Akutagawa, T.; Kakiuchi, K.; Hasegawa, T.; Noro, S.-i.; Nakamura, T.; Hasegawa, H.; Mashiko, S.; Becher, J. Angew. Chem., Int. Ed. 2005, 44, 7283–7287. doi:10.1002/anie.200502336
Return to citation in text: [1]
| 52. | Joergensen, M.; Bechgaard, K.; Bjoernholm, T.; Sommer-Larsen, P.; Hansen, L. G.; Schaumburg, K. J. Org. Chem. 1994, 59, 5877–5882. doi:10.1021/jo00099a012 |
| 53. | Le Gall, T.; Pearson, C.; Bryce, M. R.; Petty, M. C.; Dahlgaard, H.; Becher, J. Eur. J. Org. Chem. 2003, 3562–3568. doi:10.1002/ejoc.200300286 |
| 83. | Akutagawa, T.; Ohta, T.; Hasegawa, T.; Nakamura, T.; Christensen, C. A.; Becher, J. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 5028–5033. doi:10.1073/pnas.082644299 |
| 84. | Akutagawa, T.; Kakiuchi, K.; Hasegawa, T.; Noro, S.-i.; Nakamura, T.; Hasegawa, H.; Mashiko, S.; Becher, J. Angew. Chem., Int. Ed. 2005, 44, 7283–7287. doi:10.1002/anie.200502336 |
| 54. | Kitamura, T.; Nakaso, S.; Mizoshita, N.; Tochigi, Y.; Shimomura, T.; Moriyama, M.; Ito, K.; Kato, T. J. Am. Chem. Soc. 2005, 127, 14769–14775. doi:10.1021/ja053496z |
| 55. | Kitahara, T.; Shirakawa, M.; Kawano, S.-i.; Beginn, U.; Fujita, N.; Shinkai, S. J. Am. Chem. Soc. 2005, 127, 14980–14981. doi:10.1021/ja0552038 |
| 56. | Wang, C.; Zhang, D.; Zhu, D. J. Am. Chem. Soc. 2005, 127, 16372–16373. doi:10.1021/ja055800u |
| 22. | Hasegawa, M.; Enozawa, H.; Kawabata, Y.; Iyoda, M. J. Am. Chem. Soc. 2007, 129, 3072–3073. doi:10.1021/ja069025+ |
| 78. | Tatewaki, Y.; Hatanaka, T.; Tsunashima, R.; Nakamura, T.; Kimura, M.; Shirai, H. Chem. – Asian J. 2009, 4, 1474–1479. doi:10.1002/asia.200900044 |
| 79. | Tanaka, K.; Kunita, T.; Ishiguro, F.; Naka, K.; Chujo, Y. Langmuir 2009, 25, 6929–6933. doi:10.1021/la900219b |
| 80. | Ahn, S.; Kim, Y.; Beak, S.; Ishimoto, S.; Enozawa, H.; Isomura, E.; Hasegawa, M.; Iyoda, M.; Park, Y. J. Mater. Chem. 2010, 20, 10817–10823. doi:10.1039/c0jm02628j |
| 81. | Tanaka, K.; Matsumoto, T.; Ishiguro, F.; Chujo, Y. J. Mater. Chem. 2011, 21, 9603–9607. doi:10.1039/c1jm11161b |
| 82. | Jain, A.; Rao, K. V.; Mogera, U.; Sagade, A. A.; George, S. J. Chem. – Eur. J. 2011, 17, 12355–12361. doi:10.1002/chem.201101813 |
| 77. | Nalluri, S. K. M.; Shivarova, N.; Kanibolotsky, A. L.; Zelzer, M.; Gupta, S.; Frederix, P. W. J. M.; Skabara, P. J.; Gleskova, H.; Ulijn, R. V. Langmuir 2014, 30, 12429–12437. doi:10.1021/la503459y |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 60. | Iyoda, M.; Kuwatani, Y.; Ueno, U.; Oda, M. Chem. Commun. 1992, 158–159. doi:10.1039/c39920000158 |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 59. | Iyoda, M.; Hasegawa, M.; Takano, J.-i.; Hara, K.; Kuwatani, Y. Chem. Lett. 2002, 32, 590–591. doi:10.1246/cl.2002.590 |
| 59. | Iyoda, M.; Hasegawa, M.; Takano, J.-i.; Hara, K.; Kuwatani, Y. Chem. Lett. 2002, 32, 590–591. doi:10.1246/cl.2002.590 |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 58. | Honna, Y.; Isomura, E.; Enozawa, H.; Hasegawa, M.; Takase, M.; Nishinaga, T.; Iyoda, M. Tetrahedron Lett. 2010, 51, 679–682. doi:10.1016/j.tetlet.2009.11.106 |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 57. | Inokuchi, H.; Saito, G.; Seki, K.; Wu, P.; Tanf, T. B.; Mori, T.; Imaeda, K.; Enoki, T.; Higuchi, Y.; Inaka, K.; Yasuoka, N. Chem. Lett. 1986, 15, 1263–1266. doi:10.1246/cl.1986.1263 |
| 22. | Hasegawa, M.; Enozawa, H.; Kawabata, Y.; Iyoda, M. J. Am. Chem. Soc. 2007, 129, 3072–3073. doi:10.1021/ja069025+ |
| 57. | Inokuchi, H.; Saito, G.; Seki, K.; Wu, P.; Tanf, T. B.; Mori, T.; Imaeda, K.; Enoki, T.; Higuchi, Y.; Inaka, K.; Yasuoka, N. Chem. Lett. 1986, 15, 1263–1266. doi:10.1246/cl.1986.1263 |
| 61. | Rosokha, S. V.; Kochi, J. K. J. Am. Chem. Soc. 2007, 129, 828–838. doi:10.1021/ja064166x |
| 18. | Hasegawa, M.; Takano, J.-i.; Enozawa, H.; Kuwatani, Y.; Iyoda, M. Tetrahedron Lett. 2004, 45, 4109–4112. doi:10.1016/j.tetlet.2004.03.150 |
| 61. | Rosokha, S. V.; Kochi, J. K. J. Am. Chem. Soc. 2007, 129, 828–838. doi:10.1021/ja064166x |
| 71. | Hanai, Y.; Rahman, M. J.; Yamakawa, J.; Takase, M.; Nishinaga, T.; Hasegawa, M.; Kamada, K.; Iyoda, M. Chem. – Asian J. 2011, 6, 2940–2945. |
| 72. | Andersson, A. S.; Kilså, K.; Hassenkam, T.; Gisselbrecht, J.-P.; Boudon, C.; Gross, M.; Nielsen, M. B.; Diederich, F. Chem. – Eur. J. 2006, 12, 8451–8459. doi:10.1002/chem.200600986 |
| 70. | Lincke, K.; Frellsen, A. F.; Parker, C. R.; Bond, A. D.; Hammerich, O.; Nielsen, M. B. Angew. Chem., Int. Ed. 2012, 51, 6099–6102. doi:10.1002/anie.201202324 |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 20. | Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Heterocycles 2010, 80, 909–915. doi:10.3987/COM-09-S(S)122 |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 26. | Hasegawa, M.; Iyoda, M. Chem. Soc. Rev. 2010, 39, 2420–2427. doi:10.1039/b909347h |
| 66. | Enozawa, H.; Hasegawa, M.; Takamatsu, D.; Fukui, K.-i.; Iyoda, M. Org. Lett. 2006, 8, 1917–1920. doi:10.1021/ol0605530 |
| 67. | Enozawa, H.; Hasegawa, M.; Isomura, E.; Nishinaga, T.; Kato, T.; Yamato, M.; Kimura, T.; Iyoda, M. ChemPhysChem 2009, 10, 2607–2611. doi:10.1002/cphc.200900545 |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 69. | Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Chem. Commun. 2004, 2042–2043. doi:10.1039/b407200f |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 62. | Benniston, A. C. Chem. Soc. Rev. 2004, 33, 573–578. doi:10.1039/B309963F |
| 63. | Wouters, D.; Schubert, U. S. Angew. Chem., Int. Ed. 2004, 43, 2480–2495. doi:10.1002/anie.200300609 |
| 64. | Wassel, R. A.; Gorman, C. B. Angew. Chem., Int. Ed. 2004, 43, 5120–5123. doi:10.1002/anie.200301735 |
| 65. | Palermo, V.; Samorì, P. Angew. Chem., Int. Ed. 2007, 46, 4428–4432. doi:10.1002/anie.200700416 |
| 69. | Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Chem. Commun. 2004, 2042–2043. doi:10.1039/b407200f |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 6. | Lehn, J.-M. Science 2002, 295, 2400–2403. doi:10.1126/science.1071063 |
| 7. | Hoeben, F. J. M.; Jonkeijm, P.; Meijer, E. W.; Schenning, A. P. H. J. Chem. Rev. 2005, 105, 1491–1546. doi:10.1021/cr030070z |
| 8. | Whitesides, G. M.; Grzybowski, B. Science 2002, 295, 2418–2421. doi:10.1126/science.1070821 |
| 34. | Kreher, D.; Cariou, M.; Liu, S.-G.; Levillain, E.; Veciana, J.; Rovira, C.; Gorgues, A.; Hudhomme, P. J. Mater. Chem. 2002, 12, 2137–2159. doi:10.1039/b201695h |
| 35. | Mas-Torrent, M.; Rodrígues-Mias, R. A.; Solà, M.; Molins, M. A.; Pons, M.; Vidal-Gancedo, J.; Veciana, J.; Rovira, C. J. Org. Chem. 2002, 67, 566–575. doi:10.1021/jo010748f |
| 70. | Lincke, K.; Frellsen, A. F.; Parker, C. R.; Bond, A. D.; Hammerich, O.; Nielsen, M. B. Angew. Chem., Int. Ed. 2012, 51, 6099–6102. doi:10.1002/anie.201202324 |
| 5. | Desiraju, G. R. Angew. Chem., Int. Ed. Engl. 1995, 34, 2311–2327. doi:10.1002/anie.199523111 |
| 36. | Bras, Y. L.; Sallé, M.; Leriche, P.; Mingotaud, C.; Richomme, P.; Møller, J. J. Mater. Chem. 1997, 7, 2393–2396. doi:10.1039/a704005i |
| 3. | Ishiguro, T.; Yamaji, K.; Saito, G. Organic Superconductors, 2nd ed.; Springer Series in Solid-State Sciences, Vol. 88; Springer: Berlin, Germany, 1998. doi:10.1007/978-3-642-58262-2 |
| 4. | Saito, G.; Yoshida, Y. Bull. Chem. Soc. Jpn. 2007, 80, 1–137. doi:10.1246/bcsj.80.1 |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 2. | Yamada, Y.; Sugimoto, T. TTF Chemistry. Fundamentals and Applications of Tetrathiafulvalene; KODANSHA–Springer: New York, NY, U.S.A., 2004. |
| 30. | Christensen, C. A.; Bryce, M. R.; Batsanov, A. S.; Becher, J. Chem. Commun. 2000, 331–332. doi:10.1039/A909882H |
| 31. | Christensen, C. A.; Bryce, M. R.; Becher, J. Synthesis 2000, 1695–1704. doi:10.1055/s-2000-8203 |
| 32. | Zou, L.; Xu, W.; Shao, X.; Zhang, D.; Wang, Q.; Zhu, D. Org. Biomol. Chem. 2003, 1, 2157–2159. doi:10.1039/b301587d |
| 33. | Le Derf, F.; Levillain, E.; Trippé, G.; Gorgues, A.; Sallé, M.; Sebastían, R.-M.; Caminade, A.-M.; Majoral, J.-P. Angew. Chem., Int. Ed. 2001, 40, 224–227. doi:10.1002/1521-3773(20010105)40:1<224::AID-ANIE224>3.0.CO;2-O |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 24. | Jeppesen, J. O.; Nielsen, M. B.; Becher, J. Chem. Rev. 2004, 104, 5115–5131. doi:10.1021/cr030630u |
| 27. | Canevet, D.; Sallé, M.; Zhang, G.; Zhang, D.; Zhu, D. Chem. Commun. 2009, 2245–2269. doi:10.1039/b818607n |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 22. | Hasegawa, M.; Enozawa, H.; Kawabata, Y.; Iyoda, M. J. Am. Chem. Soc. 2007, 129, 3072–3073. doi:10.1021/ja069025+ |
| 23. | Takase, M.; Yoshida, N.; Nishinaga, T.; Iyoda, M. Org. Lett. 2011, 13, 3896–3899. doi:10.1021/ol2014279 |
| 28. | Pérez, E. M.; Illescas, B. M.; Herranz, M. Á.; Martín, N. New J. Chem. 2009, 33, 228–234. doi:10.1039/B816272G |
| 29. | Brunetti, F. G.; López, J. L.; Atienza, C.; Martín, N. J. Mater. Chem. 2012, 22, 4188–4205. doi:10.1039/c2jm15710a |
| 18. | Hasegawa, M.; Takano, J.-i.; Enozawa, H.; Kuwatani, Y.; Iyoda, M. Tetrahedron Lett. 2004, 45, 4109–4112. doi:10.1016/j.tetlet.2004.03.150 |
| 73. | Hasegawa, M.; Daigoku, K.; Hashimoto, K.; Nishikawa, H.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 51–60. doi:10.1246/bcsj.20110224 |
| 12. | Bryce, M. R.; Marshallsay, G. J.; Moore, A. J. J. Org. Chem. 1992, 57, 4859–4862. doi:10.1021/jo00044a020 |
| 13. | Formigué, M.; Johannsen, I.; Boubekeur, K.; Nelson, C.; Batail, P. J. Am. Chem. Soc. 1993, 115, 3752–3759. doi:10.1021/ja00062a047 |
| 14. | Iyoda, M.; Fukuda, M.; Yoshida, M.; Sasaki, S. Synth. Met. 1995, 70, 1171–1172. doi:10.1016/0379-6779(94)02806-A |
| 15. | Iyoda, M.; Fukuda, M.; Yoshida, M.; Sasaki, S. Chem. Lett. 1994, 23, 2369–2372. doi:10.1246/cl.1994.2369 |
| 16. | González, A.; Segura, J. K.; Martín, N. Tetrahedron Lett. 2000, 41, 3083–3086. doi:10.1016/S0040-4039(00)00344-0 |
| 17. | Kanibolotsky, A.; Roquet, S.; Cariou, M.; Leriche, P.; Turrin, C.-O.; de Bettingnies, R.; Caminade, A.-M.; Majoral, J.-P.; Khodorkovsky, V.; Gorgues, A. Org. Lett. 2004, 6, 2109–2112. doi:10.1021/ol049648x |
| 18. | Hasegawa, M.; Takano, J.-i.; Enozawa, H.; Kuwatani, Y.; Iyoda, M. Tetrahedron Lett. 2004, 45, 4109–4112. doi:10.1016/j.tetlet.2004.03.150 |
| 19. | Jia, H.-P.; Liu, S.-X.; Sanguine, L.; Levillain, E.; Decurtins, S. J. Org. Chem. 2009, 74, 5727–5729. doi:10.1021/jo901054b |
| 20. | Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Heterocycles 2010, 80, 909–915. doi:10.3987/COM-09-S(S)122 |
| 21. | Pop, F.; Melan, C.; Danila, I.; Linares, M.; Beljonne, D.; Amabilino, D. B.; Avarvari, N. Chem. – Eur. J. 2014, 20, 17443–17453. doi:10.1002/chem.201404753 |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 9. | Nayak, S.; Lyon, L. A. Angew. Chem., Int. Ed. 2005, 44, 7686–7708. doi:10.1002/anie.200501321 |
| 10. | Gomar-Nadal, E.; Puigmartí-Luis, J.; Amabilino, D. B. Chem. Soc. Rev. 2008, 37, 490–504. doi:10.1039/B703825A |
| 11. | Li, C.; Bai, H.; Shi, G. Chem. Soc. Rev. 2009, 38, 2397–2409. doi:10.1039/b816681c |
| 25. | Iyoda, M.; Hasegawa, M.; Miyake, Y. Chem. Rev. 2004, 104, 5085–5113. doi:10.1021/cr030651o |
| 26. | Hasegawa, M.; Iyoda, M. Chem. Soc. Rev. 2010, 39, 2420–2427. doi:10.1039/b909347h |
| 69. | Hara, K.; Hasegawa, M.; Kuwatani, Y.; Enozawa, H.; Iyoda, M. Chem. Commun. 2004, 2042–2043. doi:10.1039/b407200f |
| 37. | Becher, J.; Brimert, T.; Jeppesen, J. O.; Pedersen, J. Z.; Zubarev, R.; Bjørnholm, T.; Reitzel, N.; Jensen, T. R.; Kjaer, K.; Levillain, E. Angew. Chem., Int. Ed. 2001, 40, 2497–2500. doi:10.1002/1521-3773(20010702)40:13<2497::AID-ANIE2497>3.0.CO;2-F |
| 37. | Becher, J.; Brimert, T.; Jeppesen, J. O.; Pedersen, J. Z.; Zubarev, R.; Bjørnholm, T.; Reitzel, N.; Jensen, T. R.; Kjaer, K.; Levillain, E. Angew. Chem., Int. Ed. 2001, 40, 2497–2500. doi:10.1002/1521-3773(20010702)40:13<2497::AID-ANIE2497>3.0.CO;2-F |
| 38. | Cook, M. J.; Cooke, G.; Jafari-Fini, A. Chem. Commun. 1996, 1925. doi:10.1039/CC9960001925 |
| 39. | Farren, C.; Christensen, C. A.; FitzGerald, S.; Bryce, M. R.; Beeby, A. J. Org. Chem. 2002, 67, 9130. doi:10.1021/jo020340y |
| 40. | Sly, J.; Kasák, P.; Gomar-Nadal, E.; Rovira, C.; Górriz, L.; Thordardarson, P.; Amabilino, D. B.; Rowan, A. E.; Nolte, R. J. M. Chem. Commun. 2005, 1255–1257. doi:10.1039/b416034g |
| 67. | Enozawa, H.; Hasegawa, M.; Isomura, E.; Nishinaga, T.; Kato, T.; Yamato, M.; Kimura, T.; Iyoda, M. ChemPhysChem 2009, 10, 2607–2611. doi:10.1002/cphc.200900545 |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 68. | Enozawa, H.; Takahashi, T.; Nishinaga, T.; Kato, T.; Hasegawa, M.; Iyoda, M. Bull. Chem. Soc. Jpn. 2012, 85, 1120–1137. doi:10.1246/bcsj.20120135 |
| 72. | Andersson, A. S.; Kilså, K.; Hassenkam, T.; Gisselbrecht, J.-P.; Boudon, C.; Gross, M.; Nielsen, M. B.; Diederich, F. Chem. – Eur. J. 2006, 12, 8451–8459. doi:10.1002/chem.200600986 |
| 70. | Lincke, K.; Frellsen, A. F.; Parker, C. R.; Bond, A. D.; Hammerich, O.; Nielsen, M. B. Angew. Chem., Int. Ed. 2012, 51, 6099–6102. doi:10.1002/anie.201202324 |
| 48. | Warman, J. M.; de Haas, M. P.; Dicker, G.; Grozema, F. C.; Piris, J.; Debije, M. G. Chem. Mater. 2004, 16, 4600–4609. doi:10.1021/cm049577w |
| 49. | Kroeze, J. E.; Savenije, T. J.; Vermeulen, M. J. W.; Warman, J. M. J. Phys. Chem. B 2003, 107, 7696–7705. doi:10.1021/jp0217738 |
| 50. | Kostecki, R.; Schnyder, B.; Alliata, D.; Song, X.; Kinoshita, K.; Kötz, R. Thin Solid Films 2001, 396, 36–43. doi:10.1016/S0040-6090(01)01185-3 |
| 51. | Müllen, K.; Rabe, J. P. Acc. Chem. Res. 2008, 41, 511–520. doi:10.1021/ar7001446 |
| 44. | Pérez, E. M.; Sierra, M.; Sánchez, L.; Torres, M. R.; Viruela, R.; Viruela, P. M.; Ortí, E.; Martín, N. Angew. Chem., Int. Ed. 2007, 46, 1847–1851. doi:10.1002/anie.200604327 |
| 23. | Takase, M.; Yoshida, N.; Nishinaga, T.; Iyoda, M. Org. Lett. 2011, 13, 3896–3899. doi:10.1021/ol2014279 |
| 45. | Sugimoto, T.; Awaji, H.; Misaki, Y.; Yoshida, Z.; Kai, Y.; Nakagawa, H.; Kasai, N. J. Am. Chem. Soc. 1985, 107, 5792–5793. doi:10.1021/ja00306a030 |
| 46. | Sugimoto, T.; Misaki, Y.; Arai, Y.; Yamamoto, Y.; Yoshida, Z.; Kai, Y.; Kasai, N. J. Am. Chem. Soc. 1988, 110, 628–629. doi:10.1021/ja00210a069 |
| 47. | Sugimoto, T.; Misaki, Y.; Kajita, T.; Yoshida, Z.; Kai, Y.; Kasai, N. J. Am. Chem. Soc. 1987, 109, 4106–4107. doi:10.1021/ja00247a042 |
| 76. | Inoue, R.; Hasegawa, M.; Mazaki, Y. Chem. Lett. 2015, 44, 448–450. doi:10.1246/cl.141165 |
| 43. | Davis, C. M.; Lim, J. M.; Larsen, K. R.; Kim, D. S.; Sung, Y. M.; Lyons, D. M.; Lynch, V. M.; Nielsen, K. A.; Jeppesen, J. O.; Kim, D.; Park, J. S.; Sessler, J. L. J. Am. Chem. Soc. 2014, 136, 10410–10417. doi:10.1021/ja504077f |
| 75. | Takase, M.; Yoshida, N.; Narita, T.; Fujio, F.; Nishinaga, T.; Iyoda, M. RSC Adv. 2012, 2, 3221–3224. doi:10.1039/c2ra00035k |
| 21. | Pop, F.; Melan, C.; Danila, I.; Linares, M.; Beljonne, D.; Amabilino, D. B.; Avarvari, N. Chem. – Eur. J. 2014, 20, 17443–17453. doi:10.1002/chem.201404753 |
| 23. | Takase, M.; Yoshida, N.; Nishinaga, T.; Iyoda, M. Org. Lett. 2011, 13, 3896–3899. doi:10.1021/ol2014279 |
| 40. | Sly, J.; Kasák, P.; Gomar-Nadal, E.; Rovira, C.; Górriz, L.; Thordardarson, P.; Amabilino, D. B.; Rowan, A. E.; Nolte, R. J. M. Chem. Commun. 2005, 1255–1257. doi:10.1039/b416034g |
| 26. | Hasegawa, M.; Iyoda, M. Chem. Soc. Rev. 2010, 39, 2420–2427. doi:10.1039/b909347h |
| 41. | Nielsen, K. A.; Cho, W.-S.; Jeppesen, J. O.; Lynch, V. M.; Becher, J.; Sessler, J. L. J. Am. Chem. Soc. 2004, 126, 16296–16297. doi:10.1021/ja044664a |
| 42. | Nielsen, K. A.; Bähring, S.; Jeppesen, J. O. Chem. – Eur. J. 2011, 17, 11001–11007. doi:10.1002/chem.201101266 |
| 23. | Takase, M.; Yoshida, N.; Nishinaga, T.; Iyoda, M. Org. Lett. 2011, 13, 3896–3899. doi:10.1021/ol2014279 |
© 2015 Iyoda and Hasegawa; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (http://www.beilstein-journals.org/bjoc)