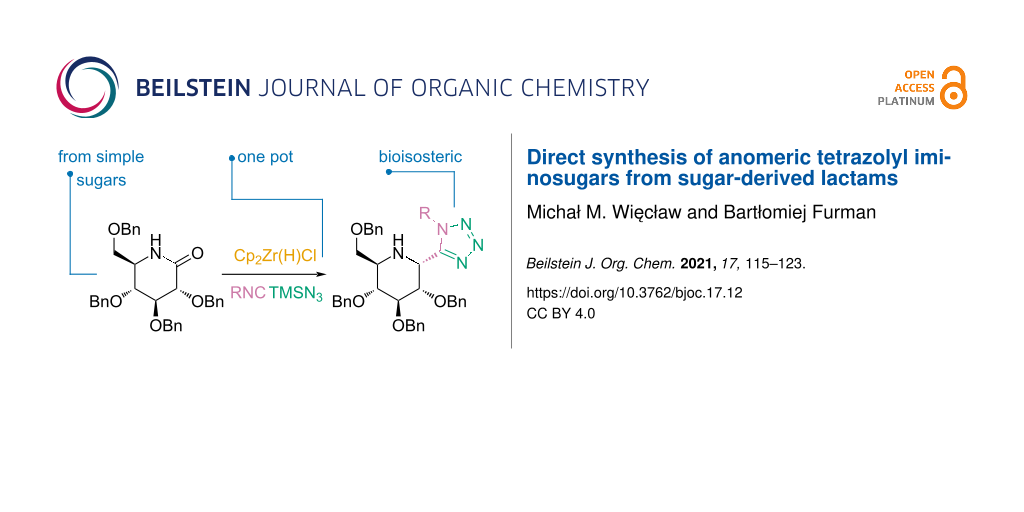
Abstract
Herein we present the direct asymmetric synthesis of tetrazole-functionalized 1-deoxynojirimycin derivatives from simple sugars via a Schwartz’s reagent-mediated reductive amide functionalization followed by a variant of the Ugi–azide multicomponent reaction. The anomeric configurations of two products were unambiguously confirmed by X-ray analysis. This work also describes examples of interesting further transformations of the title products. Finally, some surprising observations regarding the mechanism of their formation were made.

Graphical Abstract
Introduction
The transformation of an amide into another chemical moiety in a controlled manner is not a trivial task. Although the Vilsmeier–Haack reaction [1] or amide reduction with LiAlH4 are textbook examples that easily come to mind, there are not many other methods available. Simple alkyl and aryl amides, unlike other carbonyl compounds, typically do not undergo direct addition by a nucleophile, including active organometallic compounds. For this reason, it has been chemists’ long-lasting ambition to develop a reliable, mild, and selective methodology for amide functionalization [2]. Even though a tremendous amount of work has been already done towards this matter, it is still a highly active field of research. Several review articles have been written about this topic, enclosing most of the advances made to date [3-5].
A fascinating subset of these transformations encompass the reduction of amides to imines, with direct subsequent functionalization. One of the methodologies for such a modification was developed by Charette et al. In their procedure the combination of triflic anhydride and pyridine [6] (or its 2-fluoro derivative [7]) was used as an activating agent to transform amides into reactive iminium complexes. Another stoichiometric approach was presented by Georg et al. by utilization of zirconocene chloride hydride, known as Schwartz’s reagent [8]. This reduces the amide moiety, giving a complex that can be readily transformed into an imine or iminium cation [9].
It may perhaps be observed without straying too far afield from our primary focus that reduction of amides is actually not a leading use case of Schwartz’s reagent. It is employed principally for hydrozirconation of double and triple carbon–carbon bonds [10], and its application in chemistry of amides is quite recent. It can be also utilized in reduction of other unsaturated moieties, e.g., Pace et al. have shown reduction of isocyanates to formamides [11] and reduction of isothiocyanates to thioformamides [12] by its means.
There have also been some catalytic protocols developed for the reduction of amides to imines. The most notable examples incorporate iridium complexes and silanes [13,14]. Cheng and Brookhart showed that the chlorobis(cyclooctene)iridium dimer ([Ir(coe)2Cl]2) can act as the catalyst in combination with Et2SiH2 [15]. Surprisingly, they were able to obtain imines as well as amines using this methodology. Based on the works of Nagashima [16], an iridium-based protocol for tertiary amides was introduced by Dixon [17-19] and Huang [20,21]. Adolfsson expanded this by the use of molybdenum-based catalysts [22]. The reductive approach allows the issues associated with nucleophilic addition to amide carbonyl groups to be overcome and as such is finding its place in a growing number of synthetic applications [10].
The employment of these methods for modification of lactams is a challenge in its own right – there are hardly any examples of such transformations available in the literature [23]. Our group was the first to surmount this challenge by means of Schwartz’s reagent-mediated reductive functionalization. Since then, we have performed a number of different functionalizations of such cyclic systems with various complexity, and with a particular focus on the modification of sugar-derived lactams. As summarized in Scheme 1, this includes simple nucleophile addition to in situ-generated imines [23], the consecutive one-pot Mannich/Michael sequence leading to oligocyclic compounds [24], and employment in subsequent Joulié–Ugi multicomponent reactions [25].
![[1860-5397-17-12-i1]](/bjoc/content/inline/1860-5397-17-12-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Our previous efforts in the field of functionalization of sugar-derived lactams.
Scheme 1: Our previous efforts in the field of functionalization of sugar-derived lactams.
This work is an extension of these efforts and seeks to investigate the possibility of incorporating the Ugi–azide multicomponent reaction in this workflow. A molecule incorporating both an iminosugar and a tetrazole fragment is of particular interest, due to the interesting properties of both moieties (Figure 1). It is probably hard to overestimate the importance of sugar scaffolds in nature, and we believe that it speaks for itself, however, a significance of iminosugar derivatives may be less obvious. Several pharmaceuticals are based on this scaffold including the glucose-derived nojirimycin, an antibiotic and glycosidase inhibitor [26] and 1-deoxygalactonojirimycin, known under the trade name Galafold®, which is utilized for the treatment of the Fabry disease, a rare genetic condition [27]. On the other hand, the tetrazole moiety is known to have a bioisosteric relationship to carboxylic acids [28], which also makes them suitable for usage as biologically active compounds. Vasella et al., for example, have previously prepared compounds similar to reported ones in this work – fused iminosugar-tetrazoles – which have shown inhibition properties against bovine liver α-ᴅ-glucuronidase and human β-ʟ-iduronidase [29]. Moreover, there are numerous reports of the organocatalytic activity of chiral aminotriazoles and aminotetrazoles in number of reactions, such as the aldol reaction [30], Michael addition [31], Mannich reaction [32], and hydrogenation [33].
![[1860-5397-17-12-1]](/bjoc/content/figures/1860-5397-17-12-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Key concepts behind the goal of this work [34].
Figure 1: Key concepts behind the goal of this work [34].
Results and Discussion
Quite recently Xie and Dixon showed that it is possible to synthesize α-tetrazolylamines from simple and linear tertiary amides using an iridium-based catalytic protocol [19]. They have, however, only reported one example of lactam functionalization which only proceeded with moderate efficiency (1-tert-butylazepan-2-one, 41% yield of the desired product). Unfortunately, this approach cannot be utilized for the functionalization of secondary amides, like sugar-derived lactams, due to the aforementioned method’s limitation to tertiary amides. Our previous work shows that Charette’s methodology is also not applicable in this case, as it does not lead to the formation of an imine [23]. Luckily, we were able to use a formerly established strategy based on Georg’s procedure with standard Ugi–azide reaction conditions [35-39] in a one-pot, tandem process. Subjecting glucose-derived lactam 1 to such a procedure gave the desired product in good yield, but with virtually no diastereoselectivity, as shown in Scheme 2.
![[1860-5397-17-12-i2]](/bjoc/content/inline/1860-5397-17-12-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preliminary experiment in search of a procedure for the synthesis of 2-(1H-tetrazol-5-yl)-iminosugars.
Scheme 2: Preliminary experiment in search of a procedure for the synthesis of 2-(1H-tetrazol-5-yl)-iminosuga...
Optimization and scope
An initial optimization study for the proton donor for TMSN3 activation (shown in Table 1) using commonly encountered reagents for such reactions was performed. To our surprise, we observed the formation of the Ugi–azide product even in the absence of a protic additive. Moreover, the aprotic conditions proved to provide the highest yield and diastereoselectivity, thus were chosen as optimal (Table 1, entry 9.). We also tried to isolate the imine after the reduction step and carry out the second step in a solvent commonly used for the Ugi–azide reaction alone. For this, we observed a significant decrease in overall yield and suspect that the low stability of imines of type 2 may be the reason for this behavior.
Table 1: Optimization of 2-(1H-tetrazol-5-yl)-iminosugar synthesis via Schwartz’s reagent-mediated reduction of amides and Ugi–azide reaction.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-12-i10.svg?max-width=637&scale=1.0)
|
||||
| Entry | Additive | Solvent | Yield [%] | drc |
| 1 | MeOHd | THF | 65 | 43:57 |
| 2 | CF3CO2H | THF | 24 | 43:57 |
| 3 | AcOH | THF | 47 | 80:20 |
| 4 | Et3N · HCl | THF | 45 | 74:26 |
| 5 | H2O | THF | 34 | >95:5 |
| 6 | (CF3)2CHOH | THF | 35 | >95:5 |
| 7 | none | MeOH | 19e | >95:5 |
| 8 | none | DCM | 36e | >95:5 |
| 9 | none | THF | 73 | >95:5 |
a1.6 equiv Cp2Zr(H)Cl in THF under argon atmosphere; b1.6 equiv of additive (if applicable), 1.1 equiv CyNC, and 1.1 equiv TMSN3. c2-(R) to 2-(S), isolated. dAdditive used in excess. eThe imine was isolated after reduction.
The established optimal conditions were applied for the synthesis of selected examples of various 2-(1H-tetrazol-5-yl)-iminosugars (Table 2). Attempts at using this methodology to synthesize pentose-derived 2-(tetrazol-5-yl)-iminosugars, using 2,3,5-tri-O-benzyl-ᴅ-ribofuranose- and -arabinofuranose-derived lactams as substrates were made. Very unexpectedly, we failed to isolate such products although we did observe their formation via mass spectrometry of the reaction mixtures. Employing alternative procedures did not help, and none of the desired products were observed at all when applying iridium complex- or triflic anhydride-based methods.
Table 2: Synthesis of 2-(1H-tetrazol-5-yl)-iminosugars using optimized conditions. Reaction yield and dr are given.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-12-i11.svg?max-width=637&scale=1.0)
|
||||
| Entry | Product | R2 | Yield [%] | dra |
| 1 | 3a | Cy | 73 | >95:5 |
| 2 | 3b | CH2CO2Et | 49 | >95:5 |
| 3 | 3c | Bn | 18 | >95:5 |
| 4 | 3d | PMP | 29 | 79:21 |
| 5 | 3e | PMB | 42 | >95:5 |
| 6 | 3f | tert-butyl | 40 | >95:5 |
| 7 | 3g | tert-octyl | 48 | >95:5 |
| 8 | 5a | Cy | 33 | >95:5 |
| 9 | 5b | CH2CO2Et | 16 | >95:5 |
a2-(R) to 2-(S), isolated.
The methodology described here provides a pathway to new, interesting compounds, containing both an iminosugar and tetrazole moiety. Such compounds have not been seen to date, and their accessibility creates exciting synthetic opportunities. Here we present two examples of possible further transformations of the products obtained over the course of this research directed towards novel, attractive molecules.
Compound 3b underwent a cyclization reaction in the presence of benzoic acid at an elevated temperature yielding lactam 6 almost quantitatively. The deoxygenative reduction of this compound turned out to be challenging, as the typical procedure using LiAlH4 proved ineffective. We were able to obtain 7 using a Schwartz’s reagent-mediated amide activation methodology followed by NaBH4 reduction. This structure with three condensed rings can be seen as a new class of unnatural, chiral alkaloid scaffolds, potentially exhibiting pharmacological activity (Scheme 3) [40].
![[1860-5397-17-12-i3]](/bjoc/content/inline/1860-5397-17-12-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of a new class of alkaloid scaffold using the presented methodology.
Scheme 3: Synthesis of a new class of alkaloid scaffold using the presented methodology.
Various unsuccessful attempts were made to deprotect compound 3e. Unexpectedly, however, one of those experiments resulted in a rearrangement in the tetrazole ring, as shown in Scheme 4, upper path. We were able to obtain the desired aminotetrazole 9 by treating 3g with dry HCl at elevated temperature (Scheme 4, lower path). The resulting compound is particularly appealing, as similar scaffolds are widely used as organocatalysts. Such moieties are employed in a number of important synthetic transformations, including the aldol reaction [30], Michael addition [31], Mannich reaction [32], and hydrogenation [33]. We plan to test these possibilities in the near future.
![[1860-5397-17-12-i4]](/bjoc/content/inline/1860-5397-17-12-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of a new, chiral 2-(tetrazol-5-yl)-iminosugar based potential organocatalyst.
Scheme 4: Synthesis of a new, chiral 2-(tetrazol-5-yl)-iminosugar based potential organocatalyst.
Stereochemistry and configuration of products
As presented in Table 2, only one diastereomer of the desired iminosugar is obtained in almost all cases. This outstanding selectivity has been observed before and is described in our previous works devoted to the functionalization of sugar-derived lactams [23-25]. We explain it in the light of Woerpel’s model, which characterizes the direction of nucleophilic addition to oxocarbenium ions [41-43]. According to this concept, the conformational stability of the compound in question is the key property to consider when predicting the reaction’s stereoselectivity.
When the oxocarbenium ion is substituted, two diastereomeric half-chair conformers are possible: 3H4 and 4H3 (shown for a 4-substituted pyranose cation in Scheme 5). Both may undergo attack by a nucleophile in two ways: on the axial trajectory from the top or the bottom face. Such an event would result in the formation of the product as a chair (1C4, 4C1) or a skew-boat (1S3, 3S1) conformer, of which the former is favored, as it proceeds via the lower-energetic chair-like transition state. The favored path of action will result in addition syn or anti to the substituent in position 5, depending on the starting conformer. Therefore, once the ground conformer of the oxocarbenium ion is established, this logic may be used to predict the reaction’s stereochemistry.
![[1860-5397-17-12-i5]](/bjoc/content/inline/1860-5397-17-12-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Principle behind Woerpel’s model for prediction of the direction of nucleophile addition to oxocarbenium cations.
Scheme 5: Principle behind Woerpel’s model for prediction of the direction of nucleophile addition to oxocarb...
The same principle may be successfully applied to reactions of iminium cations. We have previously shown that in the case of glucose- and galactose-derived, O-benzyl-protected iminosugars the addition syn to the substituent in position 3 is favored (Scheme 6). This work proves no different, as the isolated major products were in such configuration. The experimental determination of this, however, was not straightforward in all cases.
![[1860-5397-17-12-i6]](/bjoc/content/inline/1860-5397-17-12-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Difference in conformational stability of glucose- and galactose-derived iminium cations and the major product of the nucleophile attack according to Woerpel’s model [24].
Scheme 6: Difference in conformational stability of glucose- and galactose-derived iminium cations and the maj...
We were able to determine the structure of compounds 3a and 3e unambiguously by means of X-ray analysis, as shown in Figure 2. The configuration of the remaining glucose based products 3 was easily determined by the analysis of 1H-1H coupling constants and NOE effects. Unfortunately, the same approach was not possible in the case of compounds 5, as 1H NMR spectroscopy showed indefinite results. In compound 5a the coupling constant between protons H2 and H3 has a value of 8.5 Hz. This cannot be associated with a particular relative configuration without comparison with the corresponding coupling constant in 2-epi-5a. But alas, this value is unknown, due to overlapping and broadening of the relevant signals in the 1H NMR spectrum of the compound in question. For the same reasons NOE effects present in 2-epi-5a cannot be accurately interpreted. However, analysis of the NOE effects in 5a, particularly a small effect between protons H2 and H7, suggest that it may be the diastereomer 2-(R), as shown in Figure 3. This result would be in accordance with the previously mentioned Woerpel’s model. The structure of compound 5b was assigned per analogiam, as diagnostic signals in 1H NMR spectrum were also overlapped.
![[1860-5397-17-12-2]](/bjoc/content/figures/1860-5397-17-12-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: ORTEP structures of compounds 3a and 3e obtained by X-ray analysis. Hydrogen atoms and benzyl groups are omitted for clarity. Full crystallographic data available in Supporting Information File 2 and Supporting Information File 3, and in Cambridge Crystallographic Database under CCDC-2001373 and CCDC-2001372 numbers, respectively.
Figure 2: ORTEP structures of compounds 3a and 3e obtained by X-ray analysis. Hydrogen atoms and benzyl group...
![[1860-5397-17-12-3]](/bjoc/content/figures/1860-5397-17-12-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Proposed structures of compounds 5a and 2-epi-5a with 1H-1H couplings and NOE effects shown.
Figure 3: Proposed structures of compounds 5a and 2-epi-5a with 1H-1H couplings and NOE effects shown.
We made an attempt at resolving this problem by means of the electronic circular dichroism (ECD) technique. We recorded an ECD spectrum of both compounds and compared it with simulated spectra, generated for both possible diastereomers (2-(R) and 2-(S)) using computational chemistry software. Unfortunately, we were not able to fit any of these simulations to the experimental data with sufficient certainty. For the inquisitive readers, this work is fully described in Supporting Information File 1.
Mechanism of the reaction
As mentioned previously, we observed Ugi–azide products, despite the absence of a proton donor in the reaction mixture. Intriguingly, this behavior is inconsistent with the generally accepted mechanism of this transformation, which assumes the hydrolysis of TMSN3 to HN3 and activation of the imine species by protonation. Scheme 7 presents our proposal for the possible course of the Ugi–azide reaction variant described in this work. We suppose that after reduction of amide I by Schwartz’s reagent, complex II undergoes a slow, spontaneous decomposition, yielding imine III. III then reacts with TMSN3, which acts as both, an imine activator and an azide anion source. Complex IV undergoes a subsequent addition of an isocyanide moiety (intermediate V), followed by an azide anion addition. Intermediate VI undergoes a cyclization, producing VII, a silylated derivative of the expected product. The hydrolysis of VII most likely occurs during the reaction’s work-up.
![[1860-5397-17-12-i7]](/bjoc/content/inline/1860-5397-17-12-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Proposed reaction mechanism for the described Ugi–azide reaction variant.
Scheme 7: Proposed reaction mechanism for the described Ugi–azide reaction variant.
Preliminary DTF calculations were performed on a simplified model to provisionally validate this proposed mechanism. The geometry of the intermediate species were optimized with Gaussian 09 software [44], using the B3LYP/LANL2DZ theory level for Zr and B3LYP/6-31G(d,p) for other atoms, with GD3 empirical dispersion correction. The optimization was followed by a single-point energy calculation using the larger basis set Def2TZVP with a PCM solvatation model for THF, as implemented in the Gaussian software. Energy values reported are a sum of electronic and zero-point energies.
Scheme 8 shows possible pathways for the spontaneous decomposition of the zirconium complex INT-1-A to the free imine species INT-3. This process is much more likely to occur via the 5-membered cyclic transition state TS-1-A than the alternative TS-1-B, as the energy barrier of 60.1 kcal·mol−1 is definitely too high for the reaction to take place, even at an elevated temperature. Path A with a barrier of 22.6 kcal·mol−1 is certainly more feasible. We assume that the Cp2Zr(OH)Cl species just leaves the initial complex, as this seems to be the simplest possibility in absence of any Lewis acid which could catalyze this decomposition.
![[1860-5397-17-12-i8]](/bjoc/content/inline/1860-5397-17-12-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Possible pathway for spontaneous imine formation. Values reported are in kcal·mol−1.
Scheme 8: Possible pathway for spontaneous imine formation. Values reported are in kcal·mol−1.
Scheme 9 shows the energy differences in the subsequent steps of the examined reaction. The reported energy barriers are reasonably high for a slow process taking place at room temperature. The overall barrier is not considerably different to those previously published for typical mechanisms of tetrazole formation by azide addition to nitriles [45]. It is important to note that the computational investigation of this reaction mechanism was not a primary goal of this work. That said, we consider this simple, crude DFT research to support our model of the transformation described herein.
![[1860-5397-17-12-i9]](/bjoc/content/inline/1860-5397-17-12-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: A possible path for tetrazole formation in the described conditions. Values reported are in kcal·mol−1.
Scheme 9: A possible path for tetrazole formation in the described conditions. Values reported are in kcal·mol...
Conclusion
During the course of this research we have developed a methodology for the synthesis of sugar-derived α-tetrazolylamines. Such compounds – incorporating both iminosugar and tetrazole fragments – are particularly interesting, thanks to the well-known biological and catalytic activity of these moieties. This work is the first example of using Schwartz’s reagent-mediated partial reduction of lactams and the Ugi–azide multicomponent reaction in a tandem process. Yields of the described products are moderate to good, a satisfying result for such a multistep process. We have shown that such a reaction does not necessarily requires protic conditions, in opposition to what is generally agreed upon for these type of reactions. An alternative reaction mechanism is proposed and provisionally confirmed with DFT calculations. Moreover, selected α-tetrazolyl-iminosugars were subjected to further transformations, yielding new, potentially biologically active and organocatalytic compounds.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data, ECD analyses for 5a and 2-epi-5a, calculations of appropriate ECD and UV spectra, crystallographic data for 3a and 3e, atomic coordinates, energies, and number of imaginary frequencies for computed stationary points, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 11.4 MB | Download |
| Supporting Information File 2: CCDC-2001373; X-ray crystallographic data for compound 3a. | ||
| Format: CIF | Size: 1.5 MB | Download |
| Supporting Information File 3: CCDC-2001372; X-ray crystallographic data for compound 3e. | ||
| Format: CIF | Size: 799.4 KB | Download |
References
-
Vilsmeier, A.; Haack, A. Ber. Dtsch. Chem. Ges. B 1927, 60, 119–122. doi:10.1002/cber.19270600118
Return to citation in text: [1] -
Pace, V.; Holzer, W.; Olofsson, B. Adv. Synth. Catal. 2014, 356, 3697–3736. doi:10.1002/adsc.201400630
Return to citation in text: [1] -
Chaudhari, M. B.; Gnanaprakasam, B. Chem. – Asian J. 2019, 14, 76–93. doi:10.1002/asia.201801317
Return to citation in text: [1] -
Volkov, A.; Tinnis, F.; Slagbrand, T.; Trillo, P.; Adolfsson, H. Chem. Soc. Rev. 2016, 45, 6685–6697. doi:10.1039/c6cs00244g
Return to citation in text: [1] -
Kaiser, D.; Bauer, A.; Lemmerer, M.; Maulide, N. Chem. Soc. Rev. 2018, 47, 7899–7925. doi:10.1039/c8cs00335a
Return to citation in text: [1] -
Charette, A. B.; Grenon, M. Can. J. Chem. 2001, 79, 1694–1703. doi:10.1139/v01-150
Return to citation in text: [1] -
Pelletier, G.; Bechara, W. S.; Charette, A. B. J. Am. Chem. Soc. 2010, 132, 12817–12819. doi:10.1021/ja105194s
Return to citation in text: [1] -
Leighty, M. W.; Spletstoser, J. T.; Georg, G. I. Organic Syntheses; John Wiley & Sons, 2003.
Return to citation in text: [1] -
Spletstoser, J. T.; White, J. M.; Tunoori, A. R.; Georg, G. I. J. Am. Chem. Soc. 2007, 129, 3408–3419. doi:10.1021/ja066362+
Return to citation in text: [1] -
Więcław, M. M.; Stecko, S. Eur. J. Org. Chem. 2018, 6601–6623. doi:10.1002/ejoc.201701537
Return to citation in text: [1] [2] -
Pace, V.; de la Vega-Hernández, K.; Urban, E.; Langer, T. Org. Lett. 2016, 18, 2750–2753. doi:10.1021/acs.orglett.6b01226
Return to citation in text: [1] -
de la Vega-Hernández, K.; Senatore, R.; Miele, M.; Urban, E.; Holzer, W.; Pace, V. Org. Biomol. Chem. 2019, 17, 1970–1978. doi:10.1039/c8ob02312c
Return to citation in text: [1] -
Iridium Catalysis; Andersson, P. G., Ed.; Topics in Organimetal Chemistry, Vol. 34; Springer: Berlin, Heidelberg, 2011. doi:10.1007/978-3-642-15334-1
Return to citation in text: [1] -
Matheau-Raven, D.; Gabriel, P.; Leitch, J. A.; Almehmadi, Y. A.; Yamazaki, K.; Dixon, D. J. ACS Catal. 2020, 10, 8880–8897. doi:10.1021/acscatal.0c02377
Return to citation in text: [1] -
Cheng, C.; Brookhart, M. J. Am. Chem. Soc. 2012, 134, 11304–11307. doi:10.1021/ja304547s
Return to citation in text: [1] -
Motoyama, Y.; Aoki, M.; Takaoka, N.; Aoto, R.; Nagashima, H. Chem. Commun. 2009, 1574–1576. doi:10.1039/b821317h
Return to citation in text: [1] -
Gregory, A. W.; Chambers, A.; Hawkins, A.; Jakubec, P.; Dixon, D. J. Chem. – Eur. J. 2015, 21, 111–114. doi:10.1002/chem.201405256
Return to citation in text: [1] -
Xie, L.-G.; Dixon, D. J. Chem. Sci. 2017, 8, 7492–7497. doi:10.1039/c7sc03613b
Return to citation in text: [1] -
Xie, L.-G.; Dixon, D. J. Nat. Commun. 2018, 9, 2841. doi:10.1038/s41467-018-05192-7
Return to citation in text: [1] [2] -
Xiao, K.-J.; Luo, J.-M.; Ye, K.-Y.; Wang, Y.; Huang, P.-Q. Angew. Chem., Int. Ed. 2010, 49, 3037–3040. doi:10.1002/anie.201000652
Return to citation in text: [1] -
Huang, P.-Q.; Ou, W.; Han, F. Chem. Commun. 2016, 52, 11967–11970. doi:10.1039/c6cc05318a
Return to citation in text: [1] -
Tinnis, F.; Volkov, A.; Slagbrand, T.; Adolfsson, H. Angew. Chem., Int. Ed. 2016, 55, 4562–4566. doi:10.1002/anie.201600097
Return to citation in text: [1] -
Szcześniak, P.; Stecko, S.; Staszewska-Krajewska, O.; Furman, B. Tetrahedron 2014, 70, 1880–1888. doi:10.1016/j.tet.2014.01.039
Return to citation in text: [1] [2] [3] [4] -
Szcześniak, P.; Stecko, S.; Maziarz, E.; Staszewska-Krajewska, O.; Furman, B. J. Org. Chem. 2014, 79, 10487–10503. doi:10.1021/jo502146z
Return to citation in text: [1] [2] [3] -
Szcześniak, P.; Maziarz, E.; Stecko, S.; Furman, B. J. Org. Chem. 2015, 80, 3621–3633. doi:10.1021/acs.joc.5b00335
Return to citation in text: [1] [2] -
Inouye, S.; Tsuruoka, T.; Ito, T.; Niida, T. Tetrahedron 1968, 24, 2125–2144. doi:10.1016/0040-4020(68)88115-3
Return to citation in text: [1] -
Sánchez-Fernández, E. M.; García Fernández, J. M.; Mellet, C. O. Chem. Commun. 2016, 52, 5497–5515. doi:10.1039/c6cc01564f
Return to citation in text: [1] -
Malik, M. A.; Wani, M. Y.; Al-Thabaiti, S. A.; Shiekh, R. A. J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 15–37. doi:10.1007/s10847-013-0334-x
Return to citation in text: [1] -
Pabba, J.; Rempel, B. P.; Withers, S. G.; Vasella, A. Helv. Chim. Acta 2006, 89, 635–666. doi:10.1002/hlca.200690066
Return to citation in text: [1] -
Hartikka, A.; Arvidsson, P. I. Tetrahedron: Asymmetry 2004, 15, 1831–1834. doi:10.1016/j.tetasy.2004.04.029
Return to citation in text: [1] [2] -
Chen, H.; Zhang, D.; Xue, F.; Qin, Y. Tetrahedron 2013, 69, 3141–3148. doi:10.1016/j.tet.2013.02.078
Return to citation in text: [1] [2] -
Kumar, I.; Ramaraju, P.; Mir, N. A.; Singh, D.; Gupta, V. K.; Rajnikant. Chem. Commun. 2013, 49, 5645–5647. doi:10.1039/c3cc42431f
Return to citation in text: [1] [2] -
Mirabal-Gallardo, Y.; Piérola, J.; Shankaraiah, N.; Santos, L. S. Tetrahedron Lett. 2012, 53, 3672–3675. doi:10.1016/j.tetlet.2012.05.033
Return to citation in text: [1] [2] -
Wong, B. Nat. Methods 2011, 8, 441. doi:10.1038/nmeth.1618
The color palette used in this document is optimized for color-blind individuals as proposed herein.
Return to citation in text: [1] -
Ugi, I.; Steinbrückner, C. Chem. Ber. 1961, 94, 734–742. doi:10.1002/cber.19610940323
Return to citation in text: [1] -
Ramezanpour, S.; Balalaie, S.; Rominger, F.; Alavijeh, N. S.; Bijanzadeh, H. R. Tetrahedron 2013, 69, 10718–10723. doi:10.1016/j.tet.2013.10.062
Return to citation in text: [1] -
Shmatova, O. I.; Nenajdenko, V. G. J. Org. Chem. 2013, 78, 9214–9222. doi:10.1021/jo401428q
Return to citation in text: [1] -
Safa, K. D.; Shokri, T.; Abbasi, H.; Teimuri-Mofrad, R. J. Heterocycl. Chem. 2014, 51, 80–84. doi:10.1002/jhet.1858
Return to citation in text: [1] -
Santhosh, L.; Nagamangala, S. R.; Thimmalapura, V. M.; Vommina, S. V. ChemistrySelect 2017, 2, 5497–5500. doi:10.1002/slct.201701032
Return to citation in text: [1] -
Finsinger, M.; Wucherer-Plietker, M.; Blume, B. Tetrahydro-tetrazolo[1,5-a]pyrazines as ROR-gamma inhibitors. WO Pat. Appl. WO2015090507A1, June 25, 2015.
Return to citation in text: [1] -
Larsen, C. H.; Ridgway, B. H.; Shaw, J. T.; Woerpel, K. A. J. Am. Chem. Soc. 1999, 121, 12208–12209. doi:10.1021/ja993349z
Return to citation in text: [1] -
Romero, J. A. C.; Tabacco, S. A.; Woerpel, K. A. J. Am. Chem. Soc. 2000, 122, 168–169. doi:10.1021/ja993366o
Return to citation in text: [1] -
Woods, R. J.; Andrews, C. W.; Bowen, J. P. J. Am. Chem. Soc. 1992, 114, 859–864. doi:10.1021/ja00029a008
Return to citation in text: [1] -
Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2016.
Return to citation in text: [1] -
Himo, F.; Demko, Z. P.; Noodleman, L.; Sharpless, K. B. J. Am. Chem. Soc. 2002, 124, 12210–12216. doi:10.1021/ja0206644
Return to citation in text: [1]
| 32. | Kumar, I.; Ramaraju, P.; Mir, N. A.; Singh, D.; Gupta, V. K.; Rajnikant. Chem. Commun. 2013, 49, 5645–5647. doi:10.1039/c3cc42431f |
| 33. | Mirabal-Gallardo, Y.; Piérola, J.; Shankaraiah, N.; Santos, L. S. Tetrahedron Lett. 2012, 53, 3672–3675. doi:10.1016/j.tetlet.2012.05.033 |
| 34. |
Wong, B. Nat. Methods 2011, 8, 441. doi:10.1038/nmeth.1618
The color palette used in this document is optimized for color-blind individuals as proposed herein. |
| 1. | Vilsmeier, A.; Haack, A. Ber. Dtsch. Chem. Ges. B 1927, 60, 119–122. doi:10.1002/cber.19270600118 |
| 7. | Pelletier, G.; Bechara, W. S.; Charette, A. B. J. Am. Chem. Soc. 2010, 132, 12817–12819. doi:10.1021/ja105194s |
| 20. | Xiao, K.-J.; Luo, J.-M.; Ye, K.-Y.; Wang, Y.; Huang, P.-Q. Angew. Chem., Int. Ed. 2010, 49, 3037–3040. doi:10.1002/anie.201000652 |
| 21. | Huang, P.-Q.; Ou, W.; Han, F. Chem. Commun. 2016, 52, 11967–11970. doi:10.1039/c6cc05318a |
| 32. | Kumar, I.; Ramaraju, P.; Mir, N. A.; Singh, D.; Gupta, V. K.; Rajnikant. Chem. Commun. 2013, 49, 5645–5647. doi:10.1039/c3cc42431f |
| 6. | Charette, A. B.; Grenon, M. Can. J. Chem. 2001, 79, 1694–1703. doi:10.1139/v01-150 |
| 22. | Tinnis, F.; Volkov, A.; Slagbrand, T.; Adolfsson, H. Angew. Chem., Int. Ed. 2016, 55, 4562–4566. doi:10.1002/anie.201600097 |
| 33. | Mirabal-Gallardo, Y.; Piérola, J.; Shankaraiah, N.; Santos, L. S. Tetrahedron Lett. 2012, 53, 3672–3675. doi:10.1016/j.tetlet.2012.05.033 |
| 3. | Chaudhari, M. B.; Gnanaprakasam, B. Chem. – Asian J. 2019, 14, 76–93. doi:10.1002/asia.201801317 |
| 4. | Volkov, A.; Tinnis, F.; Slagbrand, T.; Trillo, P.; Adolfsson, H. Chem. Soc. Rev. 2016, 45, 6685–6697. doi:10.1039/c6cs00244g |
| 5. | Kaiser, D.; Bauer, A.; Lemmerer, M.; Maulide, N. Chem. Soc. Rev. 2018, 47, 7899–7925. doi:10.1039/c8cs00335a |
| 16. | Motoyama, Y.; Aoki, M.; Takaoka, N.; Aoto, R.; Nagashima, H. Chem. Commun. 2009, 1574–1576. doi:10.1039/b821317h |
| 30. | Hartikka, A.; Arvidsson, P. I. Tetrahedron: Asymmetry 2004, 15, 1831–1834. doi:10.1016/j.tetasy.2004.04.029 |
| 2. | Pace, V.; Holzer, W.; Olofsson, B. Adv. Synth. Catal. 2014, 356, 3697–3736. doi:10.1002/adsc.201400630 |
| 17. | Gregory, A. W.; Chambers, A.; Hawkins, A.; Jakubec, P.; Dixon, D. J. Chem. – Eur. J. 2015, 21, 111–114. doi:10.1002/chem.201405256 |
| 18. | Xie, L.-G.; Dixon, D. J. Chem. Sci. 2017, 8, 7492–7497. doi:10.1039/c7sc03613b |
| 19. | Xie, L.-G.; Dixon, D. J. Nat. Commun. 2018, 9, 2841. doi:10.1038/s41467-018-05192-7 |
| 31. | Chen, H.; Zhang, D.; Xue, F.; Qin, Y. Tetrahedron 2013, 69, 3141–3148. doi:10.1016/j.tet.2013.02.078 |
| 11. | Pace, V.; de la Vega-Hernández, K.; Urban, E.; Langer, T. Org. Lett. 2016, 18, 2750–2753. doi:10.1021/acs.orglett.6b01226 |
| 13. | Iridium Catalysis; Andersson, P. G., Ed.; Topics in Organimetal Chemistry, Vol. 34; Springer: Berlin, Heidelberg, 2011. doi:10.1007/978-3-642-15334-1 |
| 14. | Matheau-Raven, D.; Gabriel, P.; Leitch, J. A.; Almehmadi, Y. A.; Yamazaki, K.; Dixon, D. J. ACS Catal. 2020, 10, 8880–8897. doi:10.1021/acscatal.0c02377 |
| 35. | Ugi, I.; Steinbrückner, C. Chem. Ber. 1961, 94, 734–742. doi:10.1002/cber.19610940323 |
| 36. | Ramezanpour, S.; Balalaie, S.; Rominger, F.; Alavijeh, N. S.; Bijanzadeh, H. R. Tetrahedron 2013, 69, 10718–10723. doi:10.1016/j.tet.2013.10.062 |
| 37. | Shmatova, O. I.; Nenajdenko, V. G. J. Org. Chem. 2013, 78, 9214–9222. doi:10.1021/jo401428q |
| 38. | Safa, K. D.; Shokri, T.; Abbasi, H.; Teimuri-Mofrad, R. J. Heterocycl. Chem. 2014, 51, 80–84. doi:10.1002/jhet.1858 |
| 39. | Santhosh, L.; Nagamangala, S. R.; Thimmalapura, V. M.; Vommina, S. V. ChemistrySelect 2017, 2, 5497–5500. doi:10.1002/slct.201701032 |
| 10. | Więcław, M. M.; Stecko, S. Eur. J. Org. Chem. 2018, 6601–6623. doi:10.1002/ejoc.201701537 |
| 15. | Cheng, C.; Brookhart, M. J. Am. Chem. Soc. 2012, 134, 11304–11307. doi:10.1021/ja304547s |
| 40. | Finsinger, M.; Wucherer-Plietker, M.; Blume, B. Tetrahydro-tetrazolo[1,5-a]pyrazines as ROR-gamma inhibitors. WO Pat. Appl. WO2015090507A1, June 25, 2015. |
| 9. | Spletstoser, J. T.; White, J. M.; Tunoori, A. R.; Georg, G. I. J. Am. Chem. Soc. 2007, 129, 3408–3419. doi:10.1021/ja066362+ |
| 19. | Xie, L.-G.; Dixon, D. J. Nat. Commun. 2018, 9, 2841. doi:10.1038/s41467-018-05192-7 |
| 8. | Leighty, M. W.; Spletstoser, J. T.; Georg, G. I. Organic Syntheses; John Wiley & Sons, 2003. |
| 12. | de la Vega-Hernández, K.; Senatore, R.; Miele, M.; Urban, E.; Holzer, W.; Pace, V. Org. Biomol. Chem. 2019, 17, 1970–1978. doi:10.1039/c8ob02312c |
| 23. | Szcześniak, P.; Stecko, S.; Staszewska-Krajewska, O.; Furman, B. Tetrahedron 2014, 70, 1880–1888. doi:10.1016/j.tet.2014.01.039 |
| 23. | Szcześniak, P.; Stecko, S.; Staszewska-Krajewska, O.; Furman, B. Tetrahedron 2014, 70, 1880–1888. doi:10.1016/j.tet.2014.01.039 |
| 10. | Więcław, M. M.; Stecko, S. Eur. J. Org. Chem. 2018, 6601–6623. doi:10.1002/ejoc.201701537 |
| 23. | Szcześniak, P.; Stecko, S.; Staszewska-Krajewska, O.; Furman, B. Tetrahedron 2014, 70, 1880–1888. doi:10.1016/j.tet.2014.01.039 |
| 24. | Szcześniak, P.; Stecko, S.; Maziarz, E.; Staszewska-Krajewska, O.; Furman, B. J. Org. Chem. 2014, 79, 10487–10503. doi:10.1021/jo502146z |
| 25. | Szcześniak, P.; Maziarz, E.; Stecko, S.; Furman, B. J. Org. Chem. 2015, 80, 3621–3633. doi:10.1021/acs.joc.5b00335 |
| 23. | Szcześniak, P.; Stecko, S.; Staszewska-Krajewska, O.; Furman, B. Tetrahedron 2014, 70, 1880–1888. doi:10.1016/j.tet.2014.01.039 |
| 41. | Larsen, C. H.; Ridgway, B. H.; Shaw, J. T.; Woerpel, K. A. J. Am. Chem. Soc. 1999, 121, 12208–12209. doi:10.1021/ja993349z |
| 42. | Romero, J. A. C.; Tabacco, S. A.; Woerpel, K. A. J. Am. Chem. Soc. 2000, 122, 168–169. doi:10.1021/ja993366o |
| 43. | Woods, R. J.; Andrews, C. W.; Bowen, J. P. J. Am. Chem. Soc. 1992, 114, 859–864. doi:10.1021/ja00029a008 |
| 24. | Szcześniak, P.; Stecko, S.; Maziarz, E.; Staszewska-Krajewska, O.; Furman, B. J. Org. Chem. 2014, 79, 10487–10503. doi:10.1021/jo502146z |
| 30. | Hartikka, A.; Arvidsson, P. I. Tetrahedron: Asymmetry 2004, 15, 1831–1834. doi:10.1016/j.tetasy.2004.04.029 |
| 31. | Chen, H.; Zhang, D.; Xue, F.; Qin, Y. Tetrahedron 2013, 69, 3141–3148. doi:10.1016/j.tet.2013.02.078 |
| 28. | Malik, M. A.; Wani, M. Y.; Al-Thabaiti, S. A.; Shiekh, R. A. J. Inclusion Phenom. Macrocyclic Chem. 2014, 78, 15–37. doi:10.1007/s10847-013-0334-x |
| 29. | Pabba, J.; Rempel, B. P.; Withers, S. G.; Vasella, A. Helv. Chim. Acta 2006, 89, 635–666. doi:10.1002/hlca.200690066 |
| 26. | Inouye, S.; Tsuruoka, T.; Ito, T.; Niida, T. Tetrahedron 1968, 24, 2125–2144. doi:10.1016/0040-4020(68)88115-3 |
| 27. | Sánchez-Fernández, E. M.; García Fernández, J. M.; Mellet, C. O. Chem. Commun. 2016, 52, 5497–5515. doi:10.1039/c6cc01564f |
| 24. | Szcześniak, P.; Stecko, S.; Maziarz, E.; Staszewska-Krajewska, O.; Furman, B. J. Org. Chem. 2014, 79, 10487–10503. doi:10.1021/jo502146z |
| 25. | Szcześniak, P.; Maziarz, E.; Stecko, S.; Furman, B. J. Org. Chem. 2015, 80, 3621–3633. doi:10.1021/acs.joc.5b00335 |
| 45. | Himo, F.; Demko, Z. P.; Noodleman, L.; Sharpless, K. B. J. Am. Chem. Soc. 2002, 124, 12210–12216. doi:10.1021/ja0206644 |
© 2021 Więcław and Furman; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)