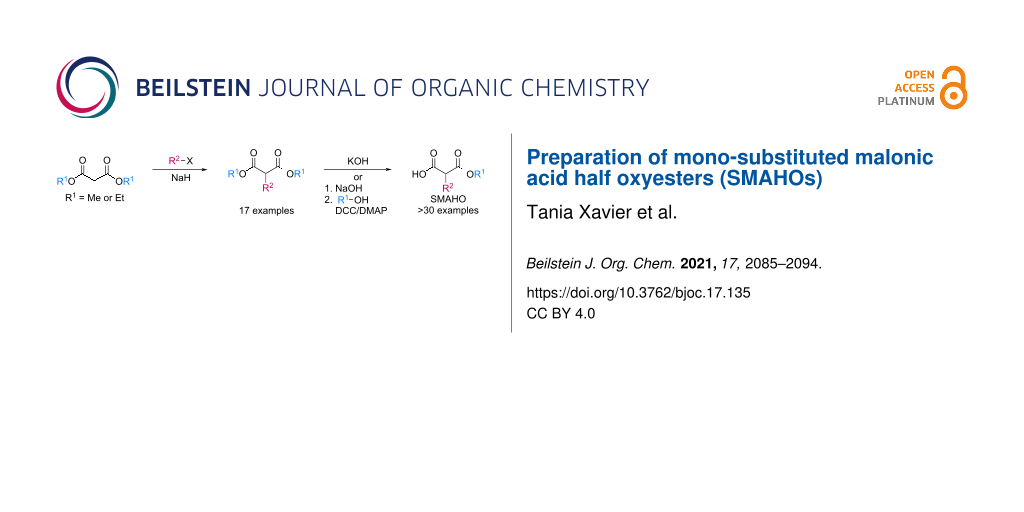
Abstract
The use of mono-substituted malonic acid half oxyesters (SMAHOs) has been hampered by the sporadic references describing their preparation. An evaluation of different approaches has been achieved, allowing to define the best strategies to introduce diversity on both the malonic position and the ester function. A classical alkylation step of a malonate by an alkyl halide followed by a monosaponification gave access to reagents bearing different substituents at the malonic position, including functionalized derivatives. On the other hand, the development of a monoesterification step of a substituted malonic acid derivative proved to be the best entry for diversity at the ester function, rather than the use of an intermediate Meldrum acid. Both these transformations are characterized by their simplicity and efficiency, allowing a straightforward access to SMAHOs from cheap starting materials.

Graphical Abstract
Introduction
Malonic acid half oxyesters (MAHOs), also known as alkyl hydrogen malonates or hemimalonates, constitute an attractive class of pronucleophiles in the design of eco-compatible syntheses [1]. Indeed, they can serve as efficient precursors of ester enolates through decarboxylation [2,3], generally using a substoichiometric amount of a weak base. Another advantage inherent to this class of reagents is their easy purification by a classical acid/base work-up thanks to the presence of the carboxylic acid moiety, even if they could also be purified by standard chromatography on silica gel. They have thus been used in a variety of reactions, the most widespread applications being aldol or Mannich-type addition reactions with carbonyl compounds and imines, respectively [4], and Galat olefination reactions of aldehydes [5-7]. Nevertheless, these developments have been mostly described with unsubstituted MAHOs, and the use of mono-substituted MAHOs (SMAHOs) [8-10], or disubstituted MAHOs [11], has been only occasionally reported.
Bibliographical data indicate that SMAHOs can be accessed by different routes (Scheme 1). The most classic one is the alkylation of a dialkyl malonate, followed by a monohydrolysis of one ester group. The alkylation is usually achieved by a nucleophilic substitution of an alkyl halide by the deprotonated malonate [12-19], but other strategies could be envisioned: Cu-catalyzed arylation reactions for aryl-substituted MAHOs [20-23]; Knoevenagel/reduction sequences for benzyl-substituted MAHOs [24-27]; and a Michael addition for 3'-oxoalkyl-substituted MAHOs [28]. As studied by Niwayama [29], the hydrolysis step is generally achieved by saponification using alcoholic KOH (or NaOH) [30-36], but other selective cleavages of one ester group are possible [37,38]. Despite its efficiency, this strategy remains limited regarding the nature of the alkoxy group of the ester function (generally Me or Et), as it arises from the parent malonate. In order to introduce diversity at this position, the privileged route is based on the opening of a Meldrum's acid derivative with an alcohol [39]. In this route, the Meldrum's acid is first alkylated, and the desired reagent is easily obtained by mixing this precursor with one equivalent of the requisite alcohol under reflux, generally in toluene [40-45]. Some reagents were also prepared by other routes [46-48], such as the α-carboxylation of an ester [49], the selective functionalization of a bromomalonate derivative [50], or the monoesterification of a malonic acid derivative [51-53].
![[1860-5397-17-135-i1]](/bjoc/content/inline/1860-5397-17-135-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
However, even if the above-mentioned strategies appear obvious for the preparation of the simplest SMAHOs, they are not sufficiently documented to cover the preparation of more complex versions of these reagents, apart from 2-amido-MAHOs for which the parent aminomalonate is commercially available. Indeed, it is very rare to find two identical procedures or stoichiometries, and these methods are used for the synthesis of a limited range of SMAHOs, presenting little diversity. Within the framework of a project pertaining to the use of SMAHOs in Galat [54] and Mannich reactions [55], we were confronted with a lack of pertinent bibliographical information, which led us to investigate the few reported synthetic routes.
Results and Discussion
Substituent modification – alkylation/saponification route
Based on the literature precedents, it appeared logical to start the preparation of SMAHOs by the alkylation of a malonate derivative followed by a monosaponification of the resulting diester. This strategy affords the advantage of being based on commercially available malonates. We thus envisioned to achieve the alkylation step by deprotonation of a malonate with NaH followed by treatment with a halogenated compound. As our first attempts revealed a strong influence of the stoichiometry and the nature of the alkylating reagent on both efficiency and selectivity, we decided to perform a quick optimization study. As a model reaction, we evaluated the influence of various parameters on the reaction between diethyl malonate (1b) and iodobutane (2d) or bromobutane (2d') in the presence of a stoichiometric amount of sodium hydride (Table 1). Using a 1:1:1 mixture of 1b/2d/NaH in DMF (c = 1.0 M) at rt, the desired monoalkylated product 3bd was isolated in 55% yield (Table 1, entry 1). By studying the stoichiometry of the reaction, we found easier to use a slight excess of the malonate to reduce the amount of the undesired dialkylated product (Table 1, entries 2 and 3). However, in all cases, the reaction mixture turned out to be very hard to stir, resulting in moderately reproducible results. We thus decided to decrease the concentration of the reaction to 0.5 M, resulting in a better 75% yield (Table 1, entry 4), whereas the use of THF led to a less efficient transformation (60%, Table 1, entry 5). The use of the brominated analog 2d' was also possible, albeit in lower 64% yield (Table 1, entry 6).
Table 1: Optimization of the alkylation step.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-135-i6.svg?max-width=637&scale=1.0)
|
|||||
| entry | x | X | y | solvent | yield (%)a |
| 1 | 1.0 | I | 1.0 | DMFb | 55 |
| 2 | 1.0 | I | 1.1 | DMFb | 68 |
| 3 | 1.1 | I | 1.0 | DMFb | 70 |
| 4 | 1.1 | I | 1.0 | DMFc | 75 |
| 5 | 1.1 | I | 1.0 | THFc | 60 |
| 6 | 1.1 | Br | 1.0 | DMFc | 64 |
aIsolated yields; bc = 1.0 M; cc = 0.5 M.
These optimized conditions were then applied to a variety of alkyl halides and the corresponding SMAHOs were obtained after monosaponification of the resulting intermediates (Table 2). In most cases, the desired products could be isolated in a pure form after a simple aqueous work-up based on their acidic properties. This sequence allowed the preparation of SMAHOs bearing different groups at the malonic position in moderate to good yields. Various primary aliphatic alkyl halides could be used (3aa, 50%; 3ab, 40%; 3ac, 75%; 3bd, 93%) and the reaction was also possible using a secondary alkyl halide (3be, 52%) with a longer reaction time. The alkylation step was obviously more efficient with primary activated alkyl halides such as allyl (3bf, 81%) or benzyl (3bh, 76%), but lower with a base-sensitive propargyl group (3bg, 58%). More functionalized lateral chains like a chlorobutyl group could be introduced efficiently (3bj, 91%) and the use of protected aminated or hydroxylated derivatives led to lower but still useful yields (3ak, 41%; 3bl, 44%; 3bm, 51%). In all these cases, the hydrolysis step delivered the expected SMAHOs in good to excellent yields. However, this reaction should be performed in a water/alcohol mixture, this latter being the same as the ester substituent to avoid undesired transesterification reactions, which somewhat limits the usefulness of this strategy. Upon application of these precautions, yields ranging from 62% to 91% were obtained in most cases. Nevertheless, the use of some protective groups is obviously a limitation of the method as this second step was incompatible with either base-sensitive or acid-sensitive substituents, as the desired SMAHOs are isolated after acidification to pH ≤ 2. It was thus impossible to prepare reagents bearing a phthalimide, an ester or an acetal group. This limitation is overcome by the other possibilities afforded by this methodology, as illustrated by the preparation of the cyano- (4bn), boron- (4ao), or silicon- (4ap) substituted SMAHOs.
Table 2: Preparation of SMAHOs from malonatesa.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-135-i7.svg?max-width=637&scale=1.0)
|
|||||||
| entry | 1 | 2 | 3 | yield (%) | 4 | yield (%) | OYb (%) |
| 1 |
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-135-i8.svg?max-width=637&scale=1.0)
1a |
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-135-i9.svg?max-width=637&scale=1.0)
2a |
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-135-i10.svg?max-width=637&scale=1.0)
3aa |
50 |
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-135-i11.svg?max-width=637&scale=1.0)
4aa |
91 | 45 |
| 2 |
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-135-i12.svg?max-width=637&scale=1.0)
1b |
2a |
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-135-i13.svg?max-width=637&scale=1.0)
3ba |
53
(76c) |
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-135-i14.svg?max-width=637&scale=1.0)
4ba |
72 | 55 |
| 3 | 1a |
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-135-i15.svg?max-width=637&scale=1.0)
2b |
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-135-i16.svg?max-width=637&scale=1.0)
3ab |
29
(40c) |
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-135-i17.svg?max-width=637&scale=1.0)
4ab |
90 | 36 |
| 4 | 1a |
![[Graphic 13]](/bjoc/content/inline/1860-5397-17-135-i18.svg?max-width=637&scale=1.0)
2c |
![[Graphic 14]](/bjoc/content/inline/1860-5397-17-135-i19.svg?max-width=637&scale=1.0)
3ac |
75 |
![[Graphic 15]](/bjoc/content/inline/1860-5397-17-135-i20.svg?max-width=637&scale=1.0)
4ac |
78 | 58 |
| 5 | 1b |
![[Graphic 16]](/bjoc/content/inline/1860-5397-17-135-i21.svg?max-width=637&scale=1.0)
2d |
![[Graphic 17]](/bjoc/content/inline/1860-5397-17-135-i22.svg?max-width=637&scale=1.0)
3bd |
93 |
![[Graphic 18]](/bjoc/content/inline/1860-5397-17-135-i23.svg?max-width=637&scale=1.0)
4bd |
72 | 67 |
| 6 | 1b |
![[Graphic 19]](/bjoc/content/inline/1860-5397-17-135-i24.svg?max-width=637&scale=1.0)
2e |
![[Graphic 20]](/bjoc/content/inline/1860-5397-17-135-i25.svg?max-width=637&scale=1.0)
3be |
52 |
![[Graphic 21]](/bjoc/content/inline/1860-5397-17-135-i26.svg?max-width=637&scale=1.0)
4be |
74 | 38 |
| 7 | 1b |
![[Graphic 22]](/bjoc/content/inline/1860-5397-17-135-i27.svg?max-width=637&scale=1.0)
2f |
![[Graphic 23]](/bjoc/content/inline/1860-5397-17-135-i28.svg?max-width=637&scale=1.0)
3bf |
81 |
![[Graphic 24]](/bjoc/content/inline/1860-5397-17-135-i29.svg?max-width=637&scale=1.0)
4bf |
71 | 57 |
| 8 | 1b |
![[Graphic 25]](/bjoc/content/inline/1860-5397-17-135-i30.svg?max-width=637&scale=1.0)
2g |
![[Graphic 26]](/bjoc/content/inline/1860-5397-17-135-i31.svg?max-width=637&scale=1.0)
3bg |
58 |
![[Graphic 27]](/bjoc/content/inline/1860-5397-17-135-i32.svg?max-width=637&scale=1.0)
4bg |
65 | 38 |
| 9 | 1b |
![[Graphic 28]](/bjoc/content/inline/1860-5397-17-135-i33.svg?max-width=637&scale=1.0)
2h |
![[Graphic 29]](/bjoc/content/inline/1860-5397-17-135-i34.svg?max-width=637&scale=1.0)
3bh |
76 |
![[Graphic 30]](/bjoc/content/inline/1860-5397-17-135-i35.svg?max-width=637&scale=1.0)
4bh |
69 | 52 |
| 10 | 1bd |
![[Graphic 31]](/bjoc/content/inline/1860-5397-17-135-i36.svg?max-width=637&scale=1.0)
2i |
![[Graphic 32]](/bjoc/content/inline/1860-5397-17-135-i37.svg?max-width=637&scale=1.0)
3bi |
30c |
![[Graphic 33]](/bjoc/content/inline/1860-5397-17-135-i38.svg?max-width=637&scale=1.0)
4bi |
71 | 21 |
| 11 | 1b |
![[Graphic 34]](/bjoc/content/inline/1860-5397-17-135-i39.svg?max-width=637&scale=1.0)
2j |
![[Graphic 35]](/bjoc/content/inline/1860-5397-17-135-i40.svg?max-width=637&scale=1.0)
3bj |
91c |
![[Graphic 36]](/bjoc/content/inline/1860-5397-17-135-i41.svg?max-width=637&scale=1.0)
4bj |
62 | 56 |
| 12 | 1a |
![[Graphic 37]](/bjoc/content/inline/1860-5397-17-135-i42.svg?max-width=637&scale=1.0)
2k |
![[Graphic 38]](/bjoc/content/inline/1860-5397-17-135-i43.svg?max-width=637&scale=1.0)
3ak |
41c |
![[Graphic 39]](/bjoc/content/inline/1860-5397-17-135-i44.svg?max-width=637&scale=1.0)
4ak |
76 | 31 |
| 13 | 1bd |
![[Graphic 40]](/bjoc/content/inline/1860-5397-17-135-i45.svg?max-width=637&scale=1.0)
2l |
![[Graphic 41]](/bjoc/content/inline/1860-5397-17-135-i46.svg?max-width=637&scale=1.0)
3bl |
44c,e |
![[Graphic 42]](/bjoc/content/inline/1860-5397-17-135-i47.svg?max-width=637&scale=1.0)
4bl |
63 | 28 |
| 14 | 1b |
![[Graphic 43]](/bjoc/content/inline/1860-5397-17-135-i48.svg?max-width=637&scale=1.0)
2m |
![[Graphic 44]](/bjoc/content/inline/1860-5397-17-135-i49.svg?max-width=637&scale=1.0)
3bm |
51c |
![[Graphic 45]](/bjoc/content/inline/1860-5397-17-135-i50.svg?max-width=637&scale=1.0)
4bm |
70 | 36 |
| 15 | 1b |
![[Graphic 46]](/bjoc/content/inline/1860-5397-17-135-i51.svg?max-width=637&scale=1.0)
2n |
![[Graphic 47]](/bjoc/content/inline/1860-5397-17-135-i52.svg?max-width=637&scale=1.0)
3bn |
24f |
![[Graphic 48]](/bjoc/content/inline/1860-5397-17-135-i53.svg?max-width=637&scale=1.0)
4bn |
65 | 16 |
| 16 | 1a |
![[Graphic 49]](/bjoc/content/inline/1860-5397-17-135-i54.svg?max-width=637&scale=1.0)
2o |
![[Graphic 50]](/bjoc/content/inline/1860-5397-17-135-i55.svg?max-width=637&scale=1.0)
3ao |
70 |
![[Graphic 51]](/bjoc/content/inline/1860-5397-17-135-i56.svg?max-width=637&scale=1.0)
4ao |
77 | 54 |
| 17 | 1a |
![[Graphic 52]](/bjoc/content/inline/1860-5397-17-135-i57.svg?max-width=637&scale=1.0)
2p |
![[Graphic 53]](/bjoc/content/inline/1860-5397-17-135-i58.svg?max-width=637&scale=1.0)
3ap |
21e,f,g |
![[Graphic 54]](/bjoc/content/inline/1860-5397-17-135-i59.svg?max-width=637&scale=1.0)
4ap |
62 | 13 |
aYields of isolated products. Step 1: 1 (1.1 equiv), 2 (5–50 mmol), NaH (1.0 equiv), DMF (c = 0.5–1.0 M), 0 °C to rt, 2 h. Step 2: KOH (1.0–1.2 equiv), R1OH/H2O 10:1 (c = 0.5 M), rt, 2 h; boverall yield (2 steps); creaction time = 16 h; d1.0 equiv; eT = 80 °C; fsolvent = THF; reaction time = 24 h; gNaI (3.0 equiv).
The efficiency of the saponification step was also illustrated by the case of α-halomalonates 3aq and 3br. Upon application of the above-mentioned conditions, the useful reagents 4aq and 4br could be isolated in 70% and 47% yield, respectively (Scheme 2).
![[1860-5397-17-135-i2]](/bjoc/content/inline/1860-5397-17-135-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Ester modification
Meldrum's acid route
We next turned our attention to the diversification of the ester group of SMAHOs. Our first approach was the use of substituted malonates, protected as Meldrum's acid derivatives, as the parent compound 5 is commercially available or easily accessible on a relatively large scale (Scheme 3) [56]. Therefore, we first achieved an alkylation step of 5 using a Knoevenagel reaction with benzaldehyde, followed by an in situ reduction of the resulting alkylidene [57,58]. Even if the reaction worked well with complete conversion, the isolation of the pure product was less straightforward and required a recrystallization from a methanol/water mixture, leading to a moderate yield (56%) due to a partial solubilization of the product 7h. This complication was yet balanced by the practicality of the second step, the opening of the Meldrum's acid moiety with an alcohol 8. Indeed, this reaction was achieved under simple conditions and led to the desired products in useful yields, only limited by partial in situ decarboxylation. This strategy thus allowed the preparation of the isopropyl (4ch, 93%) and benzyl (4dh, 57%, corrected isolated yield) ester derivatives but remains limited to reagents bearing a benzyl substituent at the malonic position.
![[1860-5397-17-135-i3]](/bjoc/content/inline/1860-5397-17-135-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of SMAHOs from Meldrum's acid.
Scheme 3: Preparation of SMAHOs from Meldrum's acid.
In an alternative strategy to circumvent the difficulties linked to the isolation of substituted Meldrum's acid derivatives, we envisioned a complementary route where the substituent was introduced prior to the protection step. Commercially available substituted malonic acids 9 were first protected with acetone under acid conditions [46], and the desired intermediates 7 were simply isolated after aqueous work-up, usually in good yields (Table 3). The opening of these latter with alcohols 8 was efficient again and allowed the preparation of new reagents in good yields, after a second aqueous work-up. Using this approach, SMAHOs bearing a benzyl (4da) or a (−)-menthyl group (4ea) were easily prepared with good overall yields (49–71%). Despite its efficiency, this second strategy required an additional step of hydrolysis of a substituted malonate to prepare the requisite substituted malonic acid.
Table 3: Preparation of SMAHOs from malonic acids.
![[Graphic 55]](/bjoc/content/inline/1860-5397-17-135-i60.svg?max-width=637&scale=1.0)
|
|||||
| entry | 7 | yield (%) | 4 | yield (%) | OYa (%) |
| 1 |
![[Graphic 56]](/bjoc/content/inline/1860-5397-17-135-i61.svg?max-width=637&scale=1.0)
7a |
84 |
![[Graphic 57]](/bjoc/content/inline/1860-5397-17-135-i62.svg?max-width=637&scale=1.0)
4da |
85 | 71 |
| 2 |
![[Graphic 58]](/bjoc/content/inline/1860-5397-17-135-i63.svg?max-width=637&scale=1.0)
4ea |
58 | 49 | ||
| 3 |
![[Graphic 59]](/bjoc/content/inline/1860-5397-17-135-i64.svg?max-width=637&scale=1.0)
7s |
84 |
![[Graphic 60]](/bjoc/content/inline/1860-5397-17-135-i65.svg?max-width=637&scale=1.0)
4bs |
65b | 55 |
aOverall yield (2 steps); bin toluene/EtOH 1:1.
Monoesterification route
In order to take advantage of the preparation of substituted malonates 3, we decided to explore a more straightforward strategy, the mono-esterification of substituted malonic acids. Indeed, this approach has been only used by Fukumoto for the preparation of (−)-menthyl-derived SMAHOs [51,52]. We began this study by preparing various substituted malonic acids 9 through the saponification of the corresponding substituted malonates 3 (Scheme 4). These standard reactions were easy and delivered the expected products 9 in good to excellent yields.
![[1860-5397-17-135-i4]](/bjoc/content/inline/1860-5397-17-135-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Saponification of substituted malonates.
Scheme 4: Saponification of substituted malonates.
We then performed a quick optimization study of the reaction between 2-methylmalonic acid (9a) and benzyl alcohol (8d) (Table 4). Starting from classical Steglich's conditions (1.0 equiv of DCC and 5 mol % of DMAP in CH2Cl2, Table 4, entry 1), the desired product was isolated in an encouraging yield of 22% after chromatography on silica gel. By adding a solution of DCC in CH2Cl2 to a solution of the other reagents in CH3CN (Table 4, entry 2), the yield was increased to 46%. The temperature of addition was also an important parameter and we found that an initial cooling to −15 °C was optimal (Table 4, entries 3–5), presumably by limiting the decomposition of the malonic acid derivative. We also studied the influence of the stoichiometry of the different reagents (Table 4, entries 6–9) and the use of a slight excess of alcohol 8d was beneficial to the reaction. Finally, carrying out the reaction in only CH3CN allows the isolation of the desired pure compound 4da after a simple aqueous work-up, although in a slightly lower 52% yield (Table 4, entry 10).
Table 4: Optimization of the mono-esterification of malonic acid 10a.
![[Graphic 61]](/bjoc/content/inline/1860-5397-17-135-i66.svg?max-width=637&scale=1.0)
|
||||||
| entry | x (equiv) | y (equiv) | z (mol %) | solvent | T | yield (%)a |
| 1 | 1.0 | 1.0 | 5 | CH2Cl2 | rt | 22 |
| 2 | 1.0 | 1.0 | 5 | CH2Cl2/CH3CN | rt | 46 |
| 3 | 1.0 | 1.0 | 5 | CH2Cl2/CH3CN | 0 °C to rt | 48 |
| 4 | 1.0 | 1.0 | 5 | CH2Cl2/CH3CN | −15 °C to rt | 59 |
| 5 | 1.0 | 1.0 | 5 | CH2Cl2/CH3CN | −78 °C to rt | 46 |
| 6 | 1.1 | 1.0 | 5 | CH2Cl2/CH3CN | −15 °C to rt | 62 |
| 7 | 1.5 | 1.0 | 5 | CH2Cl2/CH3CN | −15 °C to rt | 57 |
| 8 | 1.0 | 1,2 | 5 | CH2Cl2/CH3CN | −15 °C to rt | 53 |
| 9 | 1.0 | 1.0 | 20 | CH2Cl2/CH3CN | −15 °C to rt | 49 |
| 10 | 1.1 | 1.0 | 5 | CH3CN | −15 °C to rt | 52b |
aIsolated yields after flash chromatography on silica gel; bisolated yield after aqueous work-up.
The scope of this reaction was next explored under such optimized conditions (Table 4, entry 10), using 2-methylmalonic acid (9a) as a model substrate (Scheme 5). From a general point of view, even if the yields are moderate, the reaction is easy to set up and the desired product could be isolated after a simple aqueous work-up. SMAHOs bearing classical ester groups could be obtained in moderate yields (iPr: 51%; Bn: 53%; allyl: 52%) and the use of less nucleophilic alcohols such as t-BuOH, 2,2,2-trifluoroethanol, (−)-menthol, and phenol led to decreased yields (37%, 34%, 28%, and 17%, respectively). More functionalized alcohols such as citronellol (4ja, 58%) and (−)-ethyl lactate (4ka, 32%) could also be employed for the direct preparation of elaborated reagents.
![[1860-5397-17-135-i5]](/bjoc/content/inline/1860-5397-17-135-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Scope of the mono-esterification of substituted malonic acids. adr = 1:1.
Scheme 5: Scope of the mono-esterification of substituted malonic acids. adr = 1:1.
Conclusion
We have prepared more than 30 SMAHOs including numerous new compounds using different strategies, thus adding a consequent input to the knowledge of these reagents. In our hands, these reagents appear to be stable to air or moisture and have been stored for several months at 4 °C. The alkylation/hydrolysis of malonates afforded a straightforward access to such derivatives but the use of an alcoholic base in the saponification step precludes the presence of sensitive functional groups and limits the degree of substitution of the ester function. This last limitation could be circumvented using the mono-esterification of malonic acid derivatives, which gives access to a broad range of derivatives. Overall, this work presents the different possibilities to access these reagents, which constitute an attractive class of pronucleophiles.
Supporting Information
| Supporting Information File 1: Experimental procedures, compound characterization data, and NMR spectra for all compounds. | ||
| Format: PDF | Size: 8.9 MB | Download |
References
-
Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998.
Return to citation in text: [1] -
Rodríguez, N.; Goossen, L. J. Chem. Soc. Rev. 2011, 40, 5030–5048. doi:10.1039/c1cs15093f
Return to citation in text: [1] -
Liu, P.; Zhang, G.; Sun, P. Org. Biomol. Chem. 2016, 14, 10763–10777. doi:10.1039/c6ob02101h
Return to citation in text: [1] -
Wang, Z.-L. Adv. Synth. Catal. 2013, 355, 2745–2755. doi:10.1002/adsc.201300375
Return to citation in text: [1] -
Galat, A. J. Am. Chem. Soc. 1946, 68, 376–377. doi:10.1021/ja01207a009
Return to citation in text: [1] -
Rodriguez, J.; Waegell, B. Synthesis 1988, 534–535. doi:10.1055/s-1988-27626
Return to citation in text: [1] -
List, B.; Doehring, A.; Hechavarria Fonseca, M. T.; Job, A.; Rios Torres, R. Tetrahedron 2006, 62, 476–482. doi:10.1016/j.tet.2005.09.081
Return to citation in text: [1] -
Ireland, R. E.; Marshall, J. A. J. Am. Chem. Soc. 1959, 81, 2907–2908. doi:10.1021/ja01520a073
Return to citation in text: [1] -
Blaquiere, N.; Shore, D. G.; Rousseaux, S.; Fagnou, K. J. Org. Chem. 2009, 74, 6190–6198. doi:10.1021/jo901022j
Return to citation in text: [1] -
Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.-C.; Rouden, J. Green Chem. 2010, 12, 252–259. doi:10.1039/b915681j
Return to citation in text: [1] -
Feng, Y.-S.; Wu, W.; Xu, Z.-Q.; Li, Y.; Li, M.; Xu, H.-J. Tetrahedron 2012, 68, 2113–2120. doi:10.1016/j.tet.2012.01.032
Return to citation in text: [1] -
Kinder, D. H.; Ames, M. M. J. Org. Chem. 1987, 52, 2452–2454. doi:10.1021/jo00388a021
Return to citation in text: [1] -
Yang, K.-W.; Golich, F. C.; Sigdel, T. K.; Crowder, M. W. Bioorg. Med. Chem. Lett. 2005, 15, 5150–5153. doi:10.1016/j.bmcl.2005.08.055
Return to citation in text: [1] -
Volonterio, A.; Zanda, M. Org. Lett. 2007, 9, 841–844. doi:10.1021/ol063074+
Return to citation in text: [1] -
Jaegli, S.; Vors, J.-P.; Neuville, L.; Zhu, J. Tetrahedron 2010, 66, 8911–8921. doi:10.1016/j.tet.2010.09.056
Return to citation in text: [1] -
Staples, M. K.; Grange, R. L.; Angus, J. A.; Ziogas, J.; Tan, N. P. H.; Taylor, M. K.; Schiesser, C. H. Org. Biomol. Chem. 2011, 9, 473–479. doi:10.1039/c0ob00573h
Return to citation in text: [1] -
Qiu, G.; Mamboury, M.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2016, 55, 15377–15381. doi:10.1002/anie.201609034
Return to citation in text: [1] -
Ke, D.; Espinosa, N. Á.; Mallet-Ladeira, S.; Monot, J.; Martin-Vaca, B.; Bourissou, D. Adv. Synth. Catal. 2016, 358, 2324–2331. doi:10.1002/adsc.201600382
Return to citation in text: [1] -
Coutant, E. P.; Hervin, V.; Gagnot, G.; Ford, C.; Baatallah, R.; Janin, Y. L. Beilstein J. Org. Chem. 2018, 14, 2853–2860. doi:10.3762/bjoc.14.264
Return to citation in text: [1] -
Hennessy, E. J.; Buchwald, S. L. Org. Lett. 2002, 4, 269–272. doi:10.1021/ol017038g
Return to citation in text: [1] -
Cristau, H.-J.; Cellier, P. P.; Spindler, J.-F.; Taillefer, M. Chem. – Eur. J. 2004, 10, 5607–5622. doi:10.1002/chem.200400582
Return to citation in text: [1] -
Yip, S. F.; Cheung, H. Y.; Zhou, Z.; Kwong, F. Y. Org. Lett. 2007, 9, 3469–3472. doi:10.1021/ol701473p
Return to citation in text: [1] -
Pinhey, J. T. Pure Appl. Chem. 1996, 68, 819–824. doi:10.1351/pac199668040819
Return to citation in text: [1] -
Keenan, R. M.; Weinstock, J.; Finkelstein, J. A.; Franz, R. G.; Gaitanopoulos, D. E.; Girard, G. R.; Hill, D. T.; Morgan, T. M.; Samanen, J. M.; Peishoff, C. E.; Tucker, L. M.; Aiyar, N.; Griffin, E.; Ohlstein, E. H.; Stack, E. J.; Weidley, E. F.; Edwards, R. M. J. Med. Chem. 1993, 36, 1880–1892. doi:10.1021/jm00065a011
Return to citation in text: [1] -
Islam, I.; Bryant, J.; May, K.; Mohan, R.; Yuan, S.; Kent, L.; Morser, J.; Zhao, L.; Vergona, R.; White, K.; Adler, M.; Whitlow, M.; Buckman, B. O. Bioorg. Med. Chem. Lett. 2007, 17, 1349–1354. doi:10.1016/j.bmcl.2006.11.078
Return to citation in text: [1] -
Grange, R. L.; Ziogas, J.; North, A. J.; Angus, J. A.; Schiesser, C. H. Bioorg. Med. Chem. Lett. 2008, 18, 1241–1244. doi:10.1016/j.bmcl.2007.11.136
Return to citation in text: [1] -
Åberg, O.; Lindhe, Ö.; Hall, H.; Hellman, P.; Kihlberg, T.; Långström, B. J. Labelled Compd. Radiopharm. 2009, 52, 295–303. doi:10.1002/jlcr.1598
Return to citation in text: [1] -
Quast, H.; Janiak, R. Liebigs Ann. Chem. 1991, 1305–1308. doi:10.1002/jlac.1991199101224
Return to citation in text: [1] -
Niwayama, S.; Cho, H.; Lin, C. Tetrahedron Lett. 2008, 49, 4434–4436. doi:10.1016/j.tetlet.2008.05.007
Return to citation in text: [1] -
Leeper, F. J.; Rock, M.; Appleton, D. J. Chem. Soc., Perkin Trans. 1 1996, 2633–2642. doi:10.1039/p19960002633
Return to citation in text: [1] -
Faucher, A.-M.; Bailey, M. D.; Beaulieu, P. L.; Brochu, C.; Duceppe, J.-S.; Ferland, J.-M.; Ghiro, E.; Gorys, V.; Halmos, T.; Kawai, S. H.; Poirier, M.; Simoneau, B.; Tsantrizos, Y. S.; Llinàs-Brunet, M. Org. Lett. 2004, 6, 2901–2904. doi:10.1021/ol0489907
Return to citation in text: [1] -
Flipo, M.; Beghyn, T.; Charton, J.; Leroux, V. A.; Deprez, B. P.; Deprez-Poulain, R. F. Bioorg. Med. Chem. 2007, 15, 63–76. doi:10.1016/j.bmc.2006.10.010
Return to citation in text: [1] -
Singjunla, Y.; Baudoux, J.; Rouden, J. Org. Lett. 2013, 15, 5770–5773. doi:10.1021/ol402805f
Return to citation in text: [1] -
Mai, A. H.; Pawar, S.; De Borggraeve, W. M. Tetrahedron Lett. 2014, 55, 4664–4666. doi:10.1016/j.tetlet.2014.06.100
Return to citation in text: [1] -
Singjunla, Y.; Colombano, S.; Baudoux, J.; Rouden, J. Tetrahedron 2016, 72, 2369–2375. doi:10.1016/j.tet.2016.03.043
Return to citation in text: [1] -
Chaumont, P.; Baudoux, J.; Maddaluno, J.; Rouden, J.; Harrison-Marchand, A. J. Org. Chem. 2018, 83, 8081–8091. doi:10.1021/acs.joc.8b00901
Return to citation in text: [1] -
Aeberhard, U.; Keese, R.; Stamm, E.; Vögeli, U.-C.; Lau, W.; Kochi, J. K. Helv. Chim. Acta 1983, 66, 2740–2759. doi:10.1002/hlca.19830660843
Return to citation in text: [1] -
Kitazume, T.; Sato, T.; Ishikawa, N. Chem. Lett. 1984, 13, 1811–1814. doi:10.1246/cl.1984.1811
Return to citation in text: [1] -
Ryu, Y.; Scott, A. I. Tetrahedron Lett. 2003, 44, 7499–7502. doi:10.1016/j.tetlet.2003.08.014
Return to citation in text: [1] -
Chen, Y.-S.; Lawton, R. G. Tetrahedron Lett. 1997, 38, 5785–5788. doi:10.1016/s0040-4039(97)01319-1
Return to citation in text: [1] -
Trillo, P.; Baeza, A.; Nájera, C. J. Org. Chem. 2012, 77, 7344–7354. doi:10.1021/jo301049w
Return to citation in text: [1] -
Brookes, P. A.; Cordes, J.; White, A. J. P.; Barrett, A. G. M. Eur. J. Org. Chem. 2013, 7313–7319. doi:10.1002/ejoc.201300974
Return to citation in text: [1] -
Ha, M. W.; Hong, S.; Park, C.; Park, Y.; Lee, J.; Kim, M.-h.; Lee, J.; Park, H.-g. Org. Biomol. Chem. 2013, 11, 4030–4039. doi:10.1039/c3ob40481a
Return to citation in text: [1] -
Gao, B.-W.; Wang, X.-H.; Liu, X.; Shi, S.-P.; Tu, P.-F. Bioorg. Med. Chem. Lett. 2015, 25, 1279–1283. doi:10.1016/j.bmcl.2015.01.045
Return to citation in text: [1] -
Ad, O.; Hoffman, K. S.; Cairns, A. G.; Featherston, A. L.; Miller, S. J.; Söll, D.; Schepartz, A. ACS Cent. Sci. 2019, 5, 1289–1294. doi:10.1021/acscentsci.9b00460
Return to citation in text: [1] -
Dell‘Orco, P.; Brum, J.; Matsuoka, R.; Badlani, M.; Muske, K. Anal. Chem. (Washington, DC, U. S.) 1999, 71, 5165–5170. doi:10.1021/ac990554o
Return to citation in text: [1] [2] -
Velázquez, F.; Venkatraman, S.; Wu, W.; Blackman, M.; Prongay, A.; Girijavallabhan, V.; Shih, N.-Y.; Njoroge, F. G. Org. Lett. 2007, 9, 3061–3064. doi:10.1021/ol0711265
Return to citation in text: [1] -
Xu, F.; Zacuto, M.; Yoshikawa, N.; Desmond, R.; Hoerrner, S.; Itoh, T.; Journet, M.; Humphrey, G. R.; Cowden, C.; Strotman, N.; Devine, P. J. Org. Chem. 2010, 75, 7829–7841. doi:10.1021/jo101704b
Return to citation in text: [1] -
Murta, M. M.; de Azevedo, M. B. M.; Greene, A. E. Synth. Commun. 1993, 23, 495–503. doi:10.1080/00397919308009804
Return to citation in text: [1] -
Hassan, H. A.; Abdel-Aziz, M.; Abuo-Rahma, G. E.-D. A. A.; Farag, H. H. Bioorg. Med. Chem. 2009, 17, 1681–1692. doi:10.1016/j.bmc.2008.12.051
Return to citation in text: [1] -
Ihara, M.; Takahashi, M.; Taniguchi, N.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Chem. Commun. 1987, 619–620. doi:10.1039/c39870000619
Return to citation in text: [1] [2] -
Ihara, M.; Takahashi, M.; Taniguchi, N.; Yasui, K.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Perkin Trans. 1 1989, 897–903. doi:10.1039/p19890000897
Return to citation in text: [1] [2] -
Huang, F.; Buchwald, P.; Browne, C. E.; Farag, H. H.; Wu, W.-M.; Ji, F.; Hochhaus, G.; Bodor, N. AAPS PharmSci 2001, 3 (4), 44–56. doi:10.1208/ps030430
Return to citation in text: [1] -
Xavier, T.; Condon, S.; Pichon, C.; Le Gall, E.; Presset, M. Org. Lett. 2019, 21, 6135–6139. doi:10.1021/acs.orglett.9b02291
Return to citation in text: [1] -
Xavier, T.; Condon, S.; Pichon, C.; Le Gall, E.; Presset, M. J. Org. Chem. 2021, 86, 5452–5462. doi:10.1021/acs.joc.0c02895
Return to citation in text: [1] -
Davidson, D.; Bernhard, S. A. J. Am. Chem. Soc. 1948, 70, 3426–3428. doi:10.1021/ja01190a060
Return to citation in text: [1] -
Lou, T.; Liao, E-T.; Wilsily, A.; Fillion, E. Org. Synth. 2012, 89, 115–125. doi:10.15227/orgsyn.089.0115
Return to citation in text: [1] -
Ramachary, D. B.; Reddy, G. B. Org. Biomol. Chem. 2006, 4, 4463–4468. doi:10.1039/b612611a
Return to citation in text: [1]
| 1. | Anastas, P. T.; Warner, J. C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. |
| 8. | Ireland, R. E.; Marshall, J. A. J. Am. Chem. Soc. 1959, 81, 2907–2908. doi:10.1021/ja01520a073 |
| 9. | Blaquiere, N.; Shore, D. G.; Rousseaux, S.; Fagnou, K. J. Org. Chem. 2009, 74, 6190–6198. doi:10.1021/jo901022j |
| 10. | Baudoux, J.; Lefebvre, P.; Legay, R.; Lasne, M.-C.; Rouden, J. Green Chem. 2010, 12, 252–259. doi:10.1039/b915681j |
| 40. | Chen, Y.-S.; Lawton, R. G. Tetrahedron Lett. 1997, 38, 5785–5788. doi:10.1016/s0040-4039(97)01319-1 |
| 41. | Trillo, P.; Baeza, A.; Nájera, C. J. Org. Chem. 2012, 77, 7344–7354. doi:10.1021/jo301049w |
| 42. | Brookes, P. A.; Cordes, J.; White, A. J. P.; Barrett, A. G. M. Eur. J. Org. Chem. 2013, 7313–7319. doi:10.1002/ejoc.201300974 |
| 43. | Ha, M. W.; Hong, S.; Park, C.; Park, Y.; Lee, J.; Kim, M.-h.; Lee, J.; Park, H.-g. Org. Biomol. Chem. 2013, 11, 4030–4039. doi:10.1039/c3ob40481a |
| 44. | Gao, B.-W.; Wang, X.-H.; Liu, X.; Shi, S.-P.; Tu, P.-F. Bioorg. Med. Chem. Lett. 2015, 25, 1279–1283. doi:10.1016/j.bmcl.2015.01.045 |
| 45. | Ad, O.; Hoffman, K. S.; Cairns, A. G.; Featherston, A. L.; Miller, S. J.; Söll, D.; Schepartz, A. ACS Cent. Sci. 2019, 5, 1289–1294. doi:10.1021/acscentsci.9b00460 |
| 5. | Galat, A. J. Am. Chem. Soc. 1946, 68, 376–377. doi:10.1021/ja01207a009 |
| 6. | Rodriguez, J.; Waegell, B. Synthesis 1988, 534–535. doi:10.1055/s-1988-27626 |
| 7. | List, B.; Doehring, A.; Hechavarria Fonseca, M. T.; Job, A.; Rios Torres, R. Tetrahedron 2006, 62, 476–482. doi:10.1016/j.tet.2005.09.081 |
| 46. | Dell‘Orco, P.; Brum, J.; Matsuoka, R.; Badlani, M.; Muske, K. Anal. Chem. (Washington, DC, U. S.) 1999, 71, 5165–5170. doi:10.1021/ac990554o |
| 47. | Velázquez, F.; Venkatraman, S.; Wu, W.; Blackman, M.; Prongay, A.; Girijavallabhan, V.; Shih, N.-Y.; Njoroge, F. G. Org. Lett. 2007, 9, 3061–3064. doi:10.1021/ol0711265 |
| 48. | Xu, F.; Zacuto, M.; Yoshikawa, N.; Desmond, R.; Hoerrner, S.; Itoh, T.; Journet, M.; Humphrey, G. R.; Cowden, C.; Strotman, N.; Devine, P. J. Org. Chem. 2010, 75, 7829–7841. doi:10.1021/jo101704b |
| 4. | Wang, Z.-L. Adv. Synth. Catal. 2013, 355, 2745–2755. doi:10.1002/adsc.201300375 |
| 37. | Aeberhard, U.; Keese, R.; Stamm, E.; Vögeli, U.-C.; Lau, W.; Kochi, J. K. Helv. Chim. Acta 1983, 66, 2740–2759. doi:10.1002/hlca.19830660843 |
| 38. | Kitazume, T.; Sato, T.; Ishikawa, N. Chem. Lett. 1984, 13, 1811–1814. doi:10.1246/cl.1984.1811 |
| 2. | Rodríguez, N.; Goossen, L. J. Chem. Soc. Rev. 2011, 40, 5030–5048. doi:10.1039/c1cs15093f |
| 3. | Liu, P.; Zhang, G.; Sun, P. Org. Biomol. Chem. 2016, 14, 10763–10777. doi:10.1039/c6ob02101h |
| 39. | Ryu, Y.; Scott, A. I. Tetrahedron Lett. 2003, 44, 7499–7502. doi:10.1016/j.tetlet.2003.08.014 |
| 24. | Keenan, R. M.; Weinstock, J.; Finkelstein, J. A.; Franz, R. G.; Gaitanopoulos, D. E.; Girard, G. R.; Hill, D. T.; Morgan, T. M.; Samanen, J. M.; Peishoff, C. E.; Tucker, L. M.; Aiyar, N.; Griffin, E.; Ohlstein, E. H.; Stack, E. J.; Weidley, E. F.; Edwards, R. M. J. Med. Chem. 1993, 36, 1880–1892. doi:10.1021/jm00065a011 |
| 25. | Islam, I.; Bryant, J.; May, K.; Mohan, R.; Yuan, S.; Kent, L.; Morser, J.; Zhao, L.; Vergona, R.; White, K.; Adler, M.; Whitlow, M.; Buckman, B. O. Bioorg. Med. Chem. Lett. 2007, 17, 1349–1354. doi:10.1016/j.bmcl.2006.11.078 |
| 26. | Grange, R. L.; Ziogas, J.; North, A. J.; Angus, J. A.; Schiesser, C. H. Bioorg. Med. Chem. Lett. 2008, 18, 1241–1244. doi:10.1016/j.bmcl.2007.11.136 |
| 27. | Åberg, O.; Lindhe, Ö.; Hall, H.; Hellman, P.; Kihlberg, T.; Långström, B. J. Labelled Compd. Radiopharm. 2009, 52, 295–303. doi:10.1002/jlcr.1598 |
| 29. | Niwayama, S.; Cho, H.; Lin, C. Tetrahedron Lett. 2008, 49, 4434–4436. doi:10.1016/j.tetlet.2008.05.007 |
| 20. | Hennessy, E. J.; Buchwald, S. L. Org. Lett. 2002, 4, 269–272. doi:10.1021/ol017038g |
| 21. | Cristau, H.-J.; Cellier, P. P.; Spindler, J.-F.; Taillefer, M. Chem. – Eur. J. 2004, 10, 5607–5622. doi:10.1002/chem.200400582 |
| 22. | Yip, S. F.; Cheung, H. Y.; Zhou, Z.; Kwong, F. Y. Org. Lett. 2007, 9, 3469–3472. doi:10.1021/ol701473p |
| 23. | Pinhey, J. T. Pure Appl. Chem. 1996, 68, 819–824. doi:10.1351/pac199668040819 |
| 30. | Leeper, F. J.; Rock, M.; Appleton, D. J. Chem. Soc., Perkin Trans. 1 1996, 2633–2642. doi:10.1039/p19960002633 |
| 31. | Faucher, A.-M.; Bailey, M. D.; Beaulieu, P. L.; Brochu, C.; Duceppe, J.-S.; Ferland, J.-M.; Ghiro, E.; Gorys, V.; Halmos, T.; Kawai, S. H.; Poirier, M.; Simoneau, B.; Tsantrizos, Y. S.; Llinàs-Brunet, M. Org. Lett. 2004, 6, 2901–2904. doi:10.1021/ol0489907 |
| 32. | Flipo, M.; Beghyn, T.; Charton, J.; Leroux, V. A.; Deprez, B. P.; Deprez-Poulain, R. F. Bioorg. Med. Chem. 2007, 15, 63–76. doi:10.1016/j.bmc.2006.10.010 |
| 33. | Singjunla, Y.; Baudoux, J.; Rouden, J. Org. Lett. 2013, 15, 5770–5773. doi:10.1021/ol402805f |
| 34. | Mai, A. H.; Pawar, S.; De Borggraeve, W. M. Tetrahedron Lett. 2014, 55, 4664–4666. doi:10.1016/j.tetlet.2014.06.100 |
| 35. | Singjunla, Y.; Colombano, S.; Baudoux, J.; Rouden, J. Tetrahedron 2016, 72, 2369–2375. doi:10.1016/j.tet.2016.03.043 |
| 36. | Chaumont, P.; Baudoux, J.; Maddaluno, J.; Rouden, J.; Harrison-Marchand, A. J. Org. Chem. 2018, 83, 8081–8091. doi:10.1021/acs.joc.8b00901 |
| 12. | Kinder, D. H.; Ames, M. M. J. Org. Chem. 1987, 52, 2452–2454. doi:10.1021/jo00388a021 |
| 13. | Yang, K.-W.; Golich, F. C.; Sigdel, T. K.; Crowder, M. W. Bioorg. Med. Chem. Lett. 2005, 15, 5150–5153. doi:10.1016/j.bmcl.2005.08.055 |
| 14. | Volonterio, A.; Zanda, M. Org. Lett. 2007, 9, 841–844. doi:10.1021/ol063074+ |
| 15. | Jaegli, S.; Vors, J.-P.; Neuville, L.; Zhu, J. Tetrahedron 2010, 66, 8911–8921. doi:10.1016/j.tet.2010.09.056 |
| 16. | Staples, M. K.; Grange, R. L.; Angus, J. A.; Ziogas, J.; Tan, N. P. H.; Taylor, M. K.; Schiesser, C. H. Org. Biomol. Chem. 2011, 9, 473–479. doi:10.1039/c0ob00573h |
| 17. | Qiu, G.; Mamboury, M.; Wang, Q.; Zhu, J. Angew. Chem., Int. Ed. 2016, 55, 15377–15381. doi:10.1002/anie.201609034 |
| 18. | Ke, D.; Espinosa, N. Á.; Mallet-Ladeira, S.; Monot, J.; Martin-Vaca, B.; Bourissou, D. Adv. Synth. Catal. 2016, 358, 2324–2331. doi:10.1002/adsc.201600382 |
| 19. | Coutant, E. P.; Hervin, V.; Gagnot, G.; Ford, C.; Baatallah, R.; Janin, Y. L. Beilstein J. Org. Chem. 2018, 14, 2853–2860. doi:10.3762/bjoc.14.264 |
| 11. | Feng, Y.-S.; Wu, W.; Xu, Z.-Q.; Li, Y.; Li, M.; Xu, H.-J. Tetrahedron 2012, 68, 2113–2120. doi:10.1016/j.tet.2012.01.032 |
| 28. | Quast, H.; Janiak, R. Liebigs Ann. Chem. 1991, 1305–1308. doi:10.1002/jlac.1991199101224 |
| 51. | Ihara, M.; Takahashi, M.; Taniguchi, N.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Chem. Commun. 1987, 619–620. doi:10.1039/c39870000619 |
| 52. | Ihara, M.; Takahashi, M.; Taniguchi, N.; Yasui, K.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Perkin Trans. 1 1989, 897–903. doi:10.1039/p19890000897 |
| 53. | Huang, F.; Buchwald, P.; Browne, C. E.; Farag, H. H.; Wu, W.-M.; Ji, F.; Hochhaus, G.; Bodor, N. AAPS PharmSci 2001, 3 (4), 44–56. doi:10.1208/ps030430 |
| 49. | Murta, M. M.; de Azevedo, M. B. M.; Greene, A. E. Synth. Commun. 1993, 23, 495–503. doi:10.1080/00397919308009804 |
| 50. | Hassan, H. A.; Abdel-Aziz, M.; Abuo-Rahma, G. E.-D. A. A.; Farag, H. H. Bioorg. Med. Chem. 2009, 17, 1681–1692. doi:10.1016/j.bmc.2008.12.051 |
| 46. | Dell‘Orco, P.; Brum, J.; Matsuoka, R.; Badlani, M.; Muske, K. Anal. Chem. (Washington, DC, U. S.) 1999, 71, 5165–5170. doi:10.1021/ac990554o |
| 51. | Ihara, M.; Takahashi, M.; Taniguchi, N.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Chem. Commun. 1987, 619–620. doi:10.1039/c39870000619 |
| 52. | Ihara, M.; Takahashi, M.; Taniguchi, N.; Yasui, K.; Fukumoto, K.; Kametani, T. J. Chem. Soc., Perkin Trans. 1 1989, 897–903. doi:10.1039/p19890000897 |
| 56. | Davidson, D.; Bernhard, S. A. J. Am. Chem. Soc. 1948, 70, 3426–3428. doi:10.1021/ja01190a060 |
| 57. | Lou, T.; Liao, E-T.; Wilsily, A.; Fillion, E. Org. Synth. 2012, 89, 115–125. doi:10.15227/orgsyn.089.0115 |
| 58. | Ramachary, D. B.; Reddy, G. B. Org. Biomol. Chem. 2006, 4, 4463–4468. doi:10.1039/b612611a |
| 54. | Xavier, T.; Condon, S.; Pichon, C.; Le Gall, E.; Presset, M. Org. Lett. 2019, 21, 6135–6139. doi:10.1021/acs.orglett.9b02291 |
| 55. | Xavier, T.; Condon, S.; Pichon, C.; Le Gall, E.; Presset, M. J. Org. Chem. 2021, 86, 5452–5462. doi:10.1021/acs.joc.0c02895 |
© 2021 Xavier et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)