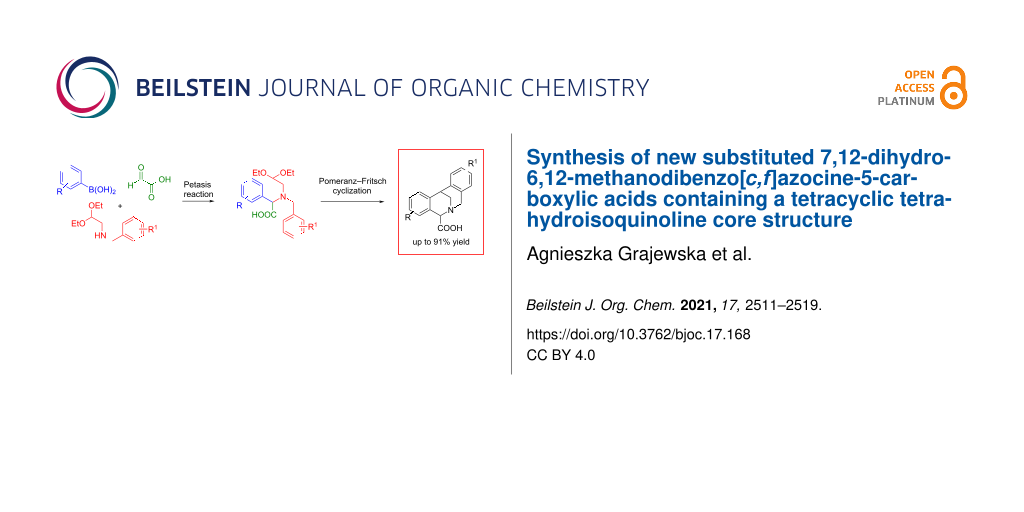
Abstract
A convenient and simple protocol has been developed for the synthesis of a series of new tetracyclic tetrahydroisoquinoline derivatives, 7,12-dihydro-6,12-methanodibenzo[c,f]-azocine-5-carboxylic acids by three component Petasis reaction with the use of aminoacetaldehyde acetals bearing substituted benzyl groups as the amine components followed by Pomeranz–Fritsch double cyclization reaction. By applying this method, several acids have been prepared in satisfactory yields. An unprecedented chemical behavior of a Petasis reaction product in diluted HCl solution leading to the formation of a phenylglycine derivative has been observed and the mechanism explaining such reactivity has been proposed.

Graphical Abstract
Introduction
The tetrahydroisoquinoline moiety is the key structure in large number of natural and synthetic biologically important molecules. Among them, isoquinoline alkaloids constitute a large family of natural products exhibiting a huge range of structural diversity and biological activity [1-3]. Within this family several structural motifs can be identified, for instance, the isopavines, constrained alkaloids possessing the 1,2,3,4-tetrahydroisoquinoline skeleton incorporated into the double cyclized azabicyclo[3.2.2]nonane, isolated from Papaveraceae and Ranunculaceae species. Natural isopavines (e.g., amurensinine (I), and O-methylthalisopavine (II), Figure 1) as well as their synthetic analogues have attracted much attention [4,5] because of their interactions with the receptors in the central nervous system promising for the treatment of nervous system disorders, such as Parkinson’s and Alzheimer’s disease [6,7].
![[1860-5397-17-168-1]](/bjoc/content/figures/1860-5397-17-168-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Natural isopavine alkaloids and synthetic derivatives of isopavine.
Figure 1: Natural isopavine alkaloids and synthetic derivatives of isopavine.
Hanessian and co-workers designed isopavines of type III (Figure 1) that act as morphinomimethics and bind strongly to human opioid receptors [8,9]. The synthesis of these compounds involved the stereocontrolled [1,2]-Stevens rearrangement of dihydromethanodibenzoazocines. Dihydromethanodibenzoazocines are example of strained, methylene-bridged tetrahydroisoquinolines (Figure 2) that have attracted much interest of scientists not only due to their resemblance to isopavines and thus their medicinal significance in the central nervous system but also as a building blocks for the synthesis of alkaloids [10,11].
![[1860-5397-17-168-2]](/bjoc/content/figures/1860-5397-17-168-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The structure and numbering of dihydromethanodibenzoazocine.
Figure 2: The structure and numbering of dihydromethanodibenzoazocine.
The classical strategy for the synthesis of dihydromethanodibenzoazocines is based on the use of N,N-dibenzylaminoacetaldehydes [8,9] and less often α-dibenzylaminoketone [12,13] derivatives in the intramolecular Friedel–Crafts double cyclization reaction and N,N-dibenzylaminoacetaldehyde dialkyl acetals [12,14-16] in the Pomeranz–Fritsch-type double cyclization reaction. There is also one example of employing p-quinol acetates as substrates for the synthesis of these compounds [17]. Recently, a methodology for the synthesis of dihydromethanodibenzoazocines based on a combination of Friedel–Crafts and Pictet–Spengler reactions was proposed by Moshkin et al. [18].
We have developed a convenient method for the preparation of C-1-substituted tetrahydroisoquinoline derivatives by using the Petasis three-component reaction followed by the Pomeranz–Fritsch–Bobbitt cyclization. The Petasis reaction between boronic acids, carbonyl derivatives, and amines, leading to the formation of amino acids and the Pomeranz–Fritsch–Bobbitt cyclization of amino acetals, leading to the construction of C-1-functionalized tetrahydroisoquinoline rings, are presented in Scheme 1.
![[1860-5397-17-168-i1]](/bjoc/content/inline/1860-5397-17-168-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The Petasis reaction and the Pomeranz–Fritsch–Bobbitt cyclization.
Scheme 1: The Petasis reaction and the Pomeranz–Fritsch–Bobbitt cyclization.
In the Petasis reaction we applied aminoacetaldehyde acetals as the amine component which enabled the preparation of the substrates for the Pomeranz–Fritsch–Bobbitt cyclization in one simple step. The Petasis reaction products were further transformed into racemic tetrahydroisoquinoline-1-carboxylic acids [19], simple isoquinoline alkaloids [20], and (+)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-1-carboxylic acid [21].
In this paper we report a straightforward synthesis of new strained tetracyclic tetrahydroisoquinoline derivatives, variously substituted 7,12-dihydro-6,12-methanodibenzo[c,f]azocine-5-carboxylic acids, via our modified method based on a combination of the Petasis reaction, in which we used aminoacetaldehyde acetals with substituted N-benzyl groups as the amine component and the Pomeranz–Fritsch double cyclization reaction (Scheme 2).
![[1860-5397-17-168-i2]](/bjoc/content/inline/1860-5397-17-168-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The synthesis of 7,12-dihydro-6,12-methanodibenzo[c,f]azocine-5-carboxylic acids via a combination of the Petasis reaction and the Pomeranz–Fritsch reaction – a retrosynthetic approach.
Scheme 2: The synthesis of 7,12-dihydro-6,12-methanodibenzo[c,f]azocine-5-carboxylic acids via a combination ...
Results and Discussion
Our investigations commenced with the synthesis of N-benzylated aminoacetaldehyde acetals 3a–e, the amine components for the Petasis reaction. The condensation of aminoacetaldehyde diethyl acetal 1 and 2,3-dimethoxybenzaldehyde (2a) was carried out at rt in anhydrous ethanol and the formed imine was then reduced in situ with sodium borohydride to give the product 3a with 98% yield. The synthesis of further N-benzylated aminoacetaldehyde acetals 3b–e was performed in these reaction conditions in EtOH or MeOH to provide the desired products with high overall yields of 85–99% as indicated in Scheme 3. The aminoacetals 3a–e were suitable for use directly in the next step of the synthesis without further purification.
![[1860-5397-17-168-i3]](/bjoc/content/inline/1860-5397-17-168-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of N-benzylated aminoacetaldehyde acetals 3a–e. Conditions: a) reaction run in EtOH; b) reaction run in MeOH.
Scheme 3: Synthesis of N-benzylated aminoacetaldehyde acetals 3a–e. Conditions: a) reaction run in EtOH; b) r...
Then, the N-benzylated aminoacetals 3a–e were subjected to the Petasis reaction with glyoxylic acid hydrate (4) and the appropriate boronic acids 5a–d carried out in DCM at rt for 24 h to afford amino acids 6a–g (Scheme 4).
![[1860-5397-17-168-i4]](/bjoc/content/inline/1860-5397-17-168-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
The condensation of N-(2,3-dimethoxybenzyl)aminoacetaldehyde acetal (3a) with glyoxylic acid hydrate (4) and 3,4-dimethoxyphenylboronic acid (5a) afforded the Petasis reaction product 6a in a high 94% yield.
The double cyclization reaction was easily performed by treatment of 6a with 20% HCl for 24 h to give dihydromethanodibenzoazocine-5-carboxylic acid (7a, Scheme 5) in 72% yield by extraction of the neutralized reaction mixture. Compound 7a was obtained in two simple steps from 3a with an overall yield of 67%.
![[1860-5397-17-168-i5]](/bjoc/content/inline/1860-5397-17-168-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of dihydromethanodibenzoazocine-5-carboxylic acids 7a–f. Conditions: a) 20% HCl, rt, 24 h; b) 70% HClO4, rt, 12 h.
Scheme 5: Synthesis of dihydromethanodibenzoazocine-5-carboxylic acids 7a–f. Conditions: a) 20% HCl, rt, 24 h...
Then the amino acid 6a was subjected to cyclization under different acidic conditions (not shown). The reaction of 6a with TiCl4 or SnCl4 in DCM at 0 °C led exclusively to the formation of 7a (TLC analysis) but it was impossible to isolate the product in a pure form probably due to the formation of the salts.
When 6a was treated with 70% HClO4 for 2 hours, the TLC analysis indicated the presence of 7a as the main product together with unreacted substrate and other difficult to separate by-products. When the reaction time was prolonged to 24 h all the substrate was consumed but a mixture of unidentified products was formed.
Then, N-2,3-methylenedioxybenzylamino acid 6b and N-3,4,5-trimethoxybenzylamino acid 6c were prepared from boronic acid 5a, glyoxylic acid hydrate (4) and aminoacetals 3b and 3c, in 99% and 90% yield, respectively (Scheme 4). Under the Pomeranz–Fritsch conditions compounds 6b and 6c were transformed into dihydromethanodibenzoazocine-5-carboxylic acids 7b with a yield of 58% and 7c with a yield of 69%. The overall yields for compounds 7b and 7c starting from 3b and 3c, were 57% and 62%, respectively (Scheme 5).
Next, 3-methoxyphenylboronic acid (5b) and 3,4-methylenedioxyphenylboronic acid (5c) reacted easily with aminoacetals 3b and 3a, respectively and glyoxylic acid hydrate (4), affording the corresponding arylglycine derivatives 6d and 6e, in satisfactory yields of 73% and 95%, respectively (Scheme 4). After cyclization in 20% HCl, compounds 6d and 6e were transformed into the desired dihydromethanodibenzoazocine-5-carboxylic acids 7d and 7e with yields of 52% and 96%, respectively. The overall yields of 7d and 7e (from 3d and 3e, respectively) were 37% and 91%, respectively (Scheme 5).
In the case of the amino acid 6f, containing insufficiently activated phenyl rings without alkoxy substituents, prepared from phenylboronic acid (5d), glyoxylic acid hydrate (4) and an N-benzyl aminoacetal 3d with good yield 79% (Scheme 4), it was necessary to perform the cyclization reaction using 70% HClO4 instead of 20% HCl. In these conditions, the desired product 7f was obtained with 67% yield (53% overall yield from 3d), while the reaction carried out in 20% HCl led to a mixture of products, among which 7f was not detected (Scheme 5).
It should be also mentioned that the amino acid 6g with a benzyloxy-substituted aromatic ring, obtained in the Petasis reaction of boronic acid 5a, glyoxylic acid hydrate (4), and O-benzylvanilin-derived aminoacetal 3e (Scheme 4), when treated with 20% HCl or 70% HClO4 gave a colored complex mixture of decomposition products, probably due to the hydrolysis of the benzyl ether.
Iwakuma et al. [15] reported the synthesis of 1,2,3,4-tetrahydroisoquinoline derivative TA-073 using substituted N-acyl aminoacetal as the key intermediate which, depending on the reaction conditions, could be transformed either into TA-073 or the appropriate double cyclized product, isopavine or pavine (not shown) (Scheme 6).
![[1860-5397-17-168-i6]](/bjoc/content/inline/1860-5397-17-168-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Inspired by these results, we next investigated the possibility of obtaining monocyclized derivatives from the Petasis reaction products of type 6. In this experiment the amino acid 6a has been chosen as a model compound and it was treated with 4% aqueous HCl in THF at reflux [15]. Surprisingly, in these conditions we isolated the N-benzylated phenylglycine derivative 8 as the sole product with 87% yield (Scheme 7). When we decreased the concentration of HCl to 2% in the reaction with 6a, the substrate was isolated unchanged even after prolonged reaction time.
![[1860-5397-17-168-i7]](/bjoc/content/inline/1860-5397-17-168-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Reaction of 6a with 4% aqueous HCl solution in THF and with 20% aqueous HCl solution. Conditions: a) 4% HCl (aq), THF, reflux; b) 20% HCl, rt.
Scheme 7: Reaction of 6a with 4% aqueous HCl solution in THF and with 20% aqueous HCl solution. Conditions: a...
In light of these results, we investigated the chemical behavior of compound 12, the decarboxylated analogue of 6a, under the reaction conditions that led to products 7a or 8 starting from acid 6a. The substrate for the synthesis of compound 12 was aminoacetal 3f, obtained with moderate 49% yield through the condensation of aminoacetaldehyde diethyl acetal (1) and 3,4-dimethoxybenzaldehyde (2f) carried out in refluxing toluene using a Dean–Stark apparatus followed by the reduction with NaBH4 (not shown). When the condensation of acetal 1 and aldehyde 2f and the subsequent reduction were carried out in EtOH at rt, according to our procedure applied for the synthesis of aminoacetals 3a–e, the product 3f was isolated with 65% yield (Scheme 8).
![[1860-5397-17-168-i8]](/bjoc/content/inline/1860-5397-17-168-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Three pathways of the synthesis of 12, the decarboxylated analogue of 6a.
Scheme 8: Three pathways of the synthesis of 12, the decarboxylated analogue of 6a.
In the reaction of aminoacetal 3f with 2,3-dimethoxybenzyl bromide (13) product 12 was formed, however, it was not pure enough to be used directly in the next step of the synthesis. Purification of the crude reaction mixture by column chromatography was difficult and resulted in a moderate yield of 12 (41%) due to insufficient differences in the Rf values of compound 12 and the by-product 2,3-dimethoxybenzyl alcohol (not shown), formed through the hydrolysis of 13 (Scheme 8).
In this situation N-(2,3-dimethoxybenzyl)veratrylamine 10 was chosen as the substrate for the synthesis of the decarboxylated derivative 12 and simultaneously 10 was the expected product of its reaction with 4% HCl. The amine 10 was obtained by reductive amination of veratral (2f) with 2,3-dimethoxybenzylamine (9) with 57% yield and subsequently alkylated with bromoacetaldehyde diethyl acetal (11) to give 12 with 73% yield.
Additionally, the aminoacetal 12 was also synthesized via another route involving a double reductive amination of the aldehydes 2a and 2f with aminoacetaldehyde acetal (1) with 59% yield.
The aminoacetal 12 was then treated with 4% HCl in THF at reflux but in contrast to the chemical behavior of amino acid 6a in the same conditions (see Scheme 7), no reaction was observed. The cyclization reaction of 12 carried out in 20% HCl led, as expected, to the double-cyclized derivative 14 (Scheme 9).
![[1860-5397-17-168-i9]](/bjoc/content/inline/1860-5397-17-168-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: The chemical behavior of 12 in 4% aqueous HCl solution in THF and in 20% aqueous HCl solution.
Scheme 9: The chemical behavior of 12 in 4% aqueous HCl solution in THF and in 20% aqueous HCl solution.
To explain the difference in the reactivity between 6a and 12 we proposed a plausible mechanism for the reaction of 6a with diluted HCl (Scheme 10). The mechanism consists of four major steps: the first step is an acid-catalyzed hydrolysis of the acetal function in 6a to afford aldehyde 15; the second step is the enolization of the aldehyde 15 to form the tautomeric compound 16; the next step involves the formation of the iminium ion 17, which is then hydrolyzed to compound 8 and hydroxyacetaldehyde in the last step.
![[1860-5397-17-168-i10]](/bjoc/content/inline/1860-5397-17-168-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: A plausible mechanism of the reaction of 6a with 4% aqueous HCl solution in THF.
Scheme 10: A plausible mechanism of the reaction of 6a with 4% aqueous HCl solution in THF.
Thus, the unexpected reactivity of 6a with diluted HCl may be explained by the electron-withdrawing effect exerted by the adjacent COOH group which facilitates hydrolysis of the iminium ion 17.
Conlusion
In conclusion, a straightforward and efficient method for the synthesis of new tetrahydroisoquinoline derivatives, 7,12-dihydro-6,12-methanodibenzo[c,f]azocine-5-carboxylic acids 7a–f has been developed. It is based on the Petasis reaction with the use of aminoacetaldehyde acetals bearing substituted benzyl groups as the amine components followed by Pomeranz–Fritsch double cyclization reaction. The presence of the carboxyl group in 7a–f allows synthesis of new derivatives of these unexplored tetracyclic tetrahydroisoquinolines for biochemical studies. The unusual interesting behavior of Petasis reaction product 6a in diluted HCl solution leading to phenylglycine derivative 8 has been detected and the mechanism explaining this reactivity has been proposed.
References
-
Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669–1730. doi:10.1021/cr010212u
Return to citation in text: [1] -
Bentley, K. W. Nat. Prod. Rep. 2005, 22, 249–268. doi:10.1039/b316108k
Return to citation in text: [1] -
Bentley, K. W. The Isoquinoline Alkaloids; Harwood Academic Publishers: Amsterdam, 1998.
Return to citation in text: [1] -
Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 3341–3370. doi:10.1021/cr030692k
Return to citation in text: [1] -
Chrzanowska, M.; Grajewska, A.; Rozwadowska, M. D. Chem. Rev. 2016, 116, 12369–12465. doi:10.1021/acs.chemrev.6b00315
Return to citation in text: [1] -
Weber, E.; Keana, J.; Barmettler, P. PCP Receptor ligands and the use thereof. International Patent Application WO90/12575A1, Nov 1, 1990.
Return to citation in text: [1] -
Childers, W. E., Jr..; Abou-Gharbia, M. A. 10,11-Dihydro-5-alkyl-12-substituted-10,5-(iminomethano)-5H-dibenzo[AD]cycloheptenes as Neuroprotectant Agents. U.S. Patent US4,940,789, July 10, 1990.
Return to citation in text: [1] -
Hanessian, S.; Mauduit, M. Angew. Chem., Int. Ed. 2001, 40, 3810–3813. doi:10.1002/1521-3773(20011015)40:20<3810::aid-anie3810>3.0.co;2-8
Return to citation in text: [1] [2] -
Hanessian, S.; Parthasarathy, S.; Mauduit, M.; Payza, K. J. Med. Chem. 2003, 46, 34–48. doi:10.1021/jm020164l
Return to citation in text: [1] [2] -
Hanessian, S.; Mauduit, M.; Demont, E.; Talbot, C. Tetrahedron 2002, 58, 1485–1490. doi:10.1016/s0040-4020(02)00012-1
Return to citation in text: [1] -
Nomoto, T.; Nasui, N.; Takayama, H. J. Chem. Soc., Chem. Commun. 1984, 1646–1647. doi:10.1039/c39840001646
Return to citation in text: [1] -
Suzuki, T.; Takamoto, M.; Okamoto, T.; Takayama, H. Chem. Pharm. Bull. 1986, 34, 1888–1900. doi:10.1248/cpb.34.1888
Return to citation in text: [1] [2] -
Coşkun, N.; Buyukuysal, L. Heterocycles 1998, 48, 53–59. doi:10.3987/com-97-7986
Return to citation in text: [1] -
Bobbitt, J. M.; Shibuya, S. J. Org. Chem. 1970, 35, 1181–1183. doi:10.1021/jo00829a074
Return to citation in text: [1] -
Yamada, K.; Takeda, M.; Itoh, N.; Ohtsuka, H.; Tsunashima, A.; Iwakuma, T. Chem. Pharm. Bull. 1982, 30, 3197–3201. doi:10.1248/cpb.30.3197
Return to citation in text: [1] [2] [3] -
Mottinelli, M.; Leese, M. P.; Potter, B. V. L. Beilstein J. Org. Chem. 2017, 13, 1871–1878. doi:10.3762/bjoc.13.182
Return to citation in text: [1] -
Hara, H.; Hoshino, O.; Umezawa, B. Chem. Pharm. Bull. 1985, 33, 2705–2711. doi:10.1248/cpb.33.2705
Return to citation in text: [1] -
Buev, E. M.; Stepanov, M. A.; Moshkin, V. S.; Sosnovskikh, V. Y. Org. Lett. 2020, 22, 631–635. doi:10.1021/acs.orglett.9b04401
Return to citation in text: [1] -
Chrzanowska, M.; Grajewska, A.; Meissner, Z.; Rozwadowska, M. D.; Wiatrowska, I. Tetrahedron 2012, 68, 3092–3097. doi:10.1016/j.tet.2012.02.005
Return to citation in text: [1] -
Chrzanowska, M.; Grajewska, A.; Rozwadowska, M. D. Heterocycles 2012, 86, 1119–1127. doi:10.3987/com-12-s(n)59
Return to citation in text: [1] -
Bułyszko, I.; Chrzanowska, M.; Grajewska, A.; Rozwadowska, M. D. Eur. J. Org. Chem. 2015, 383–388. doi:10.1002/ejoc.201403218
Return to citation in text: [1]
| 1. | Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669–1730. doi:10.1021/cr010212u |
| 2. | Bentley, K. W. Nat. Prod. Rep. 2005, 22, 249–268. doi:10.1039/b316108k |
| 3. | Bentley, K. W. The Isoquinoline Alkaloids; Harwood Academic Publishers: Amsterdam, 1998. |
| 10. | Hanessian, S.; Mauduit, M.; Demont, E.; Talbot, C. Tetrahedron 2002, 58, 1485–1490. doi:10.1016/s0040-4020(02)00012-1 |
| 11. | Nomoto, T.; Nasui, N.; Takayama, H. J. Chem. Soc., Chem. Commun. 1984, 1646–1647. doi:10.1039/c39840001646 |
| 15. | Yamada, K.; Takeda, M.; Itoh, N.; Ohtsuka, H.; Tsunashima, A.; Iwakuma, T. Chem. Pharm. Bull. 1982, 30, 3197–3201. doi:10.1248/cpb.30.3197 |
| 8. | Hanessian, S.; Mauduit, M. Angew. Chem., Int. Ed. 2001, 40, 3810–3813. doi:10.1002/1521-3773(20011015)40:20<3810::aid-anie3810>3.0.co;2-8 |
| 9. | Hanessian, S.; Parthasarathy, S.; Mauduit, M.; Payza, K. J. Med. Chem. 2003, 46, 34–48. doi:10.1021/jm020164l |
| 6. | Weber, E.; Keana, J.; Barmettler, P. PCP Receptor ligands and the use thereof. International Patent Application WO90/12575A1, Nov 1, 1990. |
| 7. | Childers, W. E., Jr..; Abou-Gharbia, M. A. 10,11-Dihydro-5-alkyl-12-substituted-10,5-(iminomethano)-5H-dibenzo[AD]cycloheptenes as Neuroprotectant Agents. U.S. Patent US4,940,789, July 10, 1990. |
| 21. | Bułyszko, I.; Chrzanowska, M.; Grajewska, A.; Rozwadowska, M. D. Eur. J. Org. Chem. 2015, 383–388. doi:10.1002/ejoc.201403218 |
| 4. | Chrzanowska, M.; Rozwadowska, M. D. Chem. Rev. 2004, 104, 3341–3370. doi:10.1021/cr030692k |
| 5. | Chrzanowska, M.; Grajewska, A.; Rozwadowska, M. D. Chem. Rev. 2016, 116, 12369–12465. doi:10.1021/acs.chemrev.6b00315 |
| 15. | Yamada, K.; Takeda, M.; Itoh, N.; Ohtsuka, H.; Tsunashima, A.; Iwakuma, T. Chem. Pharm. Bull. 1982, 30, 3197–3201. doi:10.1248/cpb.30.3197 |
| 17. | Hara, H.; Hoshino, O.; Umezawa, B. Chem. Pharm. Bull. 1985, 33, 2705–2711. doi:10.1248/cpb.33.2705 |
| 19. | Chrzanowska, M.; Grajewska, A.; Meissner, Z.; Rozwadowska, M. D.; Wiatrowska, I. Tetrahedron 2012, 68, 3092–3097. doi:10.1016/j.tet.2012.02.005 |
| 12. | Suzuki, T.; Takamoto, M.; Okamoto, T.; Takayama, H. Chem. Pharm. Bull. 1986, 34, 1888–1900. doi:10.1248/cpb.34.1888 |
| 14. | Bobbitt, J. M.; Shibuya, S. J. Org. Chem. 1970, 35, 1181–1183. doi:10.1021/jo00829a074 |
| 15. | Yamada, K.; Takeda, M.; Itoh, N.; Ohtsuka, H.; Tsunashima, A.; Iwakuma, T. Chem. Pharm. Bull. 1982, 30, 3197–3201. doi:10.1248/cpb.30.3197 |
| 16. | Mottinelli, M.; Leese, M. P.; Potter, B. V. L. Beilstein J. Org. Chem. 2017, 13, 1871–1878. doi:10.3762/bjoc.13.182 |
| 20. | Chrzanowska, M.; Grajewska, A.; Rozwadowska, M. D. Heterocycles 2012, 86, 1119–1127. doi:10.3987/com-12-s(n)59 |
| 12. | Suzuki, T.; Takamoto, M.; Okamoto, T.; Takayama, H. Chem. Pharm. Bull. 1986, 34, 1888–1900. doi:10.1248/cpb.34.1888 |
| 13. | Coşkun, N.; Buyukuysal, L. Heterocycles 1998, 48, 53–59. doi:10.3987/com-97-7986 |
| 8. | Hanessian, S.; Mauduit, M. Angew. Chem., Int. Ed. 2001, 40, 3810–3813. doi:10.1002/1521-3773(20011015)40:20<3810::aid-anie3810>3.0.co;2-8 |
| 9. | Hanessian, S.; Parthasarathy, S.; Mauduit, M.; Payza, K. J. Med. Chem. 2003, 46, 34–48. doi:10.1021/jm020164l |
| 18. | Buev, E. M.; Stepanov, M. A.; Moshkin, V. S.; Sosnovskikh, V. Y. Org. Lett. 2020, 22, 631–635. doi:10.1021/acs.orglett.9b04401 |
© 2021 Grajewska et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)