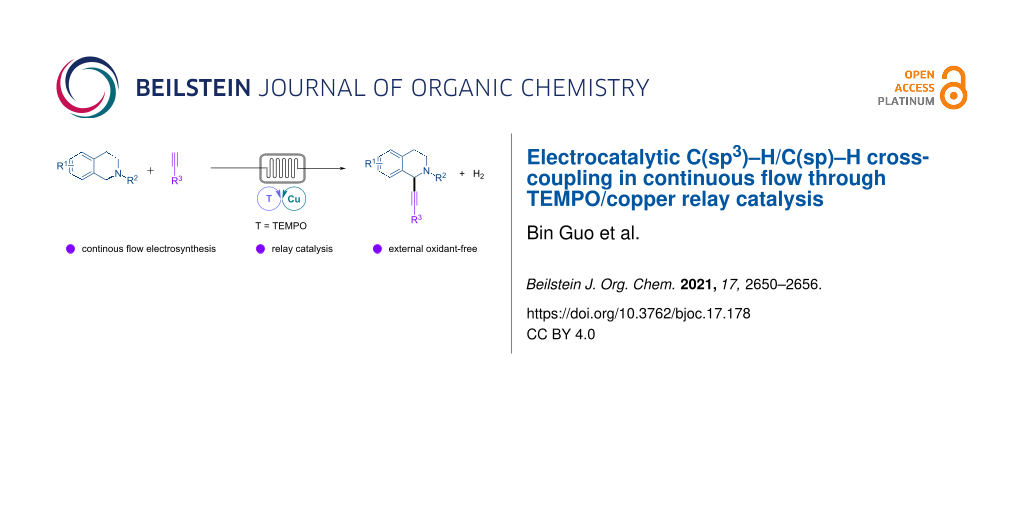
Abstract
Electrocatalytic dehydrogenative C(sp3)–H/C(sp)–H cross-coupling of tetrahydroisoquinolines with terminal alkynes has been achieved in a continuous-flow microreactor through 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)/copper relay catalysis. The reaction is easily scalable and requires low concentration of supporting electrolyte and no external chemical oxidants or ligands, providing straightforward and sustainable access to 2-functionalized tetrahydroisoquinolines.

Graphical Abstract
Introduction
The dehydrogenative cross-coupling of two C–H bonds represents an ideal strategy for the construction of C–C bonds [1,2]. In this context, few methods have been developed for the dehydrogenative cross-coupling of tetrahydroisoquinolines with terminal alkynes because of the prevalence of the tetrahydroisoquinoline moiety in natural products and bioactive molecules [3-10]. These methods proceed through the oxidation of the tetrahydroisoquinoline to an iminium intermediate with various chemical oxidants such as peroxides and DDQ followed by reaction with the copper acetylide species to deliver the 2-substituted tetrahydroisoquinoline product (Scheme 1A). These methods usually require elevated temperatures [3-5], prompting the development of mild conditions by merging photoredox catalysis with copper catalysis (Scheme 1B) [8,9]. Notwithstanding of these outstanding achievements, noble metal-based catalysts and chemical oxidants are employed under these photochemical conditions.
![[1860-5397-17-178-i1]](/bjoc/content/inline/1860-5397-17-178-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: C(sp3)–H alkynylation of tetrahydroisoquinolines. L* = chiral ligand. TEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone. BPO = benzoyl peroxide.
Scheme 1: C(sp3)–H alkynylation of tetrahydroisoquinolines. L* = chiral ligand. TEMPO = 2,2,6,6-tetramethylpi...
Organic electrochemistry is an ideal tool for promoting dehydrogenative cross-coupling reactions as no external chemical oxidants are needed [11-19]. In this context, Mei and co-workers have reported an elegant TEMPO/[L*Cu] co-catalyzed asymmetric electrochemical dehydrogenative cross-coupling reaction of tetrahydroisoquinolines with terminal alkynes (Scheme 1C) [10]. The chiral ligand was found to be critical for the stereoinduction as well as product formation for these electrochemical reactions that are conducted in batch. Continuous-flow electrochemical microreactors offer several advantages for electrosynthesis and have been employed to reduce the use of supporting electrolyte, facilitate reaction scale-up, and increase reaction efficiency [20-32]. Despite these advantages of continuous-flow electrosynthesis and the intense interests in transition-metal electrocatalysis [33-39], transition-metal electrocatalysis in continuous flow remains underexplored [40]. With our continued interests in transition-metal electrocatalysis [41,42] and continuous-flow electrosynthesis [43-48], we report herein the electrocatalytic dehydrogenative cross-coupling reaction of tetrahydroisoquinolines with terminal alkynes in continuous flow (Scheme 1D). These reactions require low loadings of supporting electrolyte and proceed through Cu/TEMPO relay catalysis without need for additional ligands.
Results and Discussion
The electrosynthesis was conducted in a microreactor equipped with two Pt electrodes as the anode and cathode and operated with a constant current (Table 1). Under the optimized conditions, a solution of tetrahydroisoquinoline 1a (1 equiv), alkyne 2 (1.5 equiv), Cu(OTf)2 (10 mol %), TEMPO (20 mol %), n-Bu4NPF6 (0.2 equiv), and TFE (3.5 equiv) in MeCN was passed through the cell at 0.2 mL min−1 to give the desired product 3 in 86% yield (Table 1, entry 1). Pleasingly, a good yield of 82% was obtained in the absence of supporting electrolyte (Table 1, entry 2). While product formation was observed without TEMPO (Table 1, entry 3) and TFE (Table 1, entry 4), albeit in low yields, the reaction failed completely without the copper salt (Table 1, entry 5). Other variations also resulted in diminished yield of 3, such as lowering the loading of Cu(OTf)2 to 5 mol % (Table 1, entry 6), replacing Cu(OTf)2 with other copper salts such as Cu(acac)2 (Table 1, entry 7), Cu(TFA)2, (Table 1, entry 8), Cu(OAc)2 (Table 1, entry 9) and replacing TFE with other protic additives including MeOH (Table 1, entry 10), EtOH (Table 1, entry 11), HFIP (Table 1, entry 12) and H2O (Table 1, entry 13).
Table 1: Optimization of reaction conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-178-i5.svg?max-width=637&scale=1.0)
|
||
| Entry | Deviation from standard conditions | Yield of 3 (%) |
| 1 | none | 86b |
| 2 | no n-Bu4NPF6 | 82 |
| 3 | no TEMPO | 35 |
| 4 | no TFE | 19 |
| 5 | no Cu(OTf)2 | 0 |
| 6 | Cu(OTf)2 (5 mol %) | 71 |
| 7 | Cu(acac)2 instead of Cu(OTf)2 | 17 |
| 8 | Cu(TFA)2 instead of Cu(OTf)2 | 77 |
| 9 | Cu(OAc)2 instead of Cu(OTf)2 | 40 |
| 10 | MeOH instead of TFE | 60 |
| 11 | EtOH instead of TFE | 50 |
| 12 | HFIP instead of TFE | 38 |
| 13 | H2O instead of TFE | 20 |
aStandard conditions: 1a (0.21 mmol), 2 (0.32 mmol, 1.5 equiv), MeCN (7 mL), Pt anode, Pt cathode, interelectrode distance = 0.25 mm, 3.1 F mol−1. Yield of product 3 is determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard. TFE, 2,2,2-trifluoroethanol. PMP, p-methoxyphenyl. HFIP, 1,1,1,3,3,3-hexafluoropropan-2-ol. TEMPO, 2,2,6,6-tetramethylpiperidine 1-oxyl. Cu(acac)2, Copper(II) acetylacetonate. Cu(TFA)2, Copper(II) trifluoroacetate. bIsolated yield.
The scope of the continuous-flow electrosynthesis was investigated by varying the substituents of the tetrahydroisoquinoline and the alkyne (Scheme 2). The N-phenyl ring of the tetrahydroisoquinoline could be substituted with groups such as OMe (4, 5), Me (6), Et (7), t-Bu (8), F (9), and Cl (10). An N-2-naphthalenyl-substituted tetrahydroisoquinoline bearing two OMe groups at 6,7-positions (11) also reacted successfully. The alkyne coupling partner also tolerated variation. The reactions were found to be compatible with arylalkynes such as phenylacetylenes bearing at the para position a H (12), Me (13), t-Bu (14, 16), or Br (15), 2-ethynylpyridine (17), alkenylalkynes (18), and alkylalkynes (19–21).
![[1860-5397-17-178-i2]](/bjoc/content/inline/1860-5397-17-178-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrate scope. Reaction conditions: Pt anode, Pt cathode, interelectrode distance 0.25 mm, 1 (0.03 M, 0.21 mmol), 2 (0.045 M, 1.5 equiv), Cu(OTf)2 (10 mol %), TEMPO (20 mol %), n-Bu4NPF6 (20 mol %), TFE (3.5 equiv), MeCN (7 mL), I = 30 mA, flow rate = 0.20 mL min−1, rt. Isolated yields are reported.
Scheme 2: Substrate scope. Reaction conditions: Pt anode, Pt cathode, interelectrode distance 0.25 mm, 1 (0.0...
The continuous-flow electrosynthesis is easily scaled up by passing more material through the reactor [43,49]. Hence, repeating the reaction under flow conditions, with a solution containing 0.98 g of tetrahydroisoquinoline 1a and 1.11 g of alkyne 22 afforded 1.05 g (61%) of product 14 in 13 h (Scheme 3). The productivity could be increased if multiple reactors were employed in parallel [43].
![[1860-5397-17-178-i3]](/bjoc/content/inline/1860-5397-17-178-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
A mechanism for the electrochemical synthesis was proposed based on reported studies (Scheme 4) [3,10]. Anodic oxidation of TEMPO generates the oxoammonium salt TEMPO+ [50,51], which reacts with tetrahydroisoquinoline 23 to generate TEMPOH and iminium ion 24 [52], TEMPOH is oxidized back to TEMPO+ on the anode. On the other hand, 24 is converted to the final product 25 through reaction with copper acetylide 26, which is generated from CuI and the alkyne 27 with the assistance of CF3CH2O−. The added CuII precatalyst is likely reduced at the cathode to produce the requisite CuI. The base CF3CH2O− is produced through cathodic reduction of TFE. The addition of TFE to the reactions helps cathodic H2 evolution and may also stabilize the iminium ion through reversible reaction with this cationic species.
![[1860-5397-17-178-i4]](/bjoc/content/inline/1860-5397-17-178-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Conclusion
In summary, we have achieved the electrochemical dehydrogenation cross-coupling of tetrahydroisoquinolines with terminal alkynes in continuous flow through Cu/TEMPO relay catalysis. This work demonstrates that continuous-flow electrochemical microreactors can be a viable tool for developing efficient transition-metal electrocatalysis.
Supporting Information
| Supporting Information File 1: General procedure, characterization data for electrolysis products and NMR spectra. | ||
| Format: PDF | Size: 5.0 MB | Download |
References
-
Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n
Return to citation in text: [1] -
Wang, H.; Gao, X.; Lv, Z.; Abdelilah, T.; Lei, A. Chem. Rev. 2019, 119, 6769–6787. doi:10.1021/acs.chemrev.9b00045
Return to citation in text: [1] -
Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763
Return to citation in text: [1] [2] [3] -
Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997–4999. doi:10.1021/ol047814v
Return to citation in text: [1] [2] -
Li, Z.; MacLeod, P. D.; Li, C.-J. Tetrahedron: Asymmetry 2006, 17, 590–597. doi:10.1016/j.tetasy.2006.02.007
Return to citation in text: [1] [2] -
Su, W.; Yu, J.; Li, Z.; Jiang, Z. J. Org. Chem. 2011, 76, 9144–9150. doi:10.1021/jo2015533
Return to citation in text: [1] -
Yu, J.; Li, Z.; Jia, K.; Jiang, Z.; Liu, M.; Su, W. Tetrahedron Lett. 2013, 54, 2006–2009. doi:10.1016/j.tetlet.2013.02.007
Return to citation in text: [1] -
Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050
Return to citation in text: [1] [2] -
Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/c4ob02138j
Return to citation in text: [1] [2] -
Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. Angew. Chem., Int. Ed. 2020, 59, 15254–15259. doi:10.1002/anie.202005099
Return to citation in text: [1] [2] [3] -
Yuan, Y.; Lei, A. Acc. Chem. Res. 2019, 52, 3309–3324. doi:10.1021/acs.accounts.9b00512
Return to citation in text: [1] -
Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397
Return to citation in text: [1] -
Moeller, K. D. Chem. Rev. 2018, 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656
Return to citation in text: [1] -
Röckl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45–61. doi:10.1021/acs.accounts.9b00511
Return to citation in text: [1] -
Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339–3350. doi:10.1021/acs.accounts.9b00472
Return to citation in text: [1] -
Chen, N.; Xu, H.-C. Green Synth. Catal. 2021, 2, 165–178. doi:10.1016/j.gresc.2021.03.002
Return to citation in text: [1] -
Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271
Return to citation in text: [1] -
Shi, S.-H.; Liang, Y.; Jiao, N. Chem. Rev. 2021, 121, 485–505. doi:10.1021/acs.chemrev.0c00335
Return to citation in text: [1] -
Ye, Z.; Zhang, F. Chin. J. Chem. 2019, 37, 513–528. doi:10.1002/cjoc.201900049
Return to citation in text: [1] -
Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497
Return to citation in text: [1] -
Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412
Return to citation in text: [1] -
Atobe, M.; Tateno, H.; Matsumura, Y. Chem. Rev. 2018, 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353
Return to citation in text: [1] -
Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360
Return to citation in text: [1] -
Laudadio, G.; Bartolomeu, A. d. A.; Verwijlen, L. M. H. M.; Cao, Y.; de Oliveira, K. T.; Noël, T. J. Am. Chem. Soc. 2019, 141, 11832–11836. doi:10.1021/jacs.9b06126
Return to citation in text: [1] -
Laudadio, G.; Barmpoutsis, E.; Schotten, C.; Struik, L.; Govaerts, S.; Browne, D. L.; Noël, T. J. Am. Chem. Soc. 2019, 141, 5664–5668. doi:10.1021/jacs.9b02266
Return to citation in text: [1] -
Amri, N.; Wirth, T. Synthesis 2020, 52, 1751–1761. doi:10.1055/s-0039-1690868
Return to citation in text: [1] -
Folgueiras-Amador, A. A.; Qian, X.-Y.; Xu, H.-C.; Wirth, T. Chem. – Eur. J. 2018, 24, 487–491. doi:10.1002/chem.201705016
Return to citation in text: [1] -
Watts, K.; Gattrell, W.; Wirth, T. Beilstein J. Org. Chem. 2011, 7, 1108–1114. doi:10.3762/bjoc.7.127
Return to citation in text: [1] -
Folgueiras-Amador, A. A.; Philipps, K.; Guilbaud, S.; Poelakker, J.; Wirth, T. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. doi:10.1002/anie.201709717
Return to citation in text: [1] -
Hielscher, M. M.; Gleede, B.; Waldvogel, S. R. Electrochim. Acta 2021, 368, 137420. doi:10.1016/j.electacta.2020.137420
Return to citation in text: [1] -
Lin, X.; Fang, Z.; Zeng, C.; Zhu, C.; Pang, X.; Liu, C.; He, W.; Duan, J.; Qin, N.; Guo, K. Chem. – Eur. J. 2020, 26, 13738–13742. doi:10.1002/chem.202001766
Return to citation in text: [1] -
Mo, Y.; Lu, Z.; Rughoobur, G.; Patil, P.; Gershenfeld, N.; Akinwande, A. I.; Buchwald, S. L.; Jensen, K. F. Science 2020, 368, 1352–1357. doi:10.1126/science.aba3823
Return to citation in text: [1] -
Qiu, Y.; Zhu, C.; Stangier, M.; Struwe, J.; Ackermann, L. CCS Chem. 2021, 3, 1529–1552. doi:10.31635/ccschem.020.202000365
Return to citation in text: [1] -
Ackermann, L. Acc. Chem. Res. 2020, 53, 84–104. doi:10.1021/acs.accounts.9b00510
Return to citation in text: [1] -
Meyer, T. H.; Finger, L. H.; Gandeepan, P.; Ackermann, L. Trends Chem. 2019, 1, 63–76. doi:10.1016/j.trechm.2019.01.011
Return to citation in text: [1] -
Gandeepan, P.; Finger, L. H.; Meyer, T. H.; Ackermann, L. Chem. Soc. Rev. 2020, 49, 4254–4272. doi:10.1039/d0cs00149j
Return to citation in text: [1] -
Ma, C.; Fang, P.; Liu, Z.-R.; Xu, S.-S.; Xu, K.; Cheng, X.; Lei, A.; Xu, H.-C.; Zeng, C.; Mei, T.-S. Sci. Bull. 2021, in press. doi:10.1016/j.scib.2021.07.011
Return to citation in text: [1] -
Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300–310. doi:10.1021/acs.accounts.9b00603
Return to citation in text: [1] -
Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338–352. doi:10.1002/cjoc.201700740
Return to citation in text: [1] -
Kong, W.-J.; Finger, L. H.; Messinis, A. M.; Kuniyil, R.; Oliveira, J. C. A.; Ackermann, L. J. Am. Chem. Soc. 2019, 141, 17198–17206. doi:10.1021/jacs.9b07763
Return to citation in text: [1] -
Xu, F.; Li, Y.-J.; Huang, C.; Xu, H.-C. ACS Catal. 2018, 8, 3820–3824. doi:10.1021/acscatal.8b00373
Return to citation in text: [1] -
Wu, Z.-J.; Su, F.; Lin, W.; Song, J.; Wen, T.-B.; Zhang, H.-J.; Xu, H.-C. Angew. Chem., Int. Ed. 2019, 58, 16770–16774. doi:10.1002/anie.201909951
Return to citation in text: [1] -
Huang, C.; Li, Z.-Y.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2021, 60, 11237–11241. doi:10.1002/anie.202101835
Return to citation in text: [1] [2] [3] -
Yan, H.; Song, J.; Zhu, S.; Xu, H.-C. CCS Chem. 2021, 3, 317–325. doi:10.31635/ccschem.021.202000743
Return to citation in text: [1] -
Yan, H.; Zhu, S.; Xu, H.-C. Org. Process Res. Dev. 2021, in press. doi:10.1021/acs.oprd.1c00038
Return to citation in text: [1] -
Huang, C.; Xu, H.-C. Sci. China: Chem. 2019, 62, 1501–1503. doi:10.1007/s11426-019-9554-1
Return to citation in text: [1] -
Huang, C.; Qian, X.-Y.; Xu, H.-C. Angew. Chem., Int. Ed. 2019, 58, 6650–6653. doi:10.1002/anie.201901610
Return to citation in text: [1] -
Xu, F.; Qian, X.-Y.; Li, Y.-J.; Xu, H.-C. Org. Lett. 2017, 19, 6332–6335. doi:10.1021/acs.orglett.7b03152
Return to citation in text: [1] -
Dong, Z.; Wen, Z.; Zhao, F.; Kuhn, S.; Noël, T. Chem. Eng. Sci.: X 2021, 10, 100097. doi:10.1016/j.cesx.2021.100097
Return to citation in text: [1] -
Nutting, J. E.; Rafiee, M.; Stahl, S. S. Chem. Rev. 2018, 118, 4834–4885. doi:10.1021/acs.chemrev.7b00763
Return to citation in text: [1] -
Xu, F.; Zhu, L.; Zhu, S.; Yan, X.; Xu, H.-C. Chem. – Eur. J. 2014, 20, 12740–12744. doi:10.1002/chem.201404078
Return to citation in text: [1] -
Wang, F.; Stahl, S. S. Acc. Chem. Res. 2020, 53, 561–574. doi:10.1021/acs.accounts.9b00544
Return to citation in text: [1]
| 1. | Li, C.-J. Acc. Chem. Res. 2009, 42, 335–344. doi:10.1021/ar800164n |
| 2. | Wang, H.; Gao, X.; Lv, Z.; Abdelilah, T.; Lei, A. Chem. Rev. 2019, 119, 6769–6787. doi:10.1021/acs.chemrev.9b00045 |
| 11. | Yuan, Y.; Lei, A. Acc. Chem. Res. 2019, 52, 3309–3324. doi:10.1021/acs.accounts.9b00512 |
| 12. | Yan, M.; Kawamata, Y.; Baran, P. S. Chem. Rev. 2017, 117, 13230–13319. doi:10.1021/acs.chemrev.7b00397 |
| 13. | Moeller, K. D. Chem. Rev. 2018, 118, 4817–4833. doi:10.1021/acs.chemrev.7b00656 |
| 14. | Röckl, J. L.; Pollok, D.; Franke, R.; Waldvogel, S. R. Acc. Chem. Res. 2020, 53, 45–61. doi:10.1021/acs.accounts.9b00511 |
| 15. | Xiong, P.; Xu, H.-C. Acc. Chem. Res. 2019, 52, 3339–3350. doi:10.1021/acs.accounts.9b00472 |
| 16. | Chen, N.; Xu, H.-C. Green Synth. Catal. 2021, 2, 165–178. doi:10.1016/j.gresc.2021.03.002 |
| 17. | Jiang, Y.; Xu, K.; Zeng, C. Chem. Rev. 2018, 118, 4485–4540. doi:10.1021/acs.chemrev.7b00271 |
| 18. | Shi, S.-H.; Liang, Y.; Jiao, N. Chem. Rev. 2021, 121, 485–505. doi:10.1021/acs.chemrev.0c00335 |
| 19. | Ye, Z.; Zhang, F. Chin. J. Chem. 2019, 37, 513–528. doi:10.1002/cjoc.201900049 |
| 50. | Nutting, J. E.; Rafiee, M.; Stahl, S. S. Chem. Rev. 2018, 118, 4834–4885. doi:10.1021/acs.chemrev.7b00763 |
| 51. | Xu, F.; Zhu, L.; Zhu, S.; Yan, X.; Xu, H.-C. Chem. – Eur. J. 2014, 20, 12740–12744. doi:10.1002/chem.201404078 |
| 8. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 9. | Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/c4ob02138j |
| 52. | Wang, F.; Stahl, S. S. Acc. Chem. Res. 2020, 53, 561–574. doi:10.1021/acs.accounts.9b00544 |
| 3. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763 |
| 4. | Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997–4999. doi:10.1021/ol047814v |
| 5. | Li, Z.; MacLeod, P. D.; Li, C.-J. Tetrahedron: Asymmetry 2006, 17, 590–597. doi:10.1016/j.tetasy.2006.02.007 |
| 43. | Huang, C.; Li, Z.-Y.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2021, 60, 11237–11241. doi:10.1002/anie.202101835 |
| 3. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763 |
| 4. | Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997–4999. doi:10.1021/ol047814v |
| 5. | Li, Z.; MacLeod, P. D.; Li, C.-J. Tetrahedron: Asymmetry 2006, 17, 590–597. doi:10.1016/j.tetasy.2006.02.007 |
| 6. | Su, W.; Yu, J.; Li, Z.; Jiang, Z. J. Org. Chem. 2011, 76, 9144–9150. doi:10.1021/jo2015533 |
| 7. | Yu, J.; Li, Z.; Jia, K.; Jiang, Z.; Liu, M.; Su, W. Tetrahedron Lett. 2013, 54, 2006–2009. doi:10.1016/j.tetlet.2013.02.007 |
| 8. | Rueping, M.; Koenigs, R. M.; Poscharny, K.; Fabry, D. C.; Leonori, D.; Vila, C. Chem. – Eur. J. 2012, 18, 5170–5174. doi:10.1002/chem.201200050 |
| 9. | Perepichka, I.; Kundu, S.; Hearne, Z.; Li, C.-J. Org. Biomol. Chem. 2015, 13, 447–451. doi:10.1039/c4ob02138j |
| 10. | Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. Angew. Chem., Int. Ed. 2020, 59, 15254–15259. doi:10.1002/anie.202005099 |
| 3. | Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810–11811. doi:10.1021/ja0460763 |
| 10. | Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. Angew. Chem., Int. Ed. 2020, 59, 15254–15259. doi:10.1002/anie.202005099 |
| 40. | Kong, W.-J.; Finger, L. H.; Messinis, A. M.; Kuniyil, R.; Oliveira, J. C. A.; Ackermann, L. J. Am. Chem. Soc. 2019, 141, 17198–17206. doi:10.1021/jacs.9b07763 |
| 43. | Huang, C.; Li, Z.-Y.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2021, 60, 11237–11241. doi:10.1002/anie.202101835 |
| 44. | Yan, H.; Song, J.; Zhu, S.; Xu, H.-C. CCS Chem. 2021, 3, 317–325. doi:10.31635/ccschem.021.202000743 |
| 45. | Yan, H.; Zhu, S.; Xu, H.-C. Org. Process Res. Dev. 2021, in press. doi:10.1021/acs.oprd.1c00038 |
| 46. | Huang, C.; Xu, H.-C. Sci. China: Chem. 2019, 62, 1501–1503. doi:10.1007/s11426-019-9554-1 |
| 47. | Huang, C.; Qian, X.-Y.; Xu, H.-C. Angew. Chem., Int. Ed. 2019, 58, 6650–6653. doi:10.1002/anie.201901610 |
| 48. | Xu, F.; Qian, X.-Y.; Li, Y.-J.; Xu, H.-C. Org. Lett. 2017, 19, 6332–6335. doi:10.1021/acs.orglett.7b03152 |
| 33. | Qiu, Y.; Zhu, C.; Stangier, M.; Struwe, J.; Ackermann, L. CCS Chem. 2021, 3, 1529–1552. doi:10.31635/ccschem.020.202000365 |
| 34. | Ackermann, L. Acc. Chem. Res. 2020, 53, 84–104. doi:10.1021/acs.accounts.9b00510 |
| 35. | Meyer, T. H.; Finger, L. H.; Gandeepan, P.; Ackermann, L. Trends Chem. 2019, 1, 63–76. doi:10.1016/j.trechm.2019.01.011 |
| 36. | Gandeepan, P.; Finger, L. H.; Meyer, T. H.; Ackermann, L. Chem. Soc. Rev. 2020, 49, 4254–4272. doi:10.1039/d0cs00149j |
| 37. | Ma, C.; Fang, P.; Liu, Z.-R.; Xu, S.-S.; Xu, K.; Cheng, X.; Lei, A.; Xu, H.-C.; Zeng, C.; Mei, T.-S. Sci. Bull. 2021, in press. doi:10.1016/j.scib.2021.07.011 |
| 38. | Jiao, K.-J.; Xing, Y.-K.; Yang, Q.-L.; Qiu, H.; Mei, T.-S. Acc. Chem. Res. 2020, 53, 300–310. doi:10.1021/acs.accounts.9b00603 |
| 39. | Yang, Q.-L.; Fang, P.; Mei, T.-S. Chin. J. Chem. 2018, 36, 338–352. doi:10.1002/cjoc.201700740 |
| 43. | Huang, C.; Li, Z.-Y.; Song, J.; Xu, H.-C. Angew. Chem., Int. Ed. 2021, 60, 11237–11241. doi:10.1002/anie.202101835 |
| 49. | Dong, Z.; Wen, Z.; Zhao, F.; Kuhn, S.; Noël, T. Chem. Eng. Sci.: X 2021, 10, 100097. doi:10.1016/j.cesx.2021.100097 |
| 20. | Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497 |
| 21. | Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412 |
| 22. | Atobe, M.; Tateno, H.; Matsumura, Y. Chem. Rev. 2018, 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353 |
| 23. | Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360 |
| 24. | Laudadio, G.; Bartolomeu, A. d. A.; Verwijlen, L. M. H. M.; Cao, Y.; de Oliveira, K. T.; Noël, T. J. Am. Chem. Soc. 2019, 141, 11832–11836. doi:10.1021/jacs.9b06126 |
| 25. | Laudadio, G.; Barmpoutsis, E.; Schotten, C.; Struik, L.; Govaerts, S.; Browne, D. L.; Noël, T. J. Am. Chem. Soc. 2019, 141, 5664–5668. doi:10.1021/jacs.9b02266 |
| 26. | Amri, N.; Wirth, T. Synthesis 2020, 52, 1751–1761. doi:10.1055/s-0039-1690868 |
| 27. | Folgueiras-Amador, A. A.; Qian, X.-Y.; Xu, H.-C.; Wirth, T. Chem. – Eur. J. 2018, 24, 487–491. doi:10.1002/chem.201705016 |
| 28. | Watts, K.; Gattrell, W.; Wirth, T. Beilstein J. Org. Chem. 2011, 7, 1108–1114. doi:10.3762/bjoc.7.127 |
| 29. | Folgueiras-Amador, A. A.; Philipps, K.; Guilbaud, S.; Poelakker, J.; Wirth, T. Angew. Chem., Int. Ed. 2017, 56, 15446–15450. doi:10.1002/anie.201709717 |
| 30. | Hielscher, M. M.; Gleede, B.; Waldvogel, S. R. Electrochim. Acta 2021, 368, 137420. doi:10.1016/j.electacta.2020.137420 |
| 31. | Lin, X.; Fang, Z.; Zeng, C.; Zhu, C.; Pang, X.; Liu, C.; He, W.; Duan, J.; Qin, N.; Guo, K. Chem. – Eur. J. 2020, 26, 13738–13742. doi:10.1002/chem.202001766 |
| 32. | Mo, Y.; Lu, Z.; Rughoobur, G.; Patil, P.; Gershenfeld, N.; Akinwande, A. I.; Buchwald, S. L.; Jensen, K. F. Science 2020, 368, 1352–1357. doi:10.1126/science.aba3823 |
| 10. | Gao, P.-S.; Weng, X.-J.; Wang, Z.-H.; Zheng, C.; Sun, B.; Chen, Z.-H.; You, S.-L.; Mei, T.-S. Angew. Chem., Int. Ed. 2020, 59, 15254–15259. doi:10.1002/anie.202005099 |
| 41. | Xu, F.; Li, Y.-J.; Huang, C.; Xu, H.-C. ACS Catal. 2018, 8, 3820–3824. doi:10.1021/acscatal.8b00373 |
| 42. | Wu, Z.-J.; Su, F.; Lin, W.; Song, J.; Wen, T.-B.; Zhang, H.-J.; Xu, H.-C. Angew. Chem., Int. Ed. 2019, 58, 16770–16774. doi:10.1002/anie.201909951 |
© 2021 Guo and Xu; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)