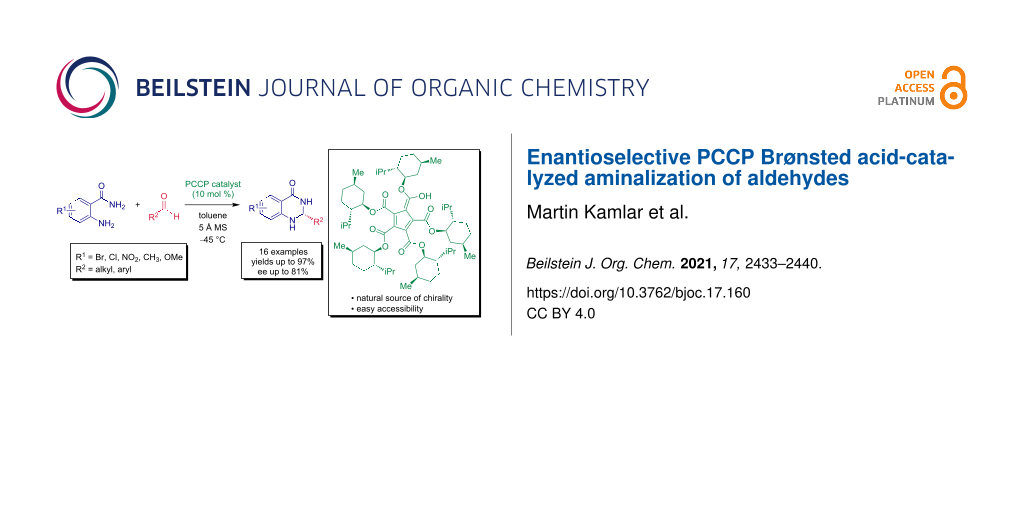
Abstract
Here we present an enantioselective aminalization of aldehydes catalyzed by Brønsted acids based on pentacarboxycyclopentadienes (PCCPs). The cyclization reaction using readily available anthranilamides as building blocks provides access to valuable 2,3-dihydroquinazolinones containing one stereogenic carbon center with good enantioselectivity (ee up to 80%) and excellent yields (up to 97%).

Graphical Abstract
Introduction
Nitrogen-containing heterocyclic compounds are commonly occurring in nature and constitute the core structures of many biologically important compounds. An important example of such heterocycles are 2,3-dihydroquinazolinones which scaffold can be found in various compounds exhibiting pharmacological properties [1-6]. Some of them are currently used to treat numerous diseases, such as the diuretic drug fenquizone used for the treatment of hypertension [7,8], or evodiamine, a stimulant used in fat reduction or inflammation [9-11]. Moreover, it was reported that both enantiomers of 2,3-dihydroquinazolinones exhibit different bioactivities [12,13]. Thus, the development of enantioselective synthetic strategies towards 2,3-dihydroquinazolinone derivatives has drawn the attention of organic chemists for a long time [14-18], even though the aminal stereocenter is sensitive to racemization [12].
The well-established and straightforward approach in the asymmetric organocatalytic synthesis of molecules with this moiety uses the reaction between aldehydes and anthranilamide building blocks. The advantage of this methodology lies in the fact that both starting materials are readily available, and the enantioselectivity of such cyclization reactions can be controlled by chiral Brønsted acids. In the scope of Brønsted acid catalysis, chiral phosphoric acids (CPA) are dominating as potent catalysts in various asymmetric transformations [19-23], although the synthesis of these catalysts is expensive and laborious [24]. One of the most frequent examples of CPAs is the binaphthol (BINOL)-derived phosphoric acid class of catalysts, firstly reported by Akiyama [25] and Terada [26]. Soon after, BINOL-derived phosphoric acids were employed in the enantioselective synthesis of 2,3-dihydroquinazolinones. The initial report in this area was made by List and co-workers, using an (S)-TRIP derivative as the chiral catalyst (Figure 1) [14]. Soon after, Rueping et al. developed a similar methodology catalyzed by other chiral BINOL-phosphoric acids [15]. However, the reaction suffered from limited scope to aromatic aldehydes without an ortho-substitution; the corresponding dihydroquinazolinones were obtained in high yields and with good enantiomeric purities. In 2013, Lin and co-workers published the application of a chiral SPINOL-phosphoric acid in the asymmetric aminalization reaction [27]. Tian´s research group developed the synthesis of dihydroquinazolinones from preformed imines instead of aldehydes catalyzed by BINOL-phosphoric acid [17]. The corresponding aminals were prepared with a wide range of substitutions using aromatic, α,β-unsaturated, or aliphatic imines. Apart from chiral phosphoric acids, chiral quaternary ammonium salts were successfully employed as catalysts in asymmetric dihydroquinazolinone synthesis [18]. Regarding the above-mentioned strategies involving chiral Brønsted acids, we envisioned that chiral pentacarboxycyclopentadiene (PCCP) derivatives could be used in the enantioselective aminalization of aldehydes with anthranilamide derivatives. PCCPs were firstly reported by Otto Diels [28,29], but recently, Lambert and co-workers introduced a new generation, chiral PCCPs (Figure 1) [30]. Due to the high stability of the aromatic cyclopentadienyl anion, PCCPs exhibit a low pKa value comparable to that of phosphoric acids. Contrary to chiral phosphoric acids, PCCPs offer less laborious and inexpensive preparation protocols [31,32], which makes them an interesting alternative for chiral Brønsted acid-catalyzed transformations [30-35].
![[1860-5397-17-160-1]](/bjoc/content/figures/1860-5397-17-160-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Synthetic strategies employing chiral Brønsted acid catalysis.
Figure 1: Synthetic strategies employing chiral Brønsted acid catalysis.
Results and Discussion
Herein, we describe our findings regarding the aminalization of aldehydes using PCCP catalysis. Our investigation commenced with the screening of the reaction between anthranilamide (1a) and isovaleraldehyde (2a) in the presence of 10 mol % of catalyst II (Table 1). First, we turned our attention to the solvent and temperature effect concerning the yield and the enantioselectivity of the aminalization reaction. While most solvents tested showed to be effective at room temperature, the enantiomeric purity of the corresponding aminal 3a was low in all cases (Table 1, entries 1–5). On the other hand, the yield of 3a was satisfactory in all reactions. In particular, when the reaction between 1a and 2a was performed in toluene, the isolated yield of 3a was almost quantitative (97%, entry 1 in Table 1). In our pursuit of better enantioselectivity, we continued with the reaction proceeded in toluene at lower temperatures. We found a temperature of −45 °C as optimal for the enantiocontrol of the model reaction, affording the product 3a in 90% yield with an enantiomeric purity of 66% ee (Table 1, entry 7). Additionally, the effect of molecular sieves on the course of the reaction was investigated and the obtained results demonstrated that molecular sieves dramatically improved the enantioselectivity (Table 1, entries 9–11). In particular, when the aminalization reaction between 1a and 2a was carried out in the presence of 5 Å molecular sieves, the corresponding product 3a was delivered in high yield (96%) and with enantiomeric purity 80% ee (Table 1, entry 11). In addition, the effect of the catalyst loading on the course of the reaction was examined. Our data clearly show that reducing the catalyst loading of II caused a significant decrease in the enantioselectivity (Table 1, entries 12 and 13). It is worth mentioning that no differences in the enantioselectivity were observed after a prolonged exposure of compound 3a to the chiral PCCP catalyst II indicating a relatively high stability of the new chiral carbon center in product 3a.
Table 1: Optimization of reaction conditions for the aminalization reaction between 1a and 2a.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-160-i3.svg?max-width=637&scale=1.0)
|
|||||||
| entry | solvent |
temperature
[°C] |
cat. loading
[mol %] |
additive | time [h] | yield [%]a | ee [%]b |
| 1 | toluene | 25 | 10 | – | 0.5 | 97 | 50 |
| 2 | THF | 25 | 10 | – | 1 | 72 | 50 |
| 3 | MTBE | 25 | 10 | – | 1 | 50 | 40 |
| 4 | DCM | 25 | 10 | – | 1 | 93 | 45 |
| 5 | EtOAc | 25 | 10 | – | 1 | 86 | 44 |
| 6 | toluene | 0 | 10 | – | 12 | 96 | 58 |
| 7 | toluene | −45 | 10 | – | 20 | 90 | 66 |
| 8 | toluene | −65 | 10 | – | 48 | 65 | 60 |
| 9 | toluene | −45 | 10 | 3 Å MS | 20 | 81 | 71 |
| 10 | toluene | −45 | 10 | 4 Å MS | 21 | 73 | 73 |
| 11 | toluene | −45 | 10 | 5 Å MS | 21 | 96 | 80 |
| 12 | toluene | −45 | 5 | 5 Å MS | 18 | 91 | 74 |
| 13 | toluene | −45 | 2 | 5 Å MS | 16 | 86 | 74 |
aIsolated yield; bdetermined by chiral HPLC.
Next, a small set of functionalized derivatives of cyclopentadienes as organocatalysts was surveyed in the model reaction (Table 2). Apart from model catalyst II, equipped with five (−)-menthol units, also the sterically less demanding amide-type catalyst III and the thiourea derivative IV were tested (Table 2). First, the diamide-type catalyst III was examined (Table 2, entry 4). Although complete conversion of 1a and 2a was achieved after a significantly prolonged time (7 days), the aminal 3a was isolated in a good yield of 60%. Unfortunately, the reaction proceeded nearly in a racemic fashion. An inefficient catalyst showed up to be the PCCP catalyst derivatized with thiourea functional units (IV); a formation of 3a was not observed even after prolonged reaction time (Table 2, entry 5). It is also worth mentioning that the non-catalyzed reaction did not deliver the corresponding product 3a even after 40 hours (Table 2, entry 1). Based on the results summarized in Table 2, the chiral PCCP catalyst II was selected as the optimal catalyst.
Table 2: Catalyst screening of the aminalization reaction between 1a and 2a.
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-160-i4.svg?max-width=637&scale=1.0)
|
||||
| entry | catalyst | time [h] | yield [%]a | ee [%]b |
| 1 | – | 40 | n.d. | n.d. |
| 2 | I | 16 | 95 | 0 |
| 3 | II | 21 | 96 | 80 |
| 4 | III | 168 | 60 | 2 |
| 5 | IV | 168 | n.d. | n.d. |
aIsolated yield; bdetermined by chiral HPLC.
With the optimized reaction conditions in our hands, we continued investigating the scope of the reaction. First, we focused on the reactivity of anthranilamide (1a) with various aldehydes 2a–j (Scheme 1). Generally, aliphatic aldehydes delivered the cyclic aminals 3a–d in excellent yields between 95–97% and enantiomeric purities between 74–80% ee. However, the sterically demanding pivalaldehyde (2c) needed a prolonged reaction time to reach the complete conversion. In addition, a significant drop in the enantioselectivity (10% ee) of 3c was observed. Also, benzaldehyde derivatives were successfully tested in the aminalization reaction. However, a decrease in reactivity and enantioselectivity was observed when compared to aliphatic aldehydes. The corresponding products 3e–j were isolated in lower yields (58–83%) with enantiomeric purities ranging from 20 to 70% ee. For example, when benzaldehydes substituted with fluorine or chlorine in the para-position were employed in catalytic reaction with anthranilamide (1a), the corresponding derivatives 3i,j were isolated in 58 and 69% yield, respectively. The rates of enantioselectivity for both reactions were lower and averaged only around 50%. In addition, the role of an electron-donating methyl group on the aromatic ring was investigated. When p-tolualdehyde (1f) was used in the cyclization reaction with anthranilamide (1a), the corresponding aminal 3f was obtained in high yield (83%) and with good enantiomeric excess of 70% ee. On the other hand, when m- or o-tolualdehyde were employed in aminalization reaction, a significant drop in the enantioselectivity was observed. Aminals 3g and 3h were obtained with 36 and 20% ee, respectively. We have also tested the reaction between anthranilamide (1a) and isovaleraldehyde (2a) in 1 mmol scale. The obtained results suggested that the reaction proceeded with slightly lower efficiency giving product 3a in 83% yield and 71% ee. On the other hand, we found that the desired product of aminalization reaction could be readily obtained in higher enantiomeric purity after crystallization from ethyl acetate. This was demonstrated for products 3a and 3f, that were obtained in enantiomeric purities of 93% and 97% ee, respectively (Scheme 1).
![[1860-5397-17-160-i1]](/bjoc/content/inline/1860-5397-17-160-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The substrate scope of the aminalization reaction for different aldehydes. aAfter recrystallization; breaction run at 1 mmol scale.
Scheme 1: The substrate scope of the aminalization reaction for different aldehydes. aAfter recrystallization...
Next, we turned our attention to the substitution of anthranilamide (Scheme 2). First, the effect of bromine as a slightly electron-withdrawing substituent on the aromatic ring was investigated. The position of bromine on the aromatic ring had a dramatic effect on the enantiomeric purity of the formed products 3k–n. When a bromine substituent is introduced in the “3” position of anthranilamide, the enantiomeric enrichment of aminal 3k reached only 30% ee. In contrast, substitution with bromine either in position “4” and “5” led to a formation of products 3l and 3m with enantiomeric purities of 70% ee and 80% ee, respectively. Finally, reaction with anthranilamide substituted with bromine in position “6” led to corresponding aminal 3n with an enantiomeric excess of 66% ee. We also increased the enantiomeric purity of 3l from 70% to 80% ee after crystallization from ethyl acetate. When anthranilamide substituted with a chlorine in the “5” position was used, the enantioselectivity of the reaction reached a value of 76% ee, and the yield of the corresponding aminal 3o exceeded 80%. Next, the effect of a strongly electron-withdrawing nitro group present on anthranilamide moiety was investigated. The reaction carried out in toluene did not reach a complete conversion even after a prolonged reaction time. When more polar THF was used as the solvent, the corresponding product 3p was obtained after 40 hours in an excellent yield of 96%; however, the enantiomeric purity of 3p was only 42% ee. Anthranilamides containing electron-donating methyl and methoxy groups were also well-tolerated in the aminalization reaction. For example, reaction with anthranilamide bearing a methyl group in the “4” position delivered product 3q in good yield (80%) and enantiopurity (69% ee). A higher yield (96%) and enantiopurity (72% ee) was reached with anthranilamide 1r, having a methyl group in the position “5”. To further broaden the scope of the aminalization reaction, we prepared 2-(2-aminophenyl)acetamide (1t) and tested it in the reaction with isovaleraldehyde (2a) to access benzodiazepinone derivatives. The reaction proceeded smoothly with complete conversion within 24 hours, yielding the desired benzodiazepinone derivative 3t in 55%. However, the enantiomeric purity dropped significantly to 35% ee. Additionally, we tested the influence of substitution of the aromatic amine and prepared the benzyl-protected anthranilamide 1u. Unfortunately, the reaction between 1u and isovaleraldehyde (2a) did not deliver the corresponding product 3u even after a prolonged reaction time.
![[1860-5397-17-160-i2]](/bjoc/content/inline/1860-5397-17-160-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The substrate scope of the intermolecular aminalization reaction for anthranilamide derivatives. aAfter recrystallization; bTHF used as a reaction solvent.
Scheme 2: The substrate scope of the intermolecular aminalization reaction for anthranilamide derivatives. aA...
To determine the absolute configuration of aminals 3a–t, derivative 3l was subjected to X-ray crystallographic analysis. The absolute configuration of the stereogenic center (C1) was assigned as R (Figure 2, for details see Supporting Information File 1) [36], which is in agreement with the configuration of aminals obtained by List and co-workers [14].
![[1860-5397-17-160-2]](/bjoc/content/figures/1860-5397-17-160-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: X-ray single-crystal structure of aminal 3l with the displacement ellipsoids drawn at the 30% probability level.
Figure 2: X-ray single-crystal structure of aminal 3l with the displacement ellipsoids drawn at the 30% proba...
Conclusion
In summary, we have reported an organocatalytic asymmetric aminalization reaction between aldehydes and anthranilamides catalyzed by a PCCP catalyst as a cheap and readily available option to conventional chiral BINOL phosphoric acids. The reaction tolerates a wide range of substitutions of anthranilamides and aromatic and aliphatic aldehydes, yielding the corresponding dihydroquinazolinones in excellent yields (up to 97%) and enantiopurities up to 80% ee. We demonstrated that bulkiness of aldehydes negatively affected the enantiocontrol of the process, and highly enantiomerically enriched dihydroquinazolinones can be achieved by crystallization (up to 97% ee). The developed methodology can also be used to form tetrahydrobenzodiazepinones; however, a significant drop in the yield and enantioselectivity was observed.
Supporting Information
| Supporting Information File 1: General synthetic procedures, characterization of compounds, X-ray experimental data, and copies of 1H and 13C NMR spectra. | ||
| Format: PDF | Size: 6.8 MB | Download |
References
-
Auti, P. S.; George, G.; Paul, A. T. RSC Adv. 2020, 10, 41353–41392. doi:10.1039/d0ra06642g
Return to citation in text: [1] -
Xu, Z.; Zhang, Y.; Fu, H.; Zhong, H.; Hong, K.; Zhu, W. Bioorg. Med. Chem. Lett. 2011, 21, 4005–4007. doi:10.1016/j.bmcl.2011.05.002
Return to citation in text: [1] -
Levin, J. I.; Chan, P. S.; Bailey, T.; Katocs, A. S., Jr.; Venkatesan, A. M. Bioorg. Med. Chem. Lett. 1994, 4, 1141–1146. doi:10.1016/s0960-894x(01)80244-4
Return to citation in text: [1] -
Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s
Return to citation in text: [1] -
Badolato, M.; Aiello, F.; Neamati, N. RSC Adv. 2018, 8, 20894–20921. doi:10.1039/c8ra02827c
Return to citation in text: [1] -
Bonola, G.; Da Re, P.; Magistretti, M. J.; Massarani, E.; Setnikar, I. J. Med. Chem. 1968, 11, 1136–1139. doi:10.1021/jm00312a007
Return to citation in text: [1] -
Selvam, T. P.; Kumar, P. V. Res. Pharm. 2011, 1 (1), 1–21.
Return to citation in text: [1] -
Angelino, P. F.; Alvino-Demartino, A.; Mappa, R.; Donati, C. Int. J. Clin. Pharmacol., Ther. Toxicol. 1985, 23, 501–505.
Return to citation in text: [1] -
Yu, H.; Jin, H.; Gong, W.; Wang, Z.; Liang, H. Molecules 2013, 18, 1826–1843. doi:10.3390/molecules18021826
Return to citation in text: [1] -
Sun, Q.; Xie, L.; Song, J.; Li, X. J. Ethnopharmacol. 2020, 262, 113164. doi:10.1016/j.jep.2020.113164
Return to citation in text: [1] -
Rebhun, J. F.; Roloff, S. J.; Velliquette, R. A.; Missler, S. R. Fitoterapia 2015, 101, 57–63. doi:10.1016/j.fitote.2014.12.009
Return to citation in text: [1] -
Chinigo, G. M.; Paige, M.; Grindrod, S.; Hamel, E.; Dakshanamurthy, S.; Chruszcz, M.; Minor, W.; Brown, M. L. J. Med. Chem. 2008, 51, 4620–4631. doi:10.1021/jm800271c
Return to citation in text: [1] [2] -
Uzunov, D. P.; Zivkovich, I.; Costa, E.; Guidotti, A.; Pirkle, W. H. J. Pharm. Sci. 1995, 84, 937–942. doi:10.1002/jps.2600840807
Return to citation in text: [1] -
Cheng, X.; Vellalath, S.; Goddard, R.; List, B. J. Am. Chem. Soc. 2008, 130, 15786–15787. doi:10.1021/ja8071034
Return to citation in text: [1] [2] [3] -
Rueping, M.; Antonchick, A. P.; Sugiono, E.; Grenader, K. Angew. Chem., Int. Ed. 2009, 48, 908–910. doi:10.1002/anie.200804770
Return to citation in text: [1] [2] -
Honjo, T.; Phipps, R. J.; Rauniyar, V.; Toste, F. D. Angew. Chem., Int. Ed. 2012, 51, 9684–9688. doi:10.1002/anie.201205383
Return to citation in text: [1] -
Cheng, D.-J.; Tian, Y.; Tian, S.-K. Adv. Synth. Catal. 2012, 354, 995–999. doi:10.1002/adsc.201100849
Return to citation in text: [1] [2] -
Ayyanar, S.; Vijaya, P. K.; Mariyappan, M.; Ashokkumar, V.; Sadhasivam, V.; Balakrishnan, S.; Chinnadurai, C.; Murugesan, S. New J. Chem. 2017, 41, 7980–7986. doi:10.1039/c7nj00538e
Return to citation in text: [1] [2] -
Li, X.; Song, Q. Chin. Chem. Lett. 2018, 29, 1181–1192. doi:10.1016/j.cclet.2018.01.045
Return to citation in text: [1] -
Terada, M. Curr. Org. Chem. 2011, 15, 2227–2256. doi:10.2174/138527211796150732
Return to citation in text: [1] -
Maji, R.; Mallojjala, S. C.; Wheeler, S. E. Chem. Soc. Rev. 2018, 47, 1142–1158. doi:10.1039/c6cs00475j
Return to citation in text: [1] -
Terada, M. Bull. Chem. Soc. Jpn. 2010, 83, 101–119. doi:10.1246/bcsj.20090268
Return to citation in text: [1] -
Wu, H.; He, Y.-P.; Shi, F. Synthesis 2015, 47, 1990–2016. doi:10.1055/s-0034-1378837
Return to citation in text: [1] -
Ahmed, I.; Clark, D. A. Org. Lett. 2014, 16, 4332–4335. doi:10.1021/ol502126r
Return to citation in text: [1] -
Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. doi:10.1002/anie.200353240
Return to citation in text: [1] -
Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356–5357. doi:10.1021/ja0491533
Return to citation in text: [1] -
Huang, D.; Li, X.; Xu, F.; Li, L.; Lin, X. ACS Catal. 2013, 3, 2244–2247. doi:10.1021/cs400591u
Return to citation in text: [1] -
Diels, O. Ber. Dtsch. Chem. Ges. A 1942, 75, 1452–1467. doi:10.1002/cber.19420751206
Return to citation in text: [1] -
Diels, O.; Kock, U. Justus Liebigs Ann. Chem. 1944, 556, 38–50. doi:10.1002/jlac.19445560105
Return to citation in text: [1] -
Gheewala, C. D.; Collins, B. E.; Lambert, T. H. Science 2016, 351, 961–965. doi:10.1126/science.aad0591
Return to citation in text: [1] [2] -
Radtke, M. A.; Dudley, C. C.; O’Leary, J. M.; Lambert, T. H. Synthesis 2019, 51, 1135–1138. doi:10.1055/s-0037-1611650
Return to citation in text: [1] [2] -
Gheewala, C. D.; Radtke, M. A.; Hui, J.; Hon, A. B.; Lambert, T. H. Org. Lett. 2017, 19, 4227–4230. doi:10.1021/acs.orglett.7b01867
Return to citation in text: [1] [2] -
Hammersley, G. R.; Nichol, M. F.; Steffens, H. C.; Delgado, J. M.; Veits, G. K.; Read de Alaniz, J. Beilstein J. Org. Chem. 2019, 15, 1569–1574. doi:10.3762/bjoc.15.160
Return to citation in text: [1] -
Gheewala, C. D.; Hirschi, J. S.; Lee, W.-H.; Paley, D. W.; Vetticatt, M. J.; Lambert, T. H. J. Am. Chem. Soc. 2018, 140, 3523–3527. doi:10.1021/jacs.8b00260
Return to citation in text: [1] -
Sui, Y.; Cui, P.; Liu, S.; Zhou, Y.; Du, P.; Zhou, H. Eur. J. Org. Chem. 2018, 215–218. doi:10.1002/ejoc.201701561
Return to citation in text: [1] -
CCDC 2081064 for 3l contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif
Return to citation in text: [1]
| 30. | Gheewala, C. D.; Collins, B. E.; Lambert, T. H. Science 2016, 351, 961–965. doi:10.1126/science.aad0591 |
| 31. | Radtke, M. A.; Dudley, C. C.; O’Leary, J. M.; Lambert, T. H. Synthesis 2019, 51, 1135–1138. doi:10.1055/s-0037-1611650 |
| 32. | Gheewala, C. D.; Radtke, M. A.; Hui, J.; Hon, A. B.; Lambert, T. H. Org. Lett. 2017, 19, 4227–4230. doi:10.1021/acs.orglett.7b01867 |
| 33. | Hammersley, G. R.; Nichol, M. F.; Steffens, H. C.; Delgado, J. M.; Veits, G. K.; Read de Alaniz, J. Beilstein J. Org. Chem. 2019, 15, 1569–1574. doi:10.3762/bjoc.15.160 |
| 34. | Gheewala, C. D.; Hirschi, J. S.; Lee, W.-H.; Paley, D. W.; Vetticatt, M. J.; Lambert, T. H. J. Am. Chem. Soc. 2018, 140, 3523–3527. doi:10.1021/jacs.8b00260 |
| 35. | Sui, Y.; Cui, P.; Liu, S.; Zhou, Y.; Du, P.; Zhou, H. Eur. J. Org. Chem. 2018, 215–218. doi:10.1002/ejoc.201701561 |
| 30. | Gheewala, C. D.; Collins, B. E.; Lambert, T. H. Science 2016, 351, 961–965. doi:10.1126/science.aad0591 |
| 31. | Radtke, M. A.; Dudley, C. C.; O’Leary, J. M.; Lambert, T. H. Synthesis 2019, 51, 1135–1138. doi:10.1055/s-0037-1611650 |
| 32. | Gheewala, C. D.; Radtke, M. A.; Hui, J.; Hon, A. B.; Lambert, T. H. Org. Lett. 2017, 19, 4227–4230. doi:10.1021/acs.orglett.7b01867 |
| 1. | Auti, P. S.; George, G.; Paul, A. T. RSC Adv. 2020, 10, 41353–41392. doi:10.1039/d0ra06642g |
| 2. | Xu, Z.; Zhang, Y.; Fu, H.; Zhong, H.; Hong, K.; Zhu, W. Bioorg. Med. Chem. Lett. 2011, 21, 4005–4007. doi:10.1016/j.bmcl.2011.05.002 |
| 3. | Levin, J. I.; Chan, P. S.; Bailey, T.; Katocs, A. S., Jr.; Venkatesan, A. M. Bioorg. Med. Chem. Lett. 1994, 4, 1141–1146. doi:10.1016/s0960-894x(01)80244-4 |
| 4. | Horton, D. A.; Bourne, G. T.; Smythe, M. L. Chem. Rev. 2003, 103, 893–930. doi:10.1021/cr020033s |
| 5. | Badolato, M.; Aiello, F.; Neamati, N. RSC Adv. 2018, 8, 20894–20921. doi:10.1039/c8ra02827c |
| 6. | Bonola, G.; Da Re, P.; Magistretti, M. J.; Massarani, E.; Setnikar, I. J. Med. Chem. 1968, 11, 1136–1139. doi:10.1021/jm00312a007 |
| 14. | Cheng, X.; Vellalath, S.; Goddard, R.; List, B. J. Am. Chem. Soc. 2008, 130, 15786–15787. doi:10.1021/ja8071034 |
| 15. | Rueping, M.; Antonchick, A. P.; Sugiono, E.; Grenader, K. Angew. Chem., Int. Ed. 2009, 48, 908–910. doi:10.1002/anie.200804770 |
| 16. | Honjo, T.; Phipps, R. J.; Rauniyar, V.; Toste, F. D. Angew. Chem., Int. Ed. 2012, 51, 9684–9688. doi:10.1002/anie.201205383 |
| 17. | Cheng, D.-J.; Tian, Y.; Tian, S.-K. Adv. Synth. Catal. 2012, 354, 995–999. doi:10.1002/adsc.201100849 |
| 18. | Ayyanar, S.; Vijaya, P. K.; Mariyappan, M.; Ashokkumar, V.; Sadhasivam, V.; Balakrishnan, S.; Chinnadurai, C.; Murugesan, S. New J. Chem. 2017, 41, 7980–7986. doi:10.1039/c7nj00538e |
| 18. | Ayyanar, S.; Vijaya, P. K.; Mariyappan, M.; Ashokkumar, V.; Sadhasivam, V.; Balakrishnan, S.; Chinnadurai, C.; Murugesan, S. New J. Chem. 2017, 41, 7980–7986. doi:10.1039/c7nj00538e |
| 12. | Chinigo, G. M.; Paige, M.; Grindrod, S.; Hamel, E.; Dakshanamurthy, S.; Chruszcz, M.; Minor, W.; Brown, M. L. J. Med. Chem. 2008, 51, 4620–4631. doi:10.1021/jm800271c |
| 13. | Uzunov, D. P.; Zivkovich, I.; Costa, E.; Guidotti, A.; Pirkle, W. H. J. Pharm. Sci. 1995, 84, 937–942. doi:10.1002/jps.2600840807 |
| 28. | Diels, O. Ber. Dtsch. Chem. Ges. A 1942, 75, 1452–1467. doi:10.1002/cber.19420751206 |
| 29. | Diels, O.; Kock, U. Justus Liebigs Ann. Chem. 1944, 556, 38–50. doi:10.1002/jlac.19445560105 |
| 9. | Yu, H.; Jin, H.; Gong, W.; Wang, Z.; Liang, H. Molecules 2013, 18, 1826–1843. doi:10.3390/molecules18021826 |
| 10. | Sun, Q.; Xie, L.; Song, J.; Li, X. J. Ethnopharmacol. 2020, 262, 113164. doi:10.1016/j.jep.2020.113164 |
| 11. | Rebhun, J. F.; Roloff, S. J.; Velliquette, R. A.; Missler, S. R. Fitoterapia 2015, 101, 57–63. doi:10.1016/j.fitote.2014.12.009 |
| 27. | Huang, D.; Li, X.; Xu, F.; Li, L.; Lin, X. ACS Catal. 2013, 3, 2244–2247. doi:10.1021/cs400591u |
| 7. | Selvam, T. P.; Kumar, P. V. Res. Pharm. 2011, 1 (1), 1–21. |
| 8. | Angelino, P. F.; Alvino-Demartino, A.; Mappa, R.; Donati, C. Int. J. Clin. Pharmacol., Ther. Toxicol. 1985, 23, 501–505. |
| 17. | Cheng, D.-J.; Tian, Y.; Tian, S.-K. Adv. Synth. Catal. 2012, 354, 995–999. doi:10.1002/adsc.201100849 |
| 25. | Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566–1568. doi:10.1002/anie.200353240 |
| 14. | Cheng, X.; Vellalath, S.; Goddard, R.; List, B. J. Am. Chem. Soc. 2008, 130, 15786–15787. doi:10.1021/ja8071034 |
| 24. | Ahmed, I.; Clark, D. A. Org. Lett. 2014, 16, 4332–4335. doi:10.1021/ol502126r |
| 15. | Rueping, M.; Antonchick, A. P.; Sugiono, E.; Grenader, K. Angew. Chem., Int. Ed. 2009, 48, 908–910. doi:10.1002/anie.200804770 |
| 19. | Li, X.; Song, Q. Chin. Chem. Lett. 2018, 29, 1181–1192. doi:10.1016/j.cclet.2018.01.045 |
| 20. | Terada, M. Curr. Org. Chem. 2011, 15, 2227–2256. doi:10.2174/138527211796150732 |
| 21. | Maji, R.; Mallojjala, S. C.; Wheeler, S. E. Chem. Soc. Rev. 2018, 47, 1142–1158. doi:10.1039/c6cs00475j |
| 22. | Terada, M. Bull. Chem. Soc. Jpn. 2010, 83, 101–119. doi:10.1246/bcsj.20090268 |
| 23. | Wu, H.; He, Y.-P.; Shi, F. Synthesis 2015, 47, 1990–2016. doi:10.1055/s-0034-1378837 |
| 36. | CCDC 2081064 for 3l contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif |
| 12. | Chinigo, G. M.; Paige, M.; Grindrod, S.; Hamel, E.; Dakshanamurthy, S.; Chruszcz, M.; Minor, W.; Brown, M. L. J. Med. Chem. 2008, 51, 4620–4631. doi:10.1021/jm800271c |
| 26. | Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356–5357. doi:10.1021/ja0491533 |
| 14. | Cheng, X.; Vellalath, S.; Goddard, R.; List, B. J. Am. Chem. Soc. 2008, 130, 15786–15787. doi:10.1021/ja8071034 |
© 2021 Kamlar et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)