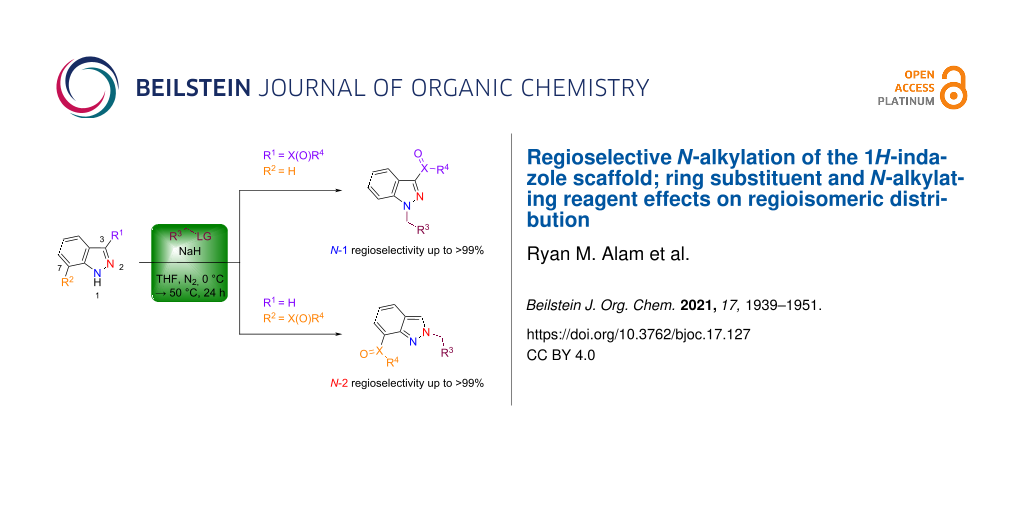
Abstract
The indazole scaffold represents a promising pharmacophore, commonly incorporated in a variety of therapeutic drugs. Although indazole-containing drugs are frequently marketed as the corresponding N-alkyl 1H- or 2H-indazole derivative, the efficient synthesis and isolation of the desired N-1 or N-2 alkylindazole regioisomer can often be challenging and adversely affect product yield. Thus, as part of a broader study focusing on the synthesis of bioactive indazole derivatives, we aimed to develop a regioselective protocol for the synthesis of N-1 alkylindazoles. Initial screening of various conditions revealed that the combination of sodium hydride (NaH) in tetrahydrofuran (THF) (in the presence of an alkyl bromide), represented a promising system for N-1 selective indazole alkylation. For example, among fourteen C-3 substituted indazoles examined, we observed > 99% N-1 regioselectivity for 3-carboxymethyl, 3-tert-butyl, 3-COMe, and 3-carboxamide indazoles. Further extension of this optimized (NaH in THF) protocol to various C-3, -4, -5, -6, and -7 substituted indazoles has highlighted the impact of steric and electronic effects on N-1/N-2 regioisomeric distribution. For example, employing C-7 NO2 or CO2Me substituted indazoles conferred excellent N-2 regioselectivity (≥ 96%). Importantly, we show that this optimized N-alkylation procedure tolerates a wide structural variety of alkylating reagents, including primary alkyl halide and secondary alkyl tosylate electrophiles, while maintaining a high degree of N-1 regioselectivity.

Graphical Abstract
Introduction
Indazole (benzo[c]pyrazole) is an aromatic bicyclic heterocycle and can be viewed as a (bio)isostere of indole [1]. While only a few naturally occurring indazoles have been reported in the literature [2-4], such as the alkaloids nigellicine (1) and nigellidine (2), there are a myriad of synthetic indazole derivatives known that display a broad range of biological activities. For example, several N-1 and N-2-substituted indazoles are currently marketed or under clinical investigation for the treatment of nausea and vomiting (granisetron (3)) [5], inflammation (benzydamine (4)) [6], or certain cancers (lonidamine (5), niraparib (6), pazopanib (7), and merestinib (8)) [7,8] (Figure 1). Considering the medicinal significance of N-substituted indazole derivatives [9], it would be of great synthetic value to further develop regioselective methods for the preparation of N-1 or N-2 substituted indazoles.
![[1860-5397-17-127-1]](/bjoc/content/figures/1860-5397-17-127-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of indazole natural products (1 and 2) and synthetic biologically active indazole derivatives (3–8).
Figure 1: Examples of indazole natural products (1 and 2) and synthetic biologically active indazole derivati...
General approaches to the synthesis of N-1 or N-2 substituted indazoles involve the incorporation of the N-substituent prior to, or following, indazole ring-closure [10,11]. For example, several reports have highlighted the use of N-alkyl or N-arylhydrazines in the regioselective synthesis of 1H-indazoles, from the corresponding ortho-haloaryl carbonyl or nitrile, in good to excellent yield (Scheme 1) [12-14].
![[1860-5397-17-127-i1]](/bjoc/content/inline/1860-5397-17-127-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic approaches to N-1 substituted indazole derivatives [12-14].
Scheme 1: Synthetic approaches to N-1 substituted indazole derivatives [12-14].
Alternative strategies to achieve regioselective indazole N-alkylation have exploited the noted difference in reactivity between the N-1 and N-2 atom of the indazole scaffold [15], as the 1H-indazole tautomer is typically considered to be more thermodynamically stable than the corresponding 2H-tautomer [16]. Using appropriate α-halo carbonyl electrophiles, Hunt et al. have shown that regioselective indazole N-alkylation can be achieved through an equilibration process which favours the thermodynamic N-1 substituted product [17].
Regioselective indazole N-acylation has been suggested to provide the N-1 substituted regioisomer, via isomerisation of the corresponding N-2 acylindazole to the more stable N-1 regioisomer [18]. Similarly, N-1 substituted indazoles have been obtained through thermodynamic equilibration, using β-halo ester electrophiles, in the presence of DMF [19]. These latter findings have been utilized to great effect by Conrow et al. to give regioselective access to N-1 alkylindazoles on kilogram scale, albeit over two steps from the corresponding N-1 acylindazole via reductive acetylation–deacetoxylation [20]. Although electronic and steric factors can influence the regiochemical outcome of indazole N-alkylation, varying reaction conditions, such as the choice of base [17,21], acid [22], solvent, and/or N-alkylating reagent may also facilitate regioselective indazole N-alkylation [23-25]. Bookser et al. have investigated the N-alkylation of related bicyclic azolo-fused-ring heterocycles, including 1H-indazole, employing NaHMDS in tetrahydrofuran (THF) or dimethyl sulfoxide (DMSO), and observed solvent-dependent regioselectivity [24]. Mechanistic hypotheses, based on elegant experimentation, were proposed to underline the roles that tight and solvent-separated ion pairs played in the observed trend in regioselectivity [24].
Our work sought to further explore the effect of C-3 substitution on N-alkylation selectivity control of the 1H-indazole scaffold. In view of these antecedents, it was envisioned that the development of a regioselective protocol for indazole N-1 alkylation would provide an improved and cost-effective approach to N-1 substituted indazole precursors as part of drug discovery and development campaigns.
Results and Discussion
Working towards the synthesis of a library of novel 1,3-disubstituted indazole derivatives necessitated us to develop a regioselective method that would permit the installation of a wide variety of alkyl sidechains at the N-1 position of methyl ester 9 (Table 1). Considering the reported influence of the reaction solvent and/or base on the regiochemical outcome of indazole N-alkylation [17,21,22,24], our initial efforts focused on examining the effect of varying these reaction parameters, using n-pentyl bromide as the prototypical N-alkylating reagent (Table 1).
Table 1: Effect of base, solvent, and temperature.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-127-i4.svg?max-width=637&scale=1.0)
|
|||||||||
| entry | base | equiv | solvent |
T
(°C) |
time
(h) |
conv.b
(%) |
ratiob
10:11 |
yield (%)c | |
| 10 | 11 | ||||||||
| 1 | Cs2CO3 | 3 | DMF | rt | 16 | 100 | 1.4:1 | 50 | 39 |
| 2 | Cs2CO3 | 3 | DMF | rt | 24 | 100 | 1.2:1 | 44 | 33 |
| 3 | Cs2CO3 | 1.5 | DMF | rt | 16 | 100 | 1.4:1 | 54 | 39 |
| 4 | K2CO3 | 1.5 | DMF | rt | 16 | 100 | 1.4:1 | 60 | 37 |
| 5 | Na2CO3 | 1.5 | DMF | rt | 16 | 34 | 1.6:1 | 17 | 10 |
| 6 | K2CO3 | 0.5 | DMF | rt | 16 | 62 | 1.5:1 | 33 | 23 |
| 7 | K2CO3 | 1.5 | THF | rt | 16 | 0 | –d | 0 | 0 |
| 8 | Na2CO3 | 1.5 | THF | rt | 16 | 0 | –d | 0 | 0 |
| 9 | Cs2CO3 | 1.5 | MeCN | rt | 16 | 67 | 1.9:1 | 29 | 15 |
| 10 | Cs2CO3 | 1.5 | MeCN | rt | 30 | 100 | 1.9:1 | 57 | 29 |
| 11 | Cs2CO3 | 1.5 | DMSO | rt | 16 | 100 | 1.6.1 | 60 | 36 |
| 12 | K2CO3 | 1.5 | DMSO | rt | 16 | 100 | 1.6:1 | –d | –d |
| 13 | Cs2CO3 | 1.5 | toluene | rt | 16 | 0 | –d | 0 | 0 |
| 14 | Cs2CO3 | 1.5 | 1,4-dioxane | rt | 16 | 0 | –d | 0 | 0 |
| 15 | DBU | 1.1 | CH2Cl2 | rt | 16 | 59 | 3.2:1 | –d | –d |
| 16 | DBU | 1.1 | THF | rt | 16 | 65 | 2.7:1 | –d | –d |
| 17 | DBU | 1.1 | DMF | rt | 16 | 72 | 1.4:1 | –d | –d |
| 18 | t-BuOK | 1.1 | THF | e | 16 | 30 | 16:1 | –d | –d |
| 19 | t-BuOK | 1.1 | THF | e | 48 | 70 | 16:1 | –d | –d |
| 20 | NaH | 1.1 | THF | e | 48 | 57 | >99:1 | 44 | 0 |
| 21 | t-BuOK | 1.1 | THF | f | 5.5 | 100 | 13:1 | –d | –d |
| 22 | NaH | 1.1 | THF | f | 24 | 100 | >99:1 | 89 | 0 |
aReaction scale = 1.4 mmol (9); bdetermined using 1H NMR (see Supporting Information File 1); cisolated yield; dnot determined; e0 °C → rt; f0 °C → 50 °C; conv. = conversion of 9 to 10 and 11 only; rt ≈ 20 °C.
Early investigations revealed that the combination of cesium carbonate (Cs2CO3) in dimethylformamide (DMF) at room temperature (≈ 20 °C) afforded a mixture of N-1 and N-2 regioisomers (10 and 11, respectively), with only partial preference for the desired N-1 regioisomer 10 (Table 1, entry 1). Furthermore, increasing the reaction time or decreasing the number of equivalents of Cs2CO3 did not appear to influence the regiochemical outcome of the reaction (Table 1, entries 2 and 3, respectively). Substituting potassium carbonate (K2CO3) for Cs2CO3 did not show any improvement in the regioisomeric distribution of 10 and 11 (ratio N-1 (10)/N-2 (11) = 1.4:1) (Table 1, entry 4). Similarly, the use of sodium carbonate under identical conditions gave a notably lower combined yield of 10 and 11 (27%), due to poor conversion (34%) (Table 1, entry 5). Attempts to reduce the amount of K2CO3 base to 0.5 equivalents, with respect to indazole 9, (Table 1, entry 6) resulted in incomplete conversion (62%) and provided no significant change in N-1 regioselectivity (ratio N-1:N-2 = 1.5:1). Importantly, using THF as the reaction solvent with potassium or sodium carbonate bases failed to give the N-alkylated products 10 or 11 (Table 1, entries 7 and 8, respectively).
Further variation of the reaction solvent revealed no significant improvement in N-1 regioselectivity, when using acetonitrile (MeCN) (Table 1, entries 9 and 10) or DMSO (Table 1, entries 11 and 12) (ratio N-1:N-2 = 1.9:1 and 1.6:1, respectively). Furthermore, employing toluene or 1,4-dioxane as the reaction solvent failed to provide regioisomer 10 or 11 (Table 1, entries 13 and 14). The latter observation may be due to the restricted solubility of Cs2CO3 in toluene and 1,4-dioxane [24]. Using K2CO3 in MeCN, Longworth et al. have obtained 10 with a similar degree of N-1 regioselectivity (ratio N-1:N-2 = 2.8:1) [26]. However, altering solvent polarity when employing 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base (Table 1, entries 15–17) positively influenced the regioselectivity, albeit with poor conversion (59–72%) when compared with other inorganic carbonate bases (vide supra).
Despite poor conversion (30%), the use of the strong alkoxide base, potassium tert-butoxide, in tetrahydrofuran (THF) gave an appreciably higher degree of N-1 regioselectivity (94%, N-1 regioselectivity) (Table 1, entry 18) than the previously investigated reaction conditions (55–76%, N-1 regioselectivity) (Table 1, entries 1–17). To improve conversion to the desired N-1 regioisomer 10, the reaction time was extended (48 h) (Table 1, entry 19) and the reaction temperature increased from room temperature (≈ 20 °C) to 50 °C (Table 1, entry 21). It was found that the latter variation facilitated the complete consumption of 9 with negligible effect on the regiochemical outcome of the reaction. Gratifyingly, sodium hydride (NaH) demonstrated excellent N-1 regioselectivity (ratio N-1:N-2 > 99:1), albeit with poor conversion (57%) (Table 1, entry 20). Again, increasing the preceding reaction temperature from room temperature to 50 °C not only facilitated the complete conversion of methyl ester 9 to desired N-1 regioisomer 10, but also maintained an excellent degree of regiocontrol (ratio N-1:N-2 > 99:1) (Table 1, entry 22). The N-alkylation of methyl ester 9 was also investigated under Mitsunobu conditions (see Supporting Information File 1), in the presence of n-pentanol (Scheme 2). Notably, the latter conditions demonstrated a strong (ca. three-fold) preference for the formation of the corresponding N-2 regioisomer 11 (ratio N-1:N-2 = 1:2.5) rather than the desired N-1 regioisomer.
![[1860-5397-17-127-i2]](/bjoc/content/inline/1860-5397-17-127-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: N-Alkylation of indazole 9 under Mitsunobu conditions shows a strong preference (ratio N-1 (10):N-2 (11) = 1:2.5) for the formation of N-2 regioisomer 11 (58%, isolated yield) over the corresponding N-1 regioisomer 10 (20% isolated yield).
Scheme 2: N-Alkylation of indazole 9 under Mitsunobu conditions shows a strong preference (ratio N-1 (10):N-2...
To assign the regiochemistry of isolated N-1 and N-2 substituted indazole isomers, a combination of one and two-dimensional NMR experiments (particularly, heteronuclear multiple bond correlation (HMBC)) was employed [27]. For example, (1H–13C) HMBC analysis of N-1 regioisomer 10 shows a 1H–13C correlation between the C-7a carbon of the indazole ring and the n-pentyl CH2 proton pair proximal to the indazole N-1 atom (Figure 2). No evident 1H–13C correlation was observed between the n-alkyl CH2 proton pair proximal to the indazole N-1 atom and the indazole C-3 atom, for N-1 substituted indazole regioisomer 10. Conversely, (1H–13C) HMBC analysis of N-2 substituted regioisomer 11, revealed a 1H–13C correlation between the alkyl CH2 proton pair (proximal to the indazole N-2 atom) and the C-3 carbon of the indazole heterocycle, while no 1H–13C correlation was observed between the alkyl CH2 proton pair and the C-7a carbon atom of the indazole ring.
![[1860-5397-17-127-2]](/bjoc/content/figures/1860-5397-17-127-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Observation of a 1H–13C correlation between the C-7a (blue circle) or C-3 (red circle) atom of the indazole ring and the N-n-pentyl CH2 proton pair proximal to N atoms of the indazole nucleus, using (1H–13C) HMBC NMR experiments, permitted the regiochemical assignment of N-1 and N-2 substituted regioisomers 10 and 11, respectively.
Figure 2: Observation of a 1H–13C correlation between the C-7a (blue circle) or C-3 (red circle) atom of the ...
With a set of optimal conditions for the regioselective N-1 alkylation of methyl ester 9 in hand (Table 1, entry 22), our attention turned to probing the influence of a variety of indazole C-3 substituents on the regiochemical N-alkylation outcome. A series of C-3 substituted indazoles (12–24) (Figure 3) were thus assembled [28-33] to investigate the effects of electronic and steric factors on indazole N-alkylation (Table 2) using our optimized conditions from Table 1 (entry 22) (referred to as “conditions A” in Table 2).
![[1860-5397-17-127-3]](/bjoc/content/figures/1860-5397-17-127-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: C-3 substituted indazole derivatives (12–24) employed to investigate C-3 substituent effects on indazole N-alkylation regioselectivity.
Figure 3: C-3 substituted indazole derivatives (12–24) employed to investigate C-3 substituent effects on ind...
As literature precedence shows, the combination of Cs2CO3 in DMF has been commonly employed to achieve indazole N-alkylation [17,34,35]. For comparison with our previously optimized N-alkylation protocol (Table 1, entry 22; conditions A), conditions that provided less favorable N-1 regioselectivity (Table 1, entry 3, henceforth referred to as “conditions B” in Table 2) were also included as a part of this investigation.
Table 2: Indazole C-3 substituent effects.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-127-i5.svg?max-width=637&scale=1.0)
|
|||||||||
| entry | R | product | cond. Aa,c | yield (%)d | cond. Bb,c | yield (%)d | |||
| N-1 | N-2 | ratio N-1:N-2e | N-1 | N-2 | ratio N-1:N-2e | N-1 | N-2 | ||
| 1 | H | 25 | 26 | 1:1.3 | 33 | 46 | 1.7:1 | 43 | 28 |
| 2 | Me | 27 | 28 | 2.3:1 | 54 | 22 | 4.6:1 | 73 | 12 |
| 3 | t-Bu | 29 | 30 | >99:1 | 85 | 0 | >99:1 | 79 | 0 |
| 4 | Ph | 31 | 32 | 7.8:1 | 54 | 11 | 14:1 | 90 | 6 |
| 5 | I | 33 | 34 | 3.8:1 | 60 | 14 | 4.4:1 | 53 | 16 |
| 6 | Br | 35 | 36 | 5.9:1 | 83 | 12 | 7.5:1 | 85 | 12 |
| 7 | Cl | 37 | 38 | 5.6:1 | 65 | 7 | 8.0:1 | 75 | 9 |
| 8 | NO2 | 39 | 40 | 83:1 | 80 | 0 | 6.4:1 | 57 | 9 |
| 9 | CN | 41 | 42 | 3.4:1 | –f | – f | 5.2:1 | – f | – f |
| 10 | CHO | 43 | 44 | 16:1 | – f | – f | 9.8:1 | – f | – f |
| 11 | COMe | 45 | 46 | >99:1 | 97g | 0g | 61:1 | 66g | 0g |
| 12 | CONHMe | 47 | 48 | >99:1 | 88 | 0 | >99:1 | 93 | 0 |
| 13 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-127-i6.svg?max-width=637&scale=1.0)
|
49 | 50 | >99:1 | 53 | 0 | >99:1 | 93 | 0 |
a1.1 equiv NaH, 1.2 equiv n-pentyl bromide, THF, N2, 0 °C → 50 °C, 24 h; b1.5 equiv Cs2CO3, 1.2 equiv n-pentyl bromide, DMF, rt, 16 h; creaction scale = 1 mmol (appropriate indazole); disolated yield; edetermined using 1H NMR (see Supporting Information File 1); fnot determined, due to the formation of an inseparable mixture (using wet flash column chromatography) of the corresponding N-1 and N-2 regioisomers; greaction scale = 0.5 mmol (22); cond. = conditions.
Increasing alkyl and aryl steric bulk at the indazolic C-3 position seems to favor N-1 regioisomer formation in the order t-Bu > Ph > Me > H, for both conditions A and B (Table 2, entries 1–4). Furthermore, having a sterically demanding t-Bu group at the indazole C-3 position (14) gave the N-1 substituted regioisomer 29 exclusively under both conditions A and B, respectively (Table 2, entry 3). Apart from the 1H-indazole scaffold [36,37], the steric influence of adjacent substituent(s) on N-alkylation regioselectivity has previously been described for other nitrogen-containing heterocycles, such as pyrazole [38], purine, and related 1,3-azoles [39]. Although the N-alkylation of indazole 12, using conditions A (NaH in THF), proceeded with poor regioselectivity (ratio N-1 (25):N-2 (26) = 1:1.3), Bookser et al. have obtained a similar regioselective outcome using a combination of NaHMDS and MeI instead of NaH and n-pentyl bromide (Table 2, entry 1), respectively [24].
The presence of a halogen atom (I, Br, or Cl) at the C-3 position of the indazole scaffold (16–18) revealed no significant trend in regioselectivity (Table 2, entries 5–7). The latter observed preference for the formation of the corresponding N-1 regioisomer (33, 35, and 37, respectively), under both conditions A and B, highlights the steric influence of the halogen C-3 substituent. Furthermore, Bookser et al. have noted comparable regioselectivity with related C-3 bromo substituted N-containing heterocycles, under similar reaction conditions [24]. Marked N-1 regioselectivity was achieved with 3-nitro substituted indazole 19, when using NaH in THF (conditions A) (ratio N-1 (39):N-2 (40) = 83:1) (Table 2, entry 8). However, the same degree of N-1 regioselectivity was not observed for indazole 19, when using Cs2CO3 in DMF (conditions B) (ratio N-1 (39):N-2 (40) = 6.4:1) (Table 2, entry 8). Interestingly, the presence of an electronegative nitrile group at the indazole C-3 position (20) provided only modest N-1 regioselectivity, under both conditions A and B (N-1 (41):N-2 (42) = 3.4:1 and 5.2:1, respectively) (Table 2, entry 9).
Remarkably, unlike methyl ester 9 (vide supra), indazoles 12, 13, 15–18, and 20 all demonstrated a higher preference for N-1 indazole alkylation under conditions B, when compared with conditions A (Table 2, entries 1, 2, 4–7, and 9). Furthermore, C-3 ketone (22) and amide (23 and 24) substituted indazoles also gave the corresponding N-1 regioisomers with a high degree of N-1 regioselectivity (ratio N-1:N-2 = 61:1 for indazole 22 and > 99:1 for indazoles 23 and 24), when employing Cs2CO3 in DMF (Table 2, entries 11–13). The latter exclusive formation of the corresponding N-1 regioisomer observed for indazoles 22 and 23 under conditions B may arise from the steric repulsion of the electrophile from the N-2 position to the N-1 atom by the indazole C-3 substituent (vide infra).
While the corresponding N-1 and N-2 regioisomers arising from the N-alkylation of C-3 substituted indazoles 12–24 were generally amenable to separation using wet flash column chromatography, the corresponding N-1- and N-2-n-pentylindazole derivatives of both indazoles 20 and 21 were largely inseparable (Table 2, entries 9 and 10, respectively). However, preparative thin-layer chromatography (PTLC) did permit the isolation of an enriched sample of the N-1-n-pentyl substituted derivative of indazole 20 for (1H–13C) HMBC NMR analysis and confirmatory regiochemical assignment. Similarly, while the regioisomeric products arising from the N-alkylation of indazole 21 could not be separated using wet flash column chromatography or PTLC, a significantly enriched sample of the corresponding N-1 regioisomer (ratio N-1 (43):N-2 (44) = 16:1; Table 2, entry 10) was obtained under conditions A.
Notably, under conditions A (NaH in THF), C-3 substituted indazoles 19 (-NO2), 21 (-CHO), 22 (-COMe), 23 (-CONHMe), and tertiary amide 24 all demonstrated a high degree of N-1 regioselectivity (ratio N-1:N-2 = 16:1 (21), 83:1 (19), > 99:1 (22, 23, and 24), respectively) (Table 2, entries 8 and 10–13). We postulate that these observed preferences for the generation of the N-1 regioisomer, under conditions A, may be due to the formation of a tight ion pair involving the indazole N-2 atom and C-3 substituents which are capable of cation chelation via the N-2 atom electron lone pair and the C-3 substituent X=O functionality, respectively [24]. Tight ion pair formation with the sodium cation and both the N-2 atom and C-3 substituents of the indazole scaffold likely hinders the approach of the electrophile to N-2 and directs alkylation to the N-1 position. Furthermore, this effect is not observed for indazoles bearing C-3 substituents that cannot participate in the formation of tight ion pairs (such as, 12–18 and 20), under conditions A (NaH in THF). The latter N-1 regioselectivity conferred through tight ion pair formation is augmented by the steric effect that the C-3 substituent enforces. The high degree of N-1 regioselectivity obtained for indazoles bearing bulky substituents at the C-3 position that are not capable of engaging in tight ion pair formation, such as 14 and 15, further highlights the influence of steric effects on regioselectivity.
To determine if the high degree of N-1 regioselectivity (ratio N-1:N-2 > 99:1) observed when employing conditions A (see Table 1 and Table 2) was due to base (NaH) or solvent (THF) effects, Cs2CO3 was substituted for NaH (Table 3, entries 1 and 2). Although the N-alkylation of indazole 9 was hindered by poor conversion (9%) when carried out at room temperature, the complete conversion of 9 to regioisomers 10 and 11 was observed and N-1 regioselectivity maintained upon increasing the reaction temperature to 50 °C (Table 3, entries 1 and 2).
Table 3: Effect of NaH and THF on N-1 regioselectivity.a
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-127-i7.svg?max-width=637&scale=1.0)
|
|||||||
| entry | base | equiv | solvent |
T
(°C) |
time
(h) |
conv.b
(%) |
ratiob
10:11 |
| 1 | Cs2CO3 | 1.5 | THF | rt | 16 | 9 | 5.8:1 |
| 2 | Cs2CO3 | 1.5 | THF | c | 24 | 100 | 6.8:1 |
| 3 | NaH | 1.1 | DMF | rt | 16 | 100 | 1.9:1 |
| 4 | NaH | 1.1 | DMF | c | 24 | 100 | 1.8:1 |
| 5 | LiH | 1.1 | THF | c | 24 | 18 | 28:1 |
| 6 | KH | 1.1 | THF | c | 24 | 100 | 11:1 |
| 7 | NaHMDS | 1.1 | THF | c | 24 | 91 | >99:1 |
| 8 | NaNH2 | 1.1 | THF | c | 24 | 69 | 62:1 |
| 9 | LDA | 1.1 | THF | c | 24 | 26 | >99:1 |
aReaction scale = 1 mmol (9); bdetermined using 1H NMR (see Supporting Information File 1); c0 °C → 50 °C; conv. = conversion of 9 to 10 and 11 only; rt ≈ 20 °C.
Employing a combination of NaH and DMF (Table 3, entries 3 and 4) caused a significant drop-off in N-1 regioselectivity (> 30%), when compared with NaH/THF (conditions A; Table 1, entry 22). Bookser et al. have reported a similar decline in N-1 regioselectivity for the alkylation of 1H-indazole with MeI when using a combination of NaHMDS and DMSO instead of THF as the reaction solvent [24]. It is likely that DMF similarly facilitates the formation of solvent-separated ion pairs which serve to diminish the high N-1 regioselectivity previously achieved when using THF (Table 3, entry 2), where tight ion pair formation between the cesium cation, N-2 atom, and chelating X=O C-3 group of the indazole ring predominates. Although the C-3 methyl carboxylate group of 9 may contribute to selective N-1 alkylation through steric effects, the use of DMF as the reaction solvent does not support tight ion pair formation and diminishes N-1 regioselectivity.
Further variation of the alkali metal cation, through the use of strong hydride bases, such as LiH or KH in THF, to examine N-1 regioselectivity (Table 3, entries 5 and 6, respectively) revealed a high preference for the formation of N-1 regioisomer 10 (ratio N-1 (10):N:2 (11) = 28:1 and 11:1, respectively). Similarly, strong amide bases, including NaHMDS, NaNH2, and LDA, also furnished N-1-alkylindazole 10 with excellent regioselectivity (up to > 98%, N-regioisomer), when using THF as the reaction solvent (Table 3, entries 7–9). These latter results indicate that the reaction solvent may play an important role in determining the regiochemical outcome of the N-alkylation. For example, the N-alkylation of methyl ester 9 under conditions B (Cs2CO3/DMF) showed no notable preference for the formation of N-1 regioisomer 10 (ratio N-1 (10):N-2 (11) = 1.4:1) (Table 1, entry 3), however, using a combination of Cs2CO3 and THF demonstrated marked N-1 regioselectivity (ratio N-1 (10):N-2 (11) = 5.8:1) (Table 3, entry 1).
To investigate the effect of the position of the indazole C-3 substituent on regiochemical outcome, several C-7 substituted indazoles (Me-, Br-, NO2-, and CO2Me) were alkylated using both conditions A (NaH in THF) and B (Cs2CO3 in DMF) (Table 4, entries 1–4). A significant reversal in regioselectivity was observed under conditions A for C-7 Me and Br-substituted indazoles, when compared with their analogous C-3 substituted counterparts (Table 2, entries 2 and 6), favouring the formation of the corresponding N-2 regioisomer (Table 4, entries 1 and 2). This latter preference for N-2 alkylation is likely due to the proximal steric bulk of both the C-7 Me and Br substituents, respectively, to the N-1 position.
Table 4: Effect of indazole benzenoid ring substituents on N-1:N-2 regioselectivity.a
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-127-i8.svg?max-width=637&scale=1.0)
|
|||||||||
| entry | R | product | cond. Ab,d | yield (%)e | cond. Bc,d | yield (%)e | |||
| N-1 | N-2 |
ratio
N-1:N-2e |
N-1 | N-2 |
ratio
N-1:N-2e |
N-1 | N-2 | ||
| 1 | 7-Me | 51 | 52 | 1:3.0 | 19 | 71 | 1:1.2 | 46 | 47 |
| 2 | 7-Br | 53 | 54 | 1:7.3 | 13 | 76 | 1:1.3 | 56 | 35 |
| 3 | 7-NO2 | 55 | 56 | 1:>99 | 0 | 81 | 1.1:1 | 50 | 44 |
| 4 | 7-CO2Me | 57 | 58 | 1:25 | 0 | 77 | 2.6:1 | 69 | 25 |
| 5 | 6-CO2Me | 59 | 60 | 1:2.2 | 22 | 55 | 1.7:1 | 61 | 35 |
| 6 | 5-CO2Me | 61 | 62 | 1:1.3 | 31 | 35 | 1.9:1 | 60 | 32 |
| 7 | 4-CO2Me | 63 | 64 | 1:1.3 | 28 | 34 | 1.3:1 | 55 | 26 |
aReaction scale = 1 mmol (appropriate indazole); b1.1 equiv NaH, 1.2 equiv n-pentyl bromide, THF, N2, 0 °C → 50 °C, 24 h; c1.5 equiv Cs2CO3, 1.2 equiv n-pentyl bromide, DMF, rt, 16 h; ddetermined using 1H NMR (see Supporting Information File 1); eisolated yield; cond. = conditions.
The presence of a nitro or methyl carboxylate group at the C-7 position of the indazole core facilitated excellent N-2 regioselectivity under conditions A (Table 4, entries 3 and 4). To further examine the positional effect of benzenoid ring substitution on N-alkylation regioselectivity, 6-, 5-, and 4-CO2Me substituted indazole derivatives were alkylated, under conditions A and B (Table 4, entries 5–7). While C-4 and C-5 substituted indazole methyl esters showed no apparent N-1 or N-2 regioselectivity under conditions A (ratio N-1:N-2 of both 61:62 and 63:64 = 1:1.3) (Table 4, entries 6 and 7), the corresponding C-6 substituted indazole methyl ester demonstrated a notable preference for N-2 alkylation (ratio N-1 (59):N-2 (60) = 1:2.2) (Table 4, entry 5). Importantly, the remarkable N-2 regioselectivity observed for C-7 NO2 and CO2Me substituted indazoles (≥ 96%) under conditions A provides further support for the role that tight ion pair formation may play in achieving regioselective N-alkylation (vide supra).
N-Alkylation of C-7 Me, Br, or NO2 substituted indazoles, using conditions B (Cs2CO3 in DMF), showed an overall loss of N-2 regioselectivity (Table 4, entries 1–3), when compared with the corresponding regiochemical outcomes obtained under conditions A (NaH in THF) (Table 4, entries 1–3). However, the N-alkylation of C-4, -5, -6, and -7 CO2Me substituted indazoles, in the presence of Cs2CO3 in DMF, all showed a preference for the formation of the corresponding N-1 regioisomer (Table 4, entries 4–7).
Mechanistically, we postulate that our optimized regioselective N-1 alkylation of the exemplar methyl ester 9 and other appropriately C-3 substituted indazoles (19, or 21–24) (under conditions A, Table 1, entry 22) involves the initial irreversible deprotonation of the indazole in the presence of NaH to initially give indazolyl salt 65 which is in equilibrium with its alternate anionic form 66 (Scheme 3). Through tautomerization, salt 66 may then form a tight ion pair with a sodium cation, via the N-2 atom and X=O containing C-3 substituent of the indazole nucleus, affording species 67 whose existence dominates in THF as solvent. The formation of 67 is then followed by the nucleophilic substitution of alkylating reagent R2–X to selectively give the desired N-1 regioisomer 68. It is likely that a mixture of the solvent-separated ion pairs 65 and 66 predominate, when using polar solvents such as DMF [24]. Furthermore, precluding tight ion pair formation through the use of DMF, may prompt indazole N-alkylation to fall predominantly under steric control, resulting in diminished N-alkylation regioselectivity.
![[1860-5397-17-127-i3]](/bjoc/content/inline/1860-5397-17-127-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Proposed mechanism for the regioselective N-1 alkylation of indazoles 9, 19, and 21–24 in the presence of NaH in THF (conditions A).
Scheme 3: Proposed mechanism for the regioselective N-1 alkylation of indazoles 9, 19, and 21–24 in the prese...
To further probe the potential influence of the alkali metal cation on the regioselective N-1 alkylation of indazole methyl ester 9, a control experiment was carried out, using 1 equivalent of the ether 15-crown-5 (with respect to NaH) (Table 5). Chelation of the sodium cation with the crown ether should disrupt the formation of tight ion pairs (Scheme 2) and attenuate N-1 regioselectivity. The presence of 15-crown-5 caused a notable reduction in N-1 regioselectivity, when compared with results obtained in the absence of the crown ether (ratio N-1 (10):N-2 (11) = 9.6:1 (Table 5, entry 1) versus > 99:1 (Table 1, entry 22)). Similarly, to disqualify any potential inherent effect of the crown ether on the regioselective outcome, increasing the number of equivalents of 15-crown-5 from one to five equivalents (with respect to NaH) gave a similar regioisomeric distribution of N-substituted indazoles 10 and 11 (Table 5, entry 2). These control experiments show that 15-crown-5 disrupts tight ion pair formation, providing further support for our mechanistic proposal that under conditions A (NaH/THF), tight ion pair formation directs regioselective N-1 alkylation.
Table 5: Effect of 15-crown-5 on the regioselective N-alkylation of indazole 9, in the presence of NaH in THF.a
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-127-i9.svg?max-width=637&scale=1.0)
|
|||||
| entry | equiv 15-C-5 |
conv.b
(%) |
ratio
10:11b |
yield (%)c | |
| 10 | 11 | ||||
| 1 | 1.1 | 100 | 9.6:1 | 77 | 6 |
| 2 | 5.5 | 100 | 6.0:1 | 71 | 10 |
aReaction scale = 1 mmol (9); bdetermined using 1H NMR (see Supporting Information File 1); cisolated yield; conv. = conversion of 9 to 10 and 11 only.
To demonstrate the scope of our optimized N-1 regioselective N-alkylation protocol (conditions A), methyl ester-substituted indazole 9 was subjected to a series of alkylating reagents under both conditions A and B (Table 6). The high selectivity observed for N-1 alkylation using NaH in THF (conditions A) was mainly effective using primary halide and tosylate compounds as electrophiles. Similar to the regiospecificity observed when employing n-pentyl bromide (ratio N-1 (10):N-2 (11) > 99:1, Table 1, entry 22), its tosylate counterpart gave the corresponding N-1 regioisomer 10 with a high degree of N-1 regioselectivity (ratio N-1 (10):N-2 (11) = 76:1) under conditions A (Table 6, entry 1). Furthermore, conditions A could be successfully applied to the synthesis of benzyl and alicyclic indazole derivatives 69–74 (Table 6, entries 2–6), affording the N-1 regioisomer almost exclusively. Notwithstanding excellent N-1 regioselectivity when using conditions A (ratio N-1 (73):N-2 (74) > 99:1), the yield of the corresponding N-1 substituted cyclohexylmethylindazole 73 was significantly reduced, due to poor conversion (13%, combined N-1 and N-2 (see Supporting Information)) (Table 5, entry 5). However, employing the corresponding tosylate under identical conditions (NaH in THF (conditions A)) permitted improved conversion to the desired N-1 substituted alicyclic indazole 73 (78%, combined N-1 and N-2 (see Supporting Information File 1)), whilst maintaining excellent N-1 regioselectivity (ratio N-1 (73):N-2 (74) = 70:1) (Table 6, entry 6).
Table 6: Alkylating reagent effects on N-1/N-2 regioselectivity.a
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-127-i10.svg?max-width=637&scale=1.0)
|
||||||||||
| entry | R | X | product | cond. Ab,c | yield (%)d | cond. Bc,e | yield (%)d | |||
| N-1 | N-2 |
ratio
N-1:N-2c |
N-1 | N-2 |
ratio
N-1:N-2c |
N-1 | N-2 | |||
| 1 | n-pentyl | OTs | 10 | 11 | 76:1 | 88 | 0 | 1.6:1 | 58 | 37 |
| 2 |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-127-i11.svg?max-width=637&scale=1.0)
|
Br | 69 | 70 | >99:1 | 90 | 0 | 2.1:1 | 60 | 22 |
| 3 |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-127-i12.svg?max-width=637&scale=1.0)
|
OTs | 69 | 70 | >62:1 | 85 | 3 | 2.1:1 | 52 | 25 |
| 4 |
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-127-i13.svg?max-width=637&scale=1.0)
|
Br | 71 | 72 | >99:1 | 82 | 0 | 3.8:1 | 77 | 21 |
| 5 |
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-127-i14.svg?max-width=637&scale=1.0)
|
Br | 73 | 74 | >99:1 | 7f | 0 | 1.5:1 | 35f | 23f |
| 6 |
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-127-i15.svg?max-width=637&scale=1.0)
|
OTs | 73 | 74 | 70:1 | 63f | 0 | 2.0:1 | 40f | 16f |
| 7 |
![[Graphic 13]](/bjoc/content/inline/1860-5397-17-127-i16.svg?max-width=637&scale=1.0)
|
Br | 75 | 76 | >99:1 | 3f | 0 | 1:1.1 | 49 | 44 |
| 8 |
![[Graphic 14]](/bjoc/content/inline/1860-5397-17-127-i17.svg?max-width=637&scale=1.0)
|
OTs | 75 | 76 | 25:1 | 81 | 2 | 1.1:1 | 43 | 38 |
| 9 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-17-127-i18.svg?max-width=637&scale=1.0)
|
Br | 77 | 78 | >99:1 | 4f | 0 | 1.0:1 | 48 | 46 |
aReaction scale = 1 mmol (9); b1.1 equiv NaH, 1.2 equiv R–X, THF, N2, 0 °C → 50 °C, 24 h; cdetermined using 1H NMR (see Supporting Information File 1); disolated yield; cond. = conditions; e1.5 equiv Cs2CO3, 1.2 equiv R–X, DMF, rt, 16 h; fincomplete conversion of 9 to the corresponding N-1 and/or N-2 regioisomer.
In the presence of NaH in THF (conditions A), secondary alkyl bromides, such as 2- and 3-bromopentanes, both gave their corresponding N-1 alkylindazoles 75–78 in only trace amounts (< 5% isolated yield, Table 6, entries 7 and 9), due to poor conversion (< 5%, combined N-1 and N-2 (see Supporting Information File 1)). While the latter observation may be due to competing elimination of the alkyl halide under strongly basic conditions [40], the use of a secondary tosylate electrophile under conditions A (Table 6, entry 8) furnished the desired N-1 regioisomer 75 in very good isolated yield (81%). These latter results would suggest that secondary alkyl tosylates are more suitable than their corresponding halide counterparts, for N-1 regioselective alkylation, under these investigated conditions. Conversely, the use of Cs2CO3 in DMF (conditions B) afforded approximately equal amounts of the corresponding N-1 and N-2 regioisomers 75–78 when using the aforementioned secondary alkyl bromides, with complete consumption of indazole 9 observed (Table 6, entries 7 and 9). However, N-1 regioselectivity is absent (ratio 75:76 and 77:78 ≈ 1:1, Table 6, entries 7–9) under conditions B, most likely due to solvent-separated ion pair formation.
Conclusion
Both 1H- and 2H-indazoles represent a core heterocyclic motif in many therapeutic small molecule drugs. Thus, from a synthetic perspective, the regioselective N-alkylation of the indazole scaffold would be of great value to the pharmaceutical industry. Focusing on 3-substituted indazoles, highly selective N-1 alkylations can be achieved using NaH in THF (conditions A) and Cs2CO3 in DMF (conditions B) as applied to primary and secondary alkyl electrophiles. When compared with conditions A, the use of Cs2CO3 in DMF (conditions B) demonstrated improved regioselectivity for the corresponding N-1 regioisomers of unsubstituted (12) and C-3 methyl (13), phenyl (15), halo (16–18), and cyano (20) substituted indazoles. Investigating the effect on the regioisomeric N-1/N-2 distribution indicated that steric bulk likely plays a significant role in determining N-1 regioselectivity. For example, the ratio of N-1/N-2 increases in the order of increasing steric bulk at the indazole C-3 position (H < Me < Ph < t-Bu). However, when the former protocol (conditions A) was extended to indazoles bearing a nitro (19) or carbonyl (21–24) functional group, the desired N-1 regioisomer was obtained with a very high degree of regioselectivity. In the case of C-3 substituted indazoles 9, 19 and 21–24 (those bearing a X=O α to the indazole C-3 position), we postulate that in the presence of NaH, the corresponding indazole salt may form a tight ion pair (67) which serves to attenuate N-2 alkylation, and affords the desired N-1 regioisomer exclusively. Furthermore, the excellent N-2 regioselectivity (≥ 96%) observed for the N-alkylation of C-7 NO2 or CO2Me indazoles (Table 4, entries 3 and 4) provides further support for the important role that tight ion pair formation plays in directing N-alkylation of the indazole scaffold.
Supporting Information
| Supporting Information File 1: Compound synthesis, characterisation, and copies of spectral data pertaining to regioisomeric distribution (N-1:N-2) determination. | ||
| Format: PDF | Size: 12.9 MB | Download |
References
-
Fludzinski, P.; Evrard, D. A.; Bloomquist, W. E.; Lacefield, W. B.; Pfeifer, W.; Jones, N. D.; Deeter, J. B.; Cohen, M. L. J. Med. Chem. 1987, 30, 1535–1537. doi:10.1021/jm00392a001
Return to citation in text: [1] -
Atta-ur-Rahman; Malik, S.; Cun-heng, H.; Clardy, J. Tetrahedron Lett. 1985, 26, 2759–2762. doi:10.1016/s0040-4039(00)94904-9
Return to citation in text: [1] -
Atta-ur-Rahman; Malik, S.; Hasan, S. S.; Choudhary, M. I.; Ni, C.-Z.; Clardy, J. Tetrahedron Lett. 1995, 36, 1993–1996. doi:10.1016/0040-4039(95)00210-4
Return to citation in text: [1] -
Yuan, T.; Nahar, P.; Sharma, M.; Liu, K.; Slitt, A.; Aisa, H. A.; Seeram, N. P. J. Nat. Prod. 2014, 77, 2316–2320. doi:10.1021/np500398m
Return to citation in text: [1] -
Spartinou, A.; Nyktari, V.; Papaioannou, A. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1289–1297. doi:10.1080/17425255.2017.1396317
Return to citation in text: [1] -
Quane, P. A.; Graham, G. G.; Ziegler, J. B. Inflammopharmacology 1998, 6, 95–107. doi:10.1007/s10787-998-0026-0
Return to citation in text: [1] -
Lu, Y.; Cole, K. P.; Fennell, J. W.; Maloney, T. D.; Mitchell, D.; Subbiah, R.; Ramadas, B. Org. Process Res. Dev. 2018, 22, 409–419. doi:10.1021/acs.oprd.8b00016
Return to citation in text: [1] -
Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. ChemMedChem 2018, 13, 1490–1507. doi:10.1002/cmdc.201800253
Return to citation in text: [1] -
Denya, I.; Malan, S. F.; Joubert, J. Expert Opin. Ther. Pat. 2018, 28, 441–453. doi:10.1080/13543776.2018.1472240
Return to citation in text: [1] -
Gaikwad, D. D.; Chapolikar, A. D.; Devkate, C. G.; Warad, K. D.; Tayade, A. P.; Pawar, R. P.; Domb, A. J. Eur. J. Med. Chem. 2015, 90, 707–731. doi:10.1016/j.ejmech.2014.11.029
Return to citation in text: [1] -
Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Molecules 2018, 23, 2783. doi:10.3390/molecules23112783
Return to citation in text: [1] -
Viña, D.; del Olmo, E.; López-Pérez, J. L.; San Feliciano, A. Org. Lett. 2007, 9, 525–528. doi:10.1021/ol062890e
Return to citation in text: [1] [2] -
Liu, H.-J.; Hung, S.-F.; Chen, C.-L.; Lin, M.-H. Tetrahedron 2013, 69, 3907–3912. doi:10.1016/j.tet.2013.03.042
Return to citation in text: [1] [2] -
Veerareddy, A.; Gogireddy, S.; Dubey, P. K. J. Heterocycl. Chem. 2014, 51, 1311–1321. doi:10.1002/jhet.1717
Return to citation in text: [1] [2] -
Teixeira, F. C.; Ramos, H.; Antunes, I. F.; Curto, M. J. M.; Duarte, M. T.; Bento, I. Molecules 2006, 11, 867–889. doi:10.3390/11110867
Return to citation in text: [1] -
Catalán, J.; de Paz, J. L. G.; Elguero, J. J. Chem. Soc., Perkin Trans. 2 1996, 57–60. doi:10.1039/p29960000057
Return to citation in text: [1] -
Hunt, K. W.; Moreno, D. A.; Suiter, N.; Clark, C. T.; Kim, G. Org. Lett. 2009, 11, 5054–5057. doi:10.1021/ol902050m
Return to citation in text: [1] [2] [3] [4] -
Yamazaki, T.; Baum, G.; Shechter, H. Tetrahedron Lett. 1974, 15, 4421–4424. doi:10.1016/s0040-4039(01)92179-3
Return to citation in text: [1] -
Saenz, J.; Mitchell, M.; Bahmanyar, S.; Stankovic, N.; Perry, M.; Craig-Woods, B.; Kline, B.; Yu, S.; Albizati, K. Org. Process Res. Dev. 2007, 11, 30–38. doi:10.1021/op0601621
Return to citation in text: [1] -
Conrow, R. E.; Delgado, P.; Dean, W. D.; Callen, G. R.; Plummer, S. V. Tetrahedron Lett. 2008, 49, 2348–2350. doi:10.1016/j.tetlet.2008.02.086
Return to citation in text: [1] -
Luo, G.; Chen, L.; Dubowchik, G. J. Org. Chem. 2006, 71, 5392–5395. doi:10.1021/jo060607j
Return to citation in text: [1] [2] -
Slade, D. J.; Pelz, N. F.; Bodnar, W.; Lampe, J. W.; Watson, P. S. J. Org. Chem. 2009, 74, 6331–6334. doi:10.1021/jo9006656
Return to citation in text: [1] [2] -
Cheung, M.; Boloor, A.; Stafford, J. A. J. Org. Chem. 2003, 68, 4093–4095. doi:10.1021/jo0265434
Return to citation in text: [1] -
Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Terentjeva, S.; Muceniece, D.; Lu̅sis, V. Org. Process Res. Dev. 2019, 23, 2057–2068. doi:10.1021/acs.oprd.9b00117
Return to citation in text: [1] -
Longworth, M.; Banister, S. D.; Mack, J. B. C.; Glass, M.; Connor, M.; Kassiou, M. Forensic Toxicol. 2016, 34, 286–303. doi:10.1007/s11419-016-0316-y
Return to citation in text: [1] -
Flynn, A. J.; Ford, A.; Khandavilli, U. B. R.; Lawrence, S. E.; Maguire, A. R. Eur. J. Org. Chem. 2019, 5368–5384. doi:10.1002/ejoc.201900494
Return to citation in text: [1] -
Bartsch, R. A.; Yang, I.-W. J. Heterocycl. Chem. 1984, 21, 1063–1064. doi:10.1002/jhet.5570210428
Return to citation in text: [1] -
Youngsaye, W.; Hartland, C. L.; Morgan, B. J.; Ting, A.; Nag, P. P.; Vincent, B.; Mosher, C. A.; Bittker, J. A.; Dandapani, S.; Palmer, M.; Whitesell, L.; Lindquist, S.; Schreiber, S. L.; Munoz, B. Beilstein J. Org. Chem. 2013, 9, 1501–1507. doi:10.3762/bjoc.9.171
Return to citation in text: [1] -
Song, P.; Chen, M.; Ma, X.; Xu, L.; Liu, T.; Zhou, Y.; Hu, Y. Bioorg. Med. Chem. 2015, 23, 1858–1868. doi:10.1016/j.bmc.2015.02.004
Return to citation in text: [1] -
Kuz'menko, V. V.; Pozharskii, A. F. Chem. Heterocycl. Compd. 1996, 32, 1152–1155. doi:10.1007/bf01169225
Return to citation in text: [1] -
Chevalier, A.; Ouahrouch, A.; Arnaud, A.; Gallavardin, T.; Franck, X. RSC Adv. 2018, 8, 13121–13128. doi:10.1039/c8ra01546e
Return to citation in text: [1] -
Clausen, D.; Chen, P.; Fradera, X.; Guo, L.; Han, Y.; He, S.; Lim, J.; Martinot, T. A.; Pasternak, A.; Xiao, L.; Yu, W. Novel Arylalkyl Pyrazole Compounds as Indoleamine 2,3-dioxygenase Inhibitors. WO Patent WO/2020/081381, April 23, 2020.
Return to citation in text: [1] -
Liu, K. G.; Robichaud, A. J.; Greenfield, A. A.; Lo, J. R.; Grosanu, C.; Mattes, J. F.; Cai, Y.; Zhang, G. M.; Zhang, J. Y.; Kowal, D. M.; Smith, D. L.; Di, L.; Kerns, E. H.; Schechter, L. E.; Comery, T. A. Bioorg. Med. Chem. 2011, 19, 650–662. doi:10.1016/j.bmc.2010.10.033
Return to citation in text: [1] -
Migliorini, A.; Oliviero, C.; Gasperi, T.; Loreto, M. A. Molecules 2012, 17, 4508–4521. doi:10.3390/molecules17044508
Return to citation in text: [1] -
Kim, J.-I.; Choi, Y. H.; Jahng, Y. Arch. Pharmacal Res. 1997, 20, 158–170. doi:10.1007/bf02974004
Return to citation in text: [1] -
Patel, M. R.; Pandya, K. G.; Lau-Cam, C. A.; Singh, S.; Pino, M. A.; Billack, B.; Degenhardt, K.; Talele, T. T. Chem. Biol. Drug Des. 2012, 79, 488–496. doi:10.1111/j.1747-0285.2011.01302.x
Return to citation in text: [1] -
Malhotra, N.; Fält-Hansen, B.; Becher, J. J. Heterocycl. Chem. 1991, 28, 1837–1839. doi:10.1002/jhet.5570280805
Return to citation in text: [1] -
Chen, S.; Graceffa, R. F.; Boezio, A. A. Org. Lett. 2016, 18, 16–19. doi:10.1021/acs.orglett.5b02994
Return to citation in text: [1] -
Conner, K. M.; Gronert, S. J. Org. Chem. 2013, 78, 8606–8613. doi:10.1021/jo4013354
Return to citation in text: [1]
| 38. | Malhotra, N.; Fält-Hansen, B.; Becher, J. J. Heterocycl. Chem. 1991, 28, 1837–1839. doi:10.1002/jhet.5570280805 |
| 39. | Chen, S.; Graceffa, R. F.; Boezio, A. A. Org. Lett. 2016, 18, 16–19. doi:10.1021/acs.orglett.5b02994 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 1. | Fludzinski, P.; Evrard, D. A.; Bloomquist, W. E.; Lacefield, W. B.; Pfeifer, W.; Jones, N. D.; Deeter, J. B.; Cohen, M. L. J. Med. Chem. 1987, 30, 1535–1537. doi:10.1021/jm00392a001 |
| 7. | Lu, Y.; Cole, K. P.; Fennell, J. W.; Maloney, T. D.; Mitchell, D.; Subbiah, R.; Ramadas, B. Org. Process Res. Dev. 2018, 22, 409–419. doi:10.1021/acs.oprd.8b00016 |
| 8. | Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. ChemMedChem 2018, 13, 1490–1507. doi:10.1002/cmdc.201800253 |
| 20. | Conrow, R. E.; Delgado, P.; Dean, W. D.; Callen, G. R.; Plummer, S. V. Tetrahedron Lett. 2008, 49, 2348–2350. doi:10.1016/j.tetlet.2008.02.086 |
| 6. | Quane, P. A.; Graham, G. G.; Ziegler, J. B. Inflammopharmacology 1998, 6, 95–107. doi:10.1007/s10787-998-0026-0 |
| 17. | Hunt, K. W.; Moreno, D. A.; Suiter, N.; Clark, C. T.; Kim, G. Org. Lett. 2009, 11, 5054–5057. doi:10.1021/ol902050m |
| 21. | Luo, G.; Chen, L.; Dubowchik, G. J. Org. Chem. 2006, 71, 5392–5395. doi:10.1021/jo060607j |
| 5. | Spartinou, A.; Nyktari, V.; Papaioannou, A. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1289–1297. doi:10.1080/17425255.2017.1396317 |
| 18. | Yamazaki, T.; Baum, G.; Shechter, H. Tetrahedron Lett. 1974, 15, 4421–4424. doi:10.1016/s0040-4039(01)92179-3 |
| 40. | Conner, K. M.; Gronert, S. J. Org. Chem. 2013, 78, 8606–8613. doi:10.1021/jo4013354 |
| 2. | Atta-ur-Rahman; Malik, S.; Cun-heng, H.; Clardy, J. Tetrahedron Lett. 1985, 26, 2759–2762. doi:10.1016/s0040-4039(00)94904-9 |
| 3. | Atta-ur-Rahman; Malik, S.; Hasan, S. S.; Choudhary, M. I.; Ni, C.-Z.; Clardy, J. Tetrahedron Lett. 1995, 36, 1993–1996. doi:10.1016/0040-4039(95)00210-4 |
| 4. | Yuan, T.; Nahar, P.; Sharma, M.; Liu, K.; Slitt, A.; Aisa, H. A.; Seeram, N. P. J. Nat. Prod. 2014, 77, 2316–2320. doi:10.1021/np500398m |
| 19. | Saenz, J.; Mitchell, M.; Bahmanyar, S.; Stankovic, N.; Perry, M.; Craig-Woods, B.; Kline, B.; Yu, S.; Albizati, K. Org. Process Res. Dev. 2007, 11, 30–38. doi:10.1021/op0601621 |
| 12. | Viña, D.; del Olmo, E.; López-Pérez, J. L.; San Feliciano, A. Org. Lett. 2007, 9, 525–528. doi:10.1021/ol062890e |
| 13. | Liu, H.-J.; Hung, S.-F.; Chen, C.-L.; Lin, M.-H. Tetrahedron 2013, 69, 3907–3912. doi:10.1016/j.tet.2013.03.042 |
| 14. | Veerareddy, A.; Gogireddy, S.; Dubey, P. K. J. Heterocycl. Chem. 2014, 51, 1311–1321. doi:10.1002/jhet.1717 |
| 16. | Catalán, J.; de Paz, J. L. G.; Elguero, J. J. Chem. Soc., Perkin Trans. 2 1996, 57–60. doi:10.1039/p29960000057 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 12. | Viña, D.; del Olmo, E.; López-Pérez, J. L.; San Feliciano, A. Org. Lett. 2007, 9, 525–528. doi:10.1021/ol062890e |
| 13. | Liu, H.-J.; Hung, S.-F.; Chen, C.-L.; Lin, M.-H. Tetrahedron 2013, 69, 3907–3912. doi:10.1016/j.tet.2013.03.042 |
| 14. | Veerareddy, A.; Gogireddy, S.; Dubey, P. K. J. Heterocycl. Chem. 2014, 51, 1311–1321. doi:10.1002/jhet.1717 |
| 17. | Hunt, K. W.; Moreno, D. A.; Suiter, N.; Clark, C. T.; Kim, G. Org. Lett. 2009, 11, 5054–5057. doi:10.1021/ol902050m |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 10. | Gaikwad, D. D.; Chapolikar, A. D.; Devkate, C. G.; Warad, K. D.; Tayade, A. P.; Pawar, R. P.; Domb, A. J. Eur. J. Med. Chem. 2015, 90, 707–731. doi:10.1016/j.ejmech.2014.11.029 |
| 11. | Zhang, S.-G.; Liang, C.-G.; Zhang, W.-H. Molecules 2018, 23, 2783. doi:10.3390/molecules23112783 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 9. | Denya, I.; Malan, S. F.; Joubert, J. Expert Opin. Ther. Pat. 2018, 28, 441–453. doi:10.1080/13543776.2018.1472240 |
| 15. | Teixeira, F. C.; Ramos, H.; Antunes, I. F.; Curto, M. J. M.; Duarte, M. T.; Bento, I. Molecules 2006, 11, 867–889. doi:10.3390/11110867 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 22. | Slade, D. J.; Pelz, N. F.; Bodnar, W.; Lampe, J. W.; Watson, P. S. J. Org. Chem. 2009, 74, 6331–6334. doi:10.1021/jo9006656 |
| 23. | Cheung, M.; Boloor, A.; Stafford, J. A. J. Org. Chem. 2003, 68, 4093–4095. doi:10.1021/jo0265434 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 25. | Terentjeva, S.; Muceniece, D.; Lu̅sis, V. Org. Process Res. Dev. 2019, 23, 2057–2068. doi:10.1021/acs.oprd.9b00117 |
| 17. | Hunt, K. W.; Moreno, D. A.; Suiter, N.; Clark, C. T.; Kim, G. Org. Lett. 2009, 11, 5054–5057. doi:10.1021/ol902050m |
| 34. | Liu, K. G.; Robichaud, A. J.; Greenfield, A. A.; Lo, J. R.; Grosanu, C.; Mattes, J. F.; Cai, Y.; Zhang, G. M.; Zhang, J. Y.; Kowal, D. M.; Smith, D. L.; Di, L.; Kerns, E. H.; Schechter, L. E.; Comery, T. A. Bioorg. Med. Chem. 2011, 19, 650–662. doi:10.1016/j.bmc.2010.10.033 |
| 35. | Migliorini, A.; Oliviero, C.; Gasperi, T.; Loreto, M. A. Molecules 2012, 17, 4508–4521. doi:10.3390/molecules17044508 |
| 36. | Kim, J.-I.; Choi, Y. H.; Jahng, Y. Arch. Pharmacal Res. 1997, 20, 158–170. doi:10.1007/bf02974004 |
| 37. | Patel, M. R.; Pandya, K. G.; Lau-Cam, C. A.; Singh, S.; Pino, M. A.; Billack, B.; Degenhardt, K.; Talele, T. T. Chem. Biol. Drug Des. 2012, 79, 488–496. doi:10.1111/j.1747-0285.2011.01302.x |
| 27. | Flynn, A. J.; Ford, A.; Khandavilli, U. B. R.; Lawrence, S. E.; Maguire, A. R. Eur. J. Org. Chem. 2019, 5368–5384. doi:10.1002/ejoc.201900494 |
| 28. | Bartsch, R. A.; Yang, I.-W. J. Heterocycl. Chem. 1984, 21, 1063–1064. doi:10.1002/jhet.5570210428 |
| 29. | Youngsaye, W.; Hartland, C. L.; Morgan, B. J.; Ting, A.; Nag, P. P.; Vincent, B.; Mosher, C. A.; Bittker, J. A.; Dandapani, S.; Palmer, M.; Whitesell, L.; Lindquist, S.; Schreiber, S. L.; Munoz, B. Beilstein J. Org. Chem. 2013, 9, 1501–1507. doi:10.3762/bjoc.9.171 |
| 30. | Song, P.; Chen, M.; Ma, X.; Xu, L.; Liu, T.; Zhou, Y.; Hu, Y. Bioorg. Med. Chem. 2015, 23, 1858–1868. doi:10.1016/j.bmc.2015.02.004 |
| 31. | Kuz'menko, V. V.; Pozharskii, A. F. Chem. Heterocycl. Compd. 1996, 32, 1152–1155. doi:10.1007/bf01169225 |
| 32. | Chevalier, A.; Ouahrouch, A.; Arnaud, A.; Gallavardin, T.; Franck, X. RSC Adv. 2018, 8, 13121–13128. doi:10.1039/c8ra01546e |
| 33. | Clausen, D.; Chen, P.; Fradera, X.; Guo, L.; Han, Y.; He, S.; Lim, J.; Martinot, T. A.; Pasternak, A.; Xiao, L.; Yu, W. Novel Arylalkyl Pyrazole Compounds as Indoleamine 2,3-dioxygenase Inhibitors. WO Patent WO/2020/081381, April 23, 2020. |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 26. | Longworth, M.; Banister, S. D.; Mack, J. B. C.; Glass, M.; Connor, M.; Kassiou, M. Forensic Toxicol. 2016, 34, 286–303. doi:10.1007/s11419-016-0316-y |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
| 17. | Hunt, K. W.; Moreno, D. A.; Suiter, N.; Clark, C. T.; Kim, G. Org. Lett. 2009, 11, 5054–5057. doi:10.1021/ol902050m |
| 21. | Luo, G.; Chen, L.; Dubowchik, G. J. Org. Chem. 2006, 71, 5392–5395. doi:10.1021/jo060607j |
| 22. | Slade, D. J.; Pelz, N. F.; Bodnar, W.; Lampe, J. W.; Watson, P. S. J. Org. Chem. 2009, 74, 6331–6334. doi:10.1021/jo9006656 |
| 24. | Bookser, B. C.; Weinhouse, M. I.; Burns, A. C.; Valiere, A. N.; Valdez, L. J.; Stanczak, P.; Na, J.; Rheingold, A. L.; Moore, C. E.; Dyck, B. J. Org. Chem. 2018, 83, 6334–6353. doi:10.1021/acs.joc.8b00540 |
© 2021 Alam and Keating; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)