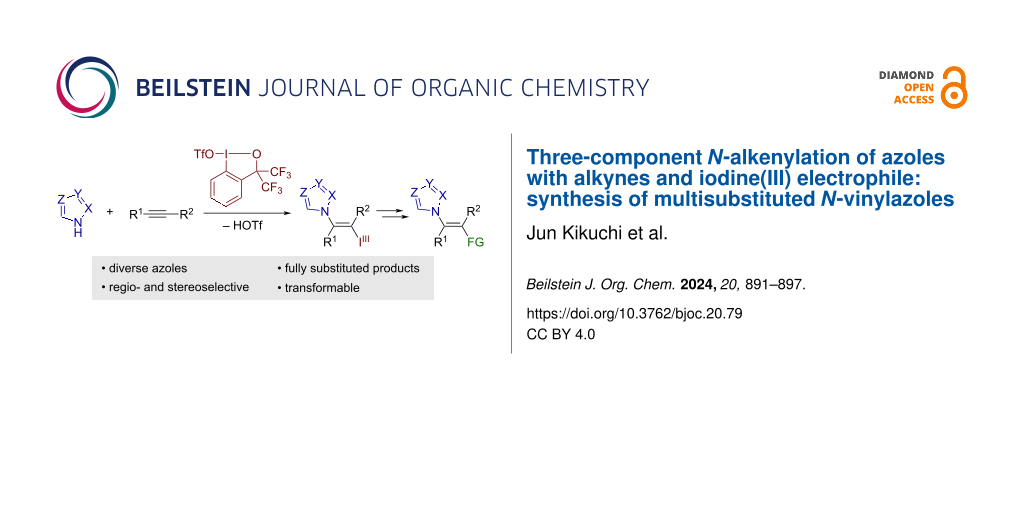
Abstract
A stereoselective N-alkenylation of azoles with alkynes and iodine(III) electrophile is reported. The reaction between various azoles and internal alkynes is mediated by benziodoxole triflate as the electrophile in a trans-fashion, affording azole-bearing vinylbenziodoxoles in moderate to good yields. The tolerable azole nuclei include pyrazole, indazole, 1,2,3-triazole, benzotriazole, and tetrazole. The iodanyl group in the product can be leveraged as a versatile synthetic handle, allowing for the preparation of hitherto inaccessible types of densely functionalized N-vinylazoles.

Graphical Abstract
Introduction
N-Functionalized azoles are prevalent in bioactive natural products and pharmaceutical agents, including antifungal drugs [1-3], and hence their selective preparation has attracted considerable attention from the synthetic community. Compared to methods for the de novo construction of azole heterocycles, direct functionalization of the azole N–H bond offers the unique merit of enabling rapid access to structurally diverse N-functionalized azoles because one versatile method would potentially apply to various azole nuclei. In this context, the N-alkenylation of azoles represents an attractive transformation due to the occurrence of the N-vinylazole motif in bioactive compounds and the synthetic utility of its olefinic C=C bond. The most extensively explored approach to this transformation is the transition metal-catalyzed C–N coupling between azoles and vinylating agents, including vinyl halides [4], boronates [5], sulfonium salts [6-8], and iodonium salts [9], which usually occurs with the retention of the stereochemistry of the vinylating agents (Scheme 1a). Nonetheless, this approach is not necessarily suited for the stereoselective preparation of densely substituted N-vinylazoles because preparing the requisite multisubstituted vinylating agents, preferably with well-defined stereochemistry, is a nontrivial task.
![[1860-5397-20-79-i1]](/bjoc/content/inline/1860-5397-20-79-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The addition of azoles to alkynes represents an alternative approach to N-vinylazoles. For example, Nolan and co-workers recently reported a gold-catalyzed addition of azoles to alkynes (hydroazolation; Scheme 1b) [10]. The gold catalysis encompassed various azoles such as pyrazole, indazole, and (benzo)triazole, exhibiting high Z-selectivity. In addition, Cao et al. reported a gold-catalyzed addition of 5-substituted tetrazoles to terminal alkynes [11]. Analogous hydroazolation reactions of alkynes have also been achieved under other metal-catalyzed conditions [12,13] or base-mediated conditions [14], with varying scopes of azoles and alkynes. Despite such advances, the hydroazolation approach is intrinsically limited to the preparation of mono- or disubstituted vinylazoles. Herein, we report on a three-component N-vinylation reaction of azoles with alkynes and iodine(III) electrophile, benziodoxole triflate (BXT, 1; Scheme 1c). Displaying exclusive trans-selectivity, the reaction tolerates a broad range of azoles, including pyrazole, 1,2,3-triazole, tetrazole, indazole, and benzotriazole, with internal alkynes as coupling partners. The resulting products represent a new class of functionalized vinylbenziodoxoles (VBXs) [15-21], which have recently emerged as unique vinylating agents [22-25]. Thus, the follow-up transformation of the iodanyl group in the present products allows for the preparation of hitherto inaccessible types of densely functionalized vinylazoles with tetrasubstituted olefinic moiety.
Results and Discussion
Our group has demonstrated benziodoxole triflate (BXT) [26] and related compounds as a versatile iodine(III) electrophile for the inter- and intramolecular difunctionalization of alkynes with various heteroatom and carbon nucleophiles [27-34]. Specifically, intermolecular trans-iodo(III)functionalization of alkynes has been achieved using oxygen nucleophiles such as alcohols [28,32], ethers [33], carboxylic acids [31], phosphate esters [31], and sulfonic acids [31]. On the other hand, nitrogen-based nucleophiles amenable to this reaction manifold have thus far been limited to nitriles in the context of Ritter-type iodo(III)amidation [29]. In light of the significance of vinylated azoles, our attention was attracted to the feasibility of iodo(III)azolation using various azoles (a single example of iodo(III)azolation using pyrazole was reported in [28]).
Table 1 summarizes the results of the optimization of the reaction conditions for the vinylation of pyrazole (2a) with 1-phenyl-1-propyne (3a) and BXT (1). Upon examination of various reaction parameters, the desired three-component N-alkenylation was found to proceed smoothly by reacting 3a with excess amounts of 1 (2 equiv) and 2a (5 equiv) in MeCN (0.2 M) at room temperature, affording the trans-difunctionalized product 4aa as a single regio- and stereoisomer in 77% yield (Table 1, entry 1). Decreasing or increasing the concentration did not improve the yield of 4aa (Table 1, entries 2 and 3). The reaction became rather sluggish in different solvents such as HFIP and Et2O (Table 1, entries 4 and 5). By reducing the equivalents of 2a to 3 equiv and 2 equiv, the yield of 4aa dropped to 66% and 55%, respectively (Table 1, entries 6 and 7). The addition of a base such as K2CO3 completely shut down the desired reaction (Table 1, entry 8). It is worth noting that the replacement of 1 with N-iodosuccinimide, a common iodine(I) electrophile, failed to promote an analogous iodoazolation reaction, highlighting the unique utility of the iodine(III) electrophile in the present alkyne difunctionalization.
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-20-79-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Deviation from standard conditions | Yield (%)b |
| 1 | none | 77c |
| 2 | c = 0.1 M | 50 |
| 3 | c = 0.4 M | 70 |
| 4 | HFIP as the solvent (0.2 M) | 16 |
| 5 | Et2O as the solvent (0.2 M) | 22 |
| 6 | 3 equiv of 2a | 66 |
| 7 | 2 equiv of 2a | 55 |
| 8 | 2 equiv of 2a, 2 equiv of K2CO3 | trace |
aThe reaction was performed on a 0.1 mmol scale; bDetermined by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard; cIsolated yield.
With the standard conditions (Table 1, entry 1) in hand, we explored the scope of the three-component N-vinylation (Scheme 2). First, various azoles were subjected to the vinylation reaction using alkyne 3a and BXT (1). 4-Bromo and 4-methylpyrazoles afforded the desired products 4ba and 4ca in 92% and 38% yields, respectively. 3-Phenylpyrazole underwent competitive alkenylation at the N1 and N2 positions, affording the N2-alkenylated product 4da and its N1-regioisomer in 84% overall yield in a ratio of 7:3. Indazole and its 4-bromo and 6-methoxycarbonyl analogues afforded the expected N1-alkenylated products 4ea–4ga in 43–68% yields. 1,2,3-Triazole and benzotriazole both smoothly participated in the reaction to give their respective products 4ha and 4ia in 60% and 81% yields, respectively. Tetrazole and 5-methyltetrazole both proved to be excellent substrates. Regardless of the presence or absence of the 5-substituent, they underwent preferential alkenylation at the N1 position over the N2 position with the identical regioselectivity of 7:3 (see 4ja and 4ka). Among other five-membered aromatic azacycles, imidazole completely failed to undergo the present N-alkenylation, whereas the reaction of 1,2,4-triazole was too sluggish to allow for the isolation and unambiguous characterization of the expected product.
![[1860-5397-20-79-i2]](/bjoc/content/inline/1860-5397-20-79-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of three-component N-alkenylation of azoles.
Scheme 2: Scope of three-component N-alkenylation of azoles.
Next, the reaction of pyrazole (2a) was explored using different alkynes. A series of (hetero)aryl(alkyl)alkynes were successfully engaged as reaction partners to give the products 4ab–4af in moderate yields, displaying tolerance to o-tolyl (4ad), 2-thienyl (4ae), and methoxymethyl (4af) groups. A dialkylalkyne such as 5-decyne smoothly underwent the difunctionalization reaction to afford the product 4ag in 62% yield. While sluggish, trimethylsilylacetylene was selectively azolated at the terminal position (see 4ah), which could be rationalized by the better stabilization of a partial positive charge at this position by the β-silyl substituent. Finally, an oxazolidinone-substituted ynamide also proved to undergo iodo(III)azolation in a regio- and stereoselective fashion to give the product 4ai in a moderate yield. Note that terminal alkynes such as phenylacetylene also took part in the reaction, albeit in a much-diminished yield (7% by 1H NMR; data not shown). Observing no byproducts originating from phenylacetylene, we speculate that the lack of reactivity stems from the relatively low electron density of the terminal alkyne, which likely leads to direct coordination of pyrazole to the iodine(III) reagent.
To probe the relative reactivity of different azoles, competition experiments were performed. The reaction of excess (5 equiv each) pyrazole (2a) and 1,2,3-triazole (2h) with the alkyne 3a and 1 afforded a mixture of the corresponding products 4aa and 4ha in a ratio of 60:40 (Scheme 3a). Performed in the same manner, the competition between 2a and tetrazole (2j) resulted in 4aa as the major adduct, accompanied by the tetrazole adducts 4ja and 4ja’ (the ratio 4aa/4ja/4ja’ = 71:21:8; Scheme 3b). Superficially, these results appear correlated with the acidity of the corresponding azoles (pKa value: pyrazole, 19.8; 1,2,3-triazole, 13.9; tetrazole, 8.2), with the lowest-acidic pyrazole being the most competitive. However, we rather surmise that the Lewis basicity of the proton-free nitrogen atom of the azole would have more direct relevance to the results of the competition experiments, where the least Lewis basic tetrazole was the least competitive. Along with this conjecture, the present reaction is proposed to involve reversible complexation between the alkyne and the cationic iodine(III) electrophile and subsequent trans-addition of the azole nucleophile, the latter step being coupled with concomitant deprotonation of the N–H bond by the triflate anion (Scheme 3c). It is important to note that the azole nucleophile preferentially adds to the carbon atom that can better stabilize a positive charge, as demonstrated by the regioselectivities observed with unsymmetrical alkynes. The failure of imidazole to participate in the iodo(III)azolation may be attributed to its much greater Lewis basicity compared to other azoles, likely killing the reactivity of the iodine(III) electrophile by direct coordination.
![[1860-5397-20-79-i3]](/bjoc/content/inline/1860-5397-20-79-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Competition experiments and plausible reaction pathway.
Scheme 3: Competition experiments and plausible reaction pathway.
The present reaction could be performed on a preparative scale. Thus, 1 mmol-scale synthesis of the vinylazoles 4aa and 4ba could be successfully performed in 68% and 74% yields, respectively (Scheme 4a). Furthermore, the iodanyl group on these products serves as a versatile handle for downstream transformations, thus allowing for the stereoselective preparation of various trisubstituted N-vinylazoles (Scheme 4b). Pd-catalyzed C–C couplings such as Suzuki–Miyaura and Sonogashira couplings on 4aa or 4ba afforded the desired products 5 and 6 in 47% and 74% yields, respectively. In the former case, the C–Br bond on the pyrazole moiety remained intact, highlighting the superior leaving group ability of the BX group. Cu-catalyzed Ullmann coupling between 4ba and 4-methoxybenzenethiol furnished the N,S-substituted olefin 7 in 59% yield. The treatment of 4aa with stoichiometric CuI and ʟ-proline effected the iodine(III)-to-iodine(I) conversion to give the vinyl iodide 8 in 76% yield. Compound 8 was used for the Ullmann coupling with imidazole, producing the vicinal N-heterocycle-substituted olefin 9 as a mixture of stereoisomers in 65% yield. Finally, 4aa proved to be a viable nucleophilic VBX for the carboiodanation of 3-methoxybenzyne [35], furnishing the new ortho-alkenylated arylbenziodoxole 10 with exclusive C–C bond formation at the distal aryne carbon [36].
![[1860-5397-20-79-i4]](/bjoc/content/inline/1860-5397-20-79-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Preparative-scale reaction and product transformations. Reaction conditions: (a) Pd(PPh3)4, 4-MeOC6H4B(OH)2, Cs2CO3, DMF/H2O, 60 °C, 18 h. (b) Pd(OAc)2, PPh3, CuI, phenylacetylene, Et3N, 50 °C, 5 h. (c) CuI, neocuproine, 4-MeOC6H4SH, NaOt-Bu, toluene, 110 °C, 13 h. (d) CuI, ʟ-proline, DMF, 80 °C, 14 h. (e) CuI, imidazole, Cs2CO3, DMF, 120 °C, 15 h. (f) 3-Methoxy-2-(trimethylsilyl)phenyl triflate, CsF, MeCN, rt, 18 h.
Scheme 4: Preparative-scale reaction and product transformations. Reaction conditions: (a) Pd(PPh3)4, 4-MeOC6H...
Conclusion
In summary, we have reported a three-component N-vinylation reaction of azoles with alkynes and iodine(III) electrophile. The present reaction represents a rare example of the installation of stereodefined trisubstituted alkenyl groups into the azole core, encompassing various azole nucleophiles including pyrazole, indazole, 1,2,3-triazole, benzotriazole, and tetrazole. The follow-up transformation of the iodanyl group provides a means to prepare hitherto inaccessible types of alkenylated azoles. Further exploration of the three-component alkenylation of nitrogen and other heteroatom nucleophiles is currently underway.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data of new compounds. | ||
| Format: PDF | Size: 6.2 MB | Download |
Acknowledgements
We thank the Central Glass Co., Ltd. for the generous donation of 1,1,1,3,3,3-hexafluoro-2-phenylpropan-2-ol (HFAB).
Funding
This work was supported by JSPS KAKENHI (Grant No. 20K23375 (N.Y.) and 19K15552 (J.K.)), The Uehara Memorial Foundation (N.Y.), Research Support Project for Life Science and Drug Discovery (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED (Grant No. JP23ama121040 (N.Y.)).
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
-
Rezaei, Z.; Khabnadideh, S.; Pakshir, K.; Hossaini, Z.; Amiri, F.; Assadpour, E. Eur. J. Med. Chem. 2009, 44, 3064–3067. doi:10.1016/j.ejmech.2008.07.012
Return to citation in text: [1] -
Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b
Return to citation in text: [1] -
Azevedo, M.-M.; Faria-Ramos, I.; Cruz, L. C.; Pina-Vaz, C.; Gonçalves Rodrigues, A. J. Agric. Food Chem. 2015, 63, 7463–7468. doi:10.1021/acs.jafc.5b02728
Return to citation in text: [1] -
Liao, Q.; Wang, Y.; Zhang, L.; Xi, C. J. Org. Chem. 2009, 74, 6371–6373. doi:10.1021/jo901105r
Return to citation in text: [1] -
Motornov, V.; Latyshev, G. V.; Kotovshchikov, Y. N.; Lukashev, N. V.; Beletskaya, I. P. Adv. Synth. Catal. 2019, 361, 3306–3311. doi:10.1002/adsc.201900225
Return to citation in text: [1] -
Zhou, M.; Tan, X.; Hu, Y.; Shen, H. C.; Qian, X. J. Org. Chem. 2018, 83, 8627–8635. doi:10.1021/acs.joc.8b00682
Return to citation in text: [1] -
Juliá, F.; Yan, J.; Paulus, F.; Ritter, T. J. Am. Chem. Soc. 2021, 143, 12992–12998. doi:10.1021/jacs.1c06632
Return to citation in text: [1] -
Chen, S.-J.; Li, J.-H.; He, Z.-Q.; Chen, G.-S.; Zhuang, Y.-Y.; Chen, C.-P.; Liu, Y.-L. J. Org. Chem. 2022, 87, 15703–15712. doi:10.1021/acs.joc.2c02323
Return to citation in text: [1] -
Csenki, J. T.; Mészáros, Á.; Gonda, Z.; Novák, Z. Chem. – Eur. J. 2021, 27, 15638–15643. doi:10.1002/chem.202102840
Return to citation in text: [1] -
Michon, C.; Gilbert, J.; Trivelli, X.; Nahra, F.; Cazin, C. S. J.; Agbossou-Niedercorn, F.; Nolan, S. P. Org. Biomol. Chem. 2019, 17, 3805–3811. doi:10.1039/c9ob00587k
Return to citation in text: [1] -
Cao, Z.; Zhao, C.; Zhu, J.; Yan, S.; Tian, L.; Sun, X.; Meng, X. ChemistrySelect 2019, 4, 11785–11789. doi:10.1002/slct.201902532
Return to citation in text: [1] -
Tsuchimoto, T.; Aoki, K.; Wagatsuma, T.; Suzuki, Y. Eur. J. Org. Chem. 2008, 4035–4040. doi:10.1002/ejoc.200800353
Return to citation in text: [1] -
Das, U. K.; Mandal, S.; Anoop, A.; Bhattacharjee, M. J. Org. Chem. 2014, 79, 9979–9991. doi:10.1021/jo502151z
Return to citation in text: [1] -
Garg, V.; Kumar, P.; Verma, A. K. J. Org. Chem. 2017, 82, 10247–10262. doi:10.1021/acs.joc.7b01746
Return to citation in text: [1] -
Declas, N.; Pisella, G.; Waser, J. Helv. Chim. Acta 2020, 103, e2000191. doi:10.1002/hlca.202000191
Return to citation in text: [1] -
Mironova, I. A.; Noskov, D. M.; Yoshimura, A.; Yusubov, M. S.; Zhdankin, V. V. Molecules 2023, 28, 2136. doi:10.3390/molecules28052136
Return to citation in text: [1] -
Stridfeldt, E.; Seemann, A.; Bouma, M. J.; Dey, C.; Ertan, A.; Olofsson, B. Chem. – Eur. J. 2016, 22, 16066–16070. doi:10.1002/chem.201603955
Return to citation in text: [1] -
Wu, J.; Deng, X.; Hirao, H.; Yoshikai, N. J. Am. Chem. Soc. 2016, 138, 9105–9108. doi:10.1021/jacs.6b06247
Return to citation in text: [1] -
Wu, J.; Xu, K.; Hirao, H.; Yoshikai, N. Chem. – Eur. J. 2017, 23, 1521–1525. doi:10.1002/chem.201605772
Return to citation in text: [1] -
Caramenti, P.; Declas, N.; Tessier, R.; Wodrich, M. D.; Waser, J. Chem. Sci. 2019, 10, 3223–3230. doi:10.1039/c8sc05573d
Return to citation in text: [1] -
Shimbo, D.; Shibata, A.; Yudasaka, M.; Maruyama, T.; Tada, N.; Uno, B.; Itoh, A. Org. Lett. 2019, 21, 9769–9773. doi:10.1021/acs.orglett.9b03990
Return to citation in text: [1] -
Pisella, G.; Gagnebin, A.; Waser, J. Org. Lett. 2020, 22, 3884–3889. doi:10.1021/acs.orglett.0c01150
Return to citation in text: [1] -
Di Tommaso, E. M.; Norrby, P.-O.; Olofsson, B. Angew. Chem., Int. Ed. 2022, 61, e202206347. doi:10.1002/anie.202206347
Return to citation in text: [1] -
Milzarek, T. M.; Waser, J. Angew. Chem., Int. Ed. 2023, 62, e202306128. doi:10.1002/anie.202306128
Return to citation in text: [1] -
Bhaskar Pal, K.; Di Tommaso, E. M.; Inge, A. K.; Olofsson, B. Angew. Chem., Int. Ed. 2023, 62, e202301368. doi:10.1002/anie.202301368
Return to citation in text: [1] -
Zhdankin, V. V.; Kuehl, C. J.; Krasutsky, A. P.; Bolz, J. T.; Simonsen, A. J. J. Org. Chem. 1996, 61, 6547–6551. doi:10.1021/jo960927a
Return to citation in text: [1] -
Wu, B.; Wu, J.; Yoshikai, N. Chem. – Asian J. 2017, 12, 3123–3127. doi:10.1002/asia.201701530
Return to citation in text: [1] -
Ding, W.; Chai, J.; Wang, C.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2020, 142, 8619–8624. doi:10.1021/jacs.0c04140
Return to citation in text: [1] [2] [3] -
Chai, J.; Ding, W.; Wang, C.; Ito, S.; Wu, J.; Yoshikai, N. Chem. Sci. 2021, 12, 15128–15133. doi:10.1039/d1sc05240c
Return to citation in text: [1] [2] -
Laskar, R. A.; Ding, W.; Yoshikai, N. Org. Lett. 2021, 23, 1113–1117. doi:10.1021/acs.orglett.1c00039
Return to citation in text: [1] -
Wang, C.-S.; Tan, P. S. L.; Ding, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 430–434. doi:10.1021/acs.orglett.1c04123
Return to citation in text: [1] [2] [3] [4] -
Kikuchi, J.; Maesaki, K.; Sasaki, S.; Wang, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 6914–6918. doi:10.1021/acs.orglett.2c02570
Return to citation in text: [1] [2] -
Chai, J.; Ding, W.; Wu, J.; Yoshikai, N. Chem. – Asian J. 2020, 15, 2166–2169. doi:10.1002/asia.202000653
Return to citation in text: [1] [2] -
Kikuchi, J.; Nagata, T.; Ito, S.; Yoshikai, N. Org. Chem. Front. 2024. doi:10.1039/d4qo00489b
Return to citation in text: [1] -
Arakawa, C.; Kanemoto, K.; Nakai, K.; Wang, C.; Morohashi, S.; Kwon, E.; Ito, S.; Yoshikai, N. J. Am. Chem. Soc. 2024, 146, 3910–3919. doi:10.1021/jacs.3c11524
Return to citation in text: [1] -
Medina, J. M.; Mackey, J. L.; Garg, N. K.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 15798–15805. doi:10.1021/ja5099935
Return to citation in text: [1]
| 29. | Chai, J.; Ding, W.; Wang, C.; Ito, S.; Wu, J.; Yoshikai, N. Chem. Sci. 2021, 12, 15128–15133. doi:10.1039/d1sc05240c |
| 31. | Wang, C.-S.; Tan, P. S. L.; Ding, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 430–434. doi:10.1021/acs.orglett.1c04123 |
| 31. | Wang, C.-S.; Tan, P. S. L.; Ding, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 430–434. doi:10.1021/acs.orglett.1c04123 |
| 1. | Rezaei, Z.; Khabnadideh, S.; Pakshir, K.; Hossaini, Z.; Amiri, F.; Assadpour, E. Eur. J. Med. Chem. 2009, 44, 3064–3067. doi:10.1016/j.ejmech.2008.07.012 |
| 2. | Vitaku, E.; Smith, D. T.; Njardarson, J. T. J. Med. Chem. 2014, 57, 10257–10274. doi:10.1021/jm501100b |
| 3. | Azevedo, M.-M.; Faria-Ramos, I.; Cruz, L. C.; Pina-Vaz, C.; Gonçalves Rodrigues, A. J. Agric. Food Chem. 2015, 63, 7463–7468. doi:10.1021/acs.jafc.5b02728 |
| 9. | Csenki, J. T.; Mészáros, Á.; Gonda, Z.; Novák, Z. Chem. – Eur. J. 2021, 27, 15638–15643. doi:10.1002/chem.202102840 |
| 33. | Chai, J.; Ding, W.; Wu, J.; Yoshikai, N. Chem. – Asian J. 2020, 15, 2166–2169. doi:10.1002/asia.202000653 |
| 6. | Zhou, M.; Tan, X.; Hu, Y.; Shen, H. C.; Qian, X. J. Org. Chem. 2018, 83, 8627–8635. doi:10.1021/acs.joc.8b00682 |
| 7. | Juliá, F.; Yan, J.; Paulus, F.; Ritter, T. J. Am. Chem. Soc. 2021, 143, 12992–12998. doi:10.1021/jacs.1c06632 |
| 8. | Chen, S.-J.; Li, J.-H.; He, Z.-Q.; Chen, G.-S.; Zhuang, Y.-Y.; Chen, C.-P.; Liu, Y.-L. J. Org. Chem. 2022, 87, 15703–15712. doi:10.1021/acs.joc.2c02323 |
| 31. | Wang, C.-S.; Tan, P. S. L.; Ding, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 430–434. doi:10.1021/acs.orglett.1c04123 |
| 5. | Motornov, V.; Latyshev, G. V.; Kotovshchikov, Y. N.; Lukashev, N. V.; Beletskaya, I. P. Adv. Synth. Catal. 2019, 361, 3306–3311. doi:10.1002/adsc.201900225 |
| 27. | Wu, B.; Wu, J.; Yoshikai, N. Chem. – Asian J. 2017, 12, 3123–3127. doi:10.1002/asia.201701530 |
| 28. | Ding, W.; Chai, J.; Wang, C.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2020, 142, 8619–8624. doi:10.1021/jacs.0c04140 |
| 29. | Chai, J.; Ding, W.; Wang, C.; Ito, S.; Wu, J.; Yoshikai, N. Chem. Sci. 2021, 12, 15128–15133. doi:10.1039/d1sc05240c |
| 30. | Laskar, R. A.; Ding, W.; Yoshikai, N. Org. Lett. 2021, 23, 1113–1117. doi:10.1021/acs.orglett.1c00039 |
| 31. | Wang, C.-S.; Tan, P. S. L.; Ding, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 430–434. doi:10.1021/acs.orglett.1c04123 |
| 32. | Kikuchi, J.; Maesaki, K.; Sasaki, S.; Wang, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 6914–6918. doi:10.1021/acs.orglett.2c02570 |
| 33. | Chai, J.; Ding, W.; Wu, J.; Yoshikai, N. Chem. – Asian J. 2020, 15, 2166–2169. doi:10.1002/asia.202000653 |
| 34. | Kikuchi, J.; Nagata, T.; Ito, S.; Yoshikai, N. Org. Chem. Front. 2024. doi:10.1039/d4qo00489b |
| 4. | Liao, Q.; Wang, Y.; Zhang, L.; Xi, C. J. Org. Chem. 2009, 74, 6371–6373. doi:10.1021/jo901105r |
| 28. | Ding, W.; Chai, J.; Wang, C.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2020, 142, 8619–8624. doi:10.1021/jacs.0c04140 |
| 32. | Kikuchi, J.; Maesaki, K.; Sasaki, S.; Wang, W.; Ito, S.; Yoshikai, N. Org. Lett. 2022, 24, 6914–6918. doi:10.1021/acs.orglett.2c02570 |
| 14. | Garg, V.; Kumar, P.; Verma, A. K. J. Org. Chem. 2017, 82, 10247–10262. doi:10.1021/acs.joc.7b01746 |
| 22. | Pisella, G.; Gagnebin, A.; Waser, J. Org. Lett. 2020, 22, 3884–3889. doi:10.1021/acs.orglett.0c01150 |
| 23. | Di Tommaso, E. M.; Norrby, P.-O.; Olofsson, B. Angew. Chem., Int. Ed. 2022, 61, e202206347. doi:10.1002/anie.202206347 |
| 24. | Milzarek, T. M.; Waser, J. Angew. Chem., Int. Ed. 2023, 62, e202306128. doi:10.1002/anie.202306128 |
| 25. | Bhaskar Pal, K.; Di Tommaso, E. M.; Inge, A. K.; Olofsson, B. Angew. Chem., Int. Ed. 2023, 62, e202301368. doi:10.1002/anie.202301368 |
| 36. | Medina, J. M.; Mackey, J. L.; Garg, N. K.; Houk, K. N. J. Am. Chem. Soc. 2014, 136, 15798–15805. doi:10.1021/ja5099935 |
| 12. | Tsuchimoto, T.; Aoki, K.; Wagatsuma, T.; Suzuki, Y. Eur. J. Org. Chem. 2008, 4035–4040. doi:10.1002/ejoc.200800353 |
| 13. | Das, U. K.; Mandal, S.; Anoop, A.; Bhattacharjee, M. J. Org. Chem. 2014, 79, 9979–9991. doi:10.1021/jo502151z |
| 26. | Zhdankin, V. V.; Kuehl, C. J.; Krasutsky, A. P.; Bolz, J. T.; Simonsen, A. J. J. Org. Chem. 1996, 61, 6547–6551. doi:10.1021/jo960927a |
| 11. | Cao, Z.; Zhao, C.; Zhu, J.; Yan, S.; Tian, L.; Sun, X.; Meng, X. ChemistrySelect 2019, 4, 11785–11789. doi:10.1002/slct.201902532 |
| 28. | Ding, W.; Chai, J.; Wang, C.; Wu, J.; Yoshikai, N. J. Am. Chem. Soc. 2020, 142, 8619–8624. doi:10.1021/jacs.0c04140 |
| 10. | Michon, C.; Gilbert, J.; Trivelli, X.; Nahra, F.; Cazin, C. S. J.; Agbossou-Niedercorn, F.; Nolan, S. P. Org. Biomol. Chem. 2019, 17, 3805–3811. doi:10.1039/c9ob00587k |
| 15. | Declas, N.; Pisella, G.; Waser, J. Helv. Chim. Acta 2020, 103, e2000191. doi:10.1002/hlca.202000191 |
| 16. | Mironova, I. A.; Noskov, D. M.; Yoshimura, A.; Yusubov, M. S.; Zhdankin, V. V. Molecules 2023, 28, 2136. doi:10.3390/molecules28052136 |
| 17. | Stridfeldt, E.; Seemann, A.; Bouma, M. J.; Dey, C.; Ertan, A.; Olofsson, B. Chem. – Eur. J. 2016, 22, 16066–16070. doi:10.1002/chem.201603955 |
| 18. | Wu, J.; Deng, X.; Hirao, H.; Yoshikai, N. J. Am. Chem. Soc. 2016, 138, 9105–9108. doi:10.1021/jacs.6b06247 |
| 19. | Wu, J.; Xu, K.; Hirao, H.; Yoshikai, N. Chem. – Eur. J. 2017, 23, 1521–1525. doi:10.1002/chem.201605772 |
| 20. | Caramenti, P.; Declas, N.; Tessier, R.; Wodrich, M. D.; Waser, J. Chem. Sci. 2019, 10, 3223–3230. doi:10.1039/c8sc05573d |
| 21. | Shimbo, D.; Shibata, A.; Yudasaka, M.; Maruyama, T.; Tada, N.; Uno, B.; Itoh, A. Org. Lett. 2019, 21, 9769–9773. doi:10.1021/acs.orglett.9b03990 |
| 35. | Arakawa, C.; Kanemoto, K.; Nakai, K.; Wang, C.; Morohashi, S.; Kwon, E.; Ito, S.; Yoshikai, N. J. Am. Chem. Soc. 2024, 146, 3910–3919. doi:10.1021/jacs.3c11524 |
© 2024 Kikuchi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.