Abstract
Chiral cyclam (1,4,8,11-tetraazacyclotetradecane) derivatives were synthesized stepwise from chiral mono-Boc-1,2-diamines and (dialkyl)malonyl dichloride via open diamide-bis(N-Boc-amino) intermediates (65–91%). Deprotection and ring closure with a second malonyl unit afforded the cyclam tetraamide precursors (80–95%). The new protocol allowed the preparation of the target cyclam derivatives (53–59%) by a final optimized hydride reduction. Both the open tetraamine intermediates and the cyclam derivatives successfully coordinated with AuCl3 to give moderate to excellent yields (50–96%) of the corresponding novel tetra-coordinated N,N,N,N-Au(III) complexes with alternating five- and six-membered chelate rings. The testing of the catalytic ability of the cyclam-based N,N,N,N-Au(III) complexes demonstrated high catalytic activity of some complexes in selected test reactions (full conversion in 1–24 h, 62–97% product yields).

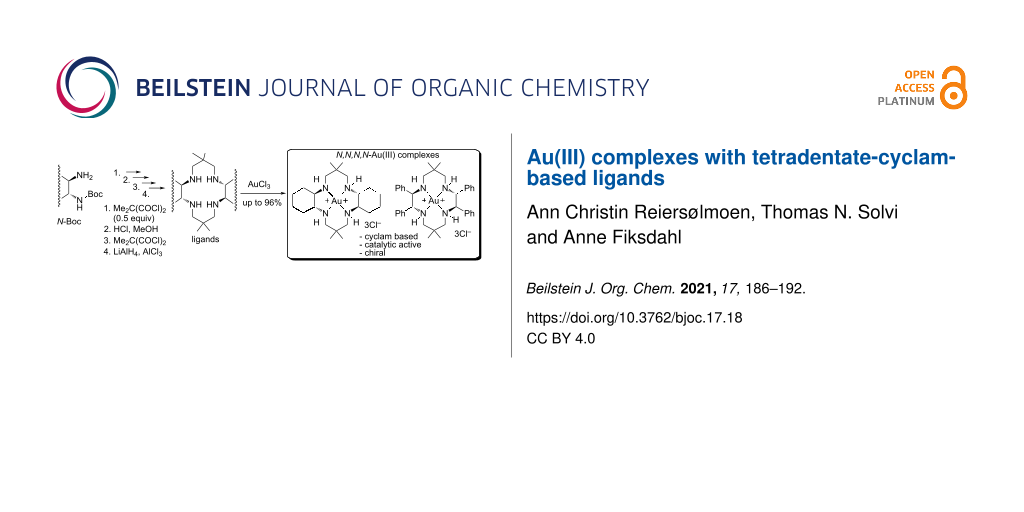
Graphical Abstract
Introduction
The importance of gold for humankind dates long back, and gold is linked to the evolution of many parts of the society. Contrary to the general fascination and importance of gold, the potential as homogenous catalyst has been neglected, compared to a range of other transition metals. The utilization of gold in synthetic organic chemistry has become a topic of interest during the last decades, as evidenced by the increasing number of review articles published in this period [1-8]. Whereas both gold(I) and gold(III) are proven to be catalytic active forms of gold, gold(I) has so far, received main attention, likely due to the higher stability, as demonstrated by the development of a high number of gold(I)-catalyzed transformations and ligated gold(I) complexes, along with improved mechanistic understanding [9-15]. In contrast, gold(III) catalysis was for a long time mostly based on inorganic salts, such as AuCl3, AuBr3, or pyridine–AuCl3 and Pic–AuCl2. However, Au(III) complexes with various coordinated ligands are about to become more explored. Different from the linear coordination mode of gold(I), gold(III) forms square planar complexes. This allows for greater steric control around the reaction center by using polydentate ligands. An interesting group of ligands which may coordinate to all the four coordination sites of gold(III), are represented by polyamine ligands, such as cyclam (1,4,8,11-tetraazacyclotetradecane), cyclen (1,4,7,10-tetraazacyclododecane)), ethylenediamine and triethylenetetraamine derivatives. Studies of Au(III)-cyclam modified complexes have been limited to arylated [16] or polymer-bound cyclams for selective uptake of Au(III) from water [17] as well as X-ray crystal structures [18-21]. Simple [Au(III)–cyclam] complexes have been investigated for various biological properties [22-29]. Particularly, studies have focused on their potential in vitro anticancer properties [24,26,29], the activity against a falciparum strain [22], the in vitro DNA binding properties [25] and reactions with bovine serum albumin [27].
Cyclam is known as a tetraamino-macrocyclic ligand, which binds strongly to give complexes with many transition metal cations. While catalytic applications of square planar cyclam complexes are reported for metals, such as Ni [30-33], Cu [34], Fe [35], catalytic properties of cyclam coordinated gold(III) complexes are not known. Trigged by this knowledge gap, we wanted to develop new chiral cyclam coordinated gold(III) complexes. Additionally, these complexes were interesting for the evaluation of the catalytic effect of the Au(III) complex upon substitution of all coordinating halides by nitrogen donors. We hereby present the synthesis of chiral cyclam ligands and related polyamino compounds, along with Au(III) coordination studies and evaluation of the catalytic ability of the successfully obtained Au(III) complexes in two model reactions.
Results and Discussion
Synthesis of potential ligands
Chiral cyclam derivatives have previously been directly synthesized from (1R,2R)-cyclohexane-1,2-diamine (A) and malonyl dichloride [36], giving 36% yield of the wanted cyclam tetraamide product 2a. Additionally, a macrocyclic byproduct (14%) was formed by condensation of three units of diamine A and malonyl dichloride. To inhibit the formation of the trimer, we decided to prepare the cyclams in an indirect way. In fact, increased yields of cyclam derivative 2a (68% yield over three steps) were obtained by malonyl reaction of the mono-Boc-protected diamine (A-Boc) followed by Boc deprotection with HCl, and final ring closure of diamide–diamine intermediate 1a with a second malonyl unit to give tetraamide product 2a (Scheme 1a). The equivalent ethyl-substituted cyclam 4a was prepared in comparable yield (63% over the three steps) by the same method with diethylmalonyl chloride. This method also allowed for isolation of the open diamide–diamine 1a (77%). In addition, the similar potential ligands 1b–e (65–95%, Scheme 1b) were likewise prepared from amines B–E. The phenyl-substituted cyclam tetraamide derivative 2b was prepared by the original direct method [36] (65%, Scheme 1c), as the mono-Boc amine B-Boc was less accessible.
![[1860-5397-17-18-i1]](/bjoc/content/inline/1860-5397-17-18-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic protocols for the preparation of potential ligands 1–4.
Scheme 1: Synthetic protocols for the preparation of potential ligands 1–4.
As the amide coordination to Au(III) in general is challenging, and not successful in our hands, as discussed below, we wanted to prepare the reduced amine products (5a,b, 6a,b) from amides 1a,b and 2a,b. Initially, by refluxing diamide–diamines 1a,b and cyclam amide precursors 2a,b in THF with LiAlH4 for 3 days [36], complex product mixtures of partly and fully reduced species were obtained for all amides except 2a. In order to activate the amides for reduction, improved reaction conditions were obtained by adding AlCl3 to the reactions. Complete reduction of polyamides 1a,b and 2a,b yielded the open tetraamine products 5a,b and the target cyclams 6a,b with four secondary amine functions in moderate to high yields (29–88%, Scheme 2) within 1–2 days.
![[1860-5397-17-18-i2]](/bjoc/content/inline/1860-5397-17-18-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reduction of diamides 1a,b and tetraamides 2a,b.
Scheme 2: Reduction of diamides 1a,b and tetraamides 2a,b.
Au(III) coordination studies
Amide-coordinated Au(III) complexes have so far scarcely been reported [37-44]. This is likely a result of the electron deficient character of the amide nitrogens. The coordination was initially tested with the cyclam tetraamide derivatives 2a,b and 4a. Judged from 1H NMR, these ligands showed no interaction with Au(III), as expected. A similar resistance to coordinate was observed for the open diamides 1c–e. The phosphorus containing ligand 1c did undergo phosphorus oxidation instead of Au(III) coordination. No effect was obtained by refluxing or by adding additives, such as silver salts, NaOH or NH4PF6.
Given the previously reported coordinating studies of unsubstituted cyclam [16,19,29], the prepared new tetraamine ligands 5a,b and 6a,b (Scheme 2) were promising candidates for Au(III) coordination. Both ligands 5a and 6a readily coordinated with AuCl3 in methanol and gave moderate to excellent yields of tetracoordinated 5a-Au(III) and 6a-Au(III) N,N,N,N-complexes with alternating five- and six-membered chelate rings (50% and 96%, respectively, Scheme 3).
![[1860-5397-17-18-i3]](/bjoc/content/inline/1860-5397-17-18-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Au(III) coordination conditions for ligands 5a,b and 6a,b. Coordination of 5b was unsuccessful.
Scheme 3: Au(III) coordination conditions for ligands 5a,b and 6a,b. Coordination of 5b was unsuccessful.
Monitoring the formation of complex 5a-Au(III), using 1H NMR, and 1H,15N-HMBC, clearly indicated a tetra-nitrogen-coordinated complex. This was evidenced by changes in NMR shift values, Δδ15Ncoord = δ15Ncomplex – δ15Nligand, by coordination. The observed Δδ15Ncoord values were in the range of 16.3–32.0 ppm for both the primary and secondary amine nitrogens, indicating a characteristic deshielding effect upon the Au(III) coordination [43,45,46], Likewise, Δδ1Hcoord 0.3–0.5 ppm for all the neighboring N–CH and N–CH2 protons indicated ligand tetra-coordination to Au(III), as well. Upon coordination of ligand 5a, four different 15N NMR values for the nitrogens were observed. This might be explained by the nitrogens becoming non-equivalent when coordinated to Au(III), as a result of the chiral centers in the ligand. The structure of 5a-Au(III) was not confirmed, due to the lack of a suitable crystal for X-ray analysis, hence, only a proposed structure for 5a-Au(III) is given (Scheme 3). Comparable effects for ligand 6a, Δδ1Hcoord 0.3–0.6 ppm, were also observed for the corresponding N–CH and N–CH2 neighboring protons by formation of complex 6a-Au(III) (Figure 1).
![[1860-5397-17-18-1]](/bjoc/content/figures/1860-5397-17-18-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR study of the formation of complex 6a-Au(III) by AuCl3 coordination to ligand 6a.
Figure 1: 1H NMR study of the formation of complex 6a-Au(III) by AuCl3 coordination to ligand 6a.
Further on, cyclam 6b readily coordinated to AuCl3 in a mixture of acetonitrile and dichloromethane, to obtain a sufficient solubility of cyclam 6b, allowing formation of 6b-Au(III) in 64% yield (Scheme 3). The corresponding Δδ1Hcoord values of 6b-Au(III) were similar to those discussed for 6a-Au(III). Surprisingly, tetraamine 5b did not behave in a similar way as the other ligands, instead giving a complex mixture, as judged by 1H NMR, when attempted coordinated to Au(III). Changing the source of Au(III) or the solvents methanol, acetonitrile and dichloromethane did not improve the outcome. Both purification and characterization of the Au(III) complexes were challenging as a result of low stability, and HRMS or elemental analysis could not be obtained, due to sample decomposition. Attempts to obtain crystals for X-ray analysis by slow diffusion of n-pentane into a DCM solution of the complexes were unsuccessful.
Catalytic activity
For evaluation of the catalytic ability of the new Au(III) complexes, alkyne carboalkoxylation [47,48] and cyclopropanation of styrene with propargyl ester [49-52] (Table 1) were selected as test reactions. These reactions have previously been studied with different gold(I) and gold(III) catalysts and a variety of substrates, thus providing a solid background for comparison. A large difference in the catalytic activity was observed for cyclam–gold complex 6a-Au(III) versus the open cyclam analogues 5a-Au(III). Complex 5a-Au(III) afforded a full conversion in the alkyne carboalkoxylation in 5.5 hours, compared to in 24 hours for complex 6a-Au(III) (Table 1, entries 1 and 2). The same trend was observed for Au(III) catalysis of the cyclopropanation reaction, where complex 5a-Au(III) and 6a-Au(III) gave full conversion in 1 hour and 12 hours, respectively (Table 1, entries 4 and 5). The cyclopropyl product 11 was obtained in >90% yield and high cis diastereoselectivity (up to 74% de), similar to our previous studies [51], which showed that JohnPhos-Au(I) and pyr-menthol-Au(III) complexes provided high amounts of the initially formed cis diastereomer in this model reaction. In contrast, some BOX-Au(III) complexes have the additional ability to rapidly transform the initially formed cis product into the isomerized trans product. Thus, the proper choice of the gold catalyst allows highly stereoselective formation of either cis or trans cyclopropanation products and facilitates the isolation of pure isomers.
Table 1: The catalytic activity of Au(III) complexes evaluated in a) alkyne carboalkoxylation and b) cyclopropanation of styrene with propargyl ester.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-18-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Complex | Reaction time | Yield product |
| a) Carboalkoxylation of alkyne | 8 | ||
| 1 | 5a-Au(III) | 5.5 h | 62% |
| 2 | 6a-Au(III) | 24 h | 80% |
| 3 | 6b-Au(III) | 15 min | 13% |
| b) Cyclopropanation | 11 (cis/trans) | ||
| 4 | 5a-Au(III) | 1 h | 90% (77:23) |
| 5 | 6a-Au(III) | 12 h | 97% (87:13) |
| 6 | 6b-Au(III) | 15 min | 58% (12:88) |
| 7 | AuCl3 | 5 min | 80% (13:87) |
Despite the chiral nature of these ligands, no enantioselectivity was observed in the test reactions. Evaluation of complex 6b-Au(III) in both reactions, revealed a large difference in catalytic activity and complex stability between the structurally similar 6a-Au(III) and 6b-Au(III) cyclam complexes, with a cyclohexyl and a diphenyl-C2 bridge between the nitrogens, respectively. In both test reactions, an immediate color change into dark red/brown took place after addition of complex 6b-Au(III), indicating a low stabilization of the coordinated diphenyl ligand and a fast release of Au. The de-coordination resulted in full conversion within 15 min in both reactions (Table 1, entries 3 and 6), compared to 24 and 12 hours for complex 6a-Au(III) (Table 1, entries 2 and 5), where the ligand seems to stabilize and deactivate the Au(III) during the reaction. Attempts to improve the 6b-Au(III) complex stability by anion exchange with less coordinating anions failed, as addition of different standard silver salts resulted in decomposition of the Au(III) complex. Consequently, the counter-anion exchange method was not possible.
Since the ligand 6b seems to de-coordinate, resulting in the cyclam Au(III) complex not being the active catalyst, and the presence of chloride anions, the activity of the 6b-Au(III) precatalyst was compared to AuCl3. AuCl3 showed slightly faster conversion into the product, 5 min vs 15 min, however, a comparable cis/trans ratio was obtained. The reduced reaction time indicates that 6b-Au(III) indeed is an precatalyst that needs some activation time before catalyzing the reaction.
Although the decomposition of complex 6b-Au(III) resulted in the rapid conversion into products, it is undesirable, as the impact of the ligand on the reaction selectivity is lost. This different stability, caused by small differences in the design of the two cyclam ligands, is in accordance with the unsuccessful Au(III) coordination of the diphenyl-C2-bridged 5b ligand, in contrast to the readily coordinating cyclohexyl-bridged 5a tetraamine, as discussed above (Scheme 3).
Conclusion
A new stepwise procedure was developed for improved preparation of chiral cyclam derivatives 5a,b and 6a,b from chiral mono-Boc-1,2-diamines and (dialkyl)malonyl dichloride. The four-step approach included ring closure of the initial open diamide–diamine intermediates 1 with a second malonyl unit, affording the cyclam tetraamides 2. The target cyclam derivatives 5 and 6 were obtained by optimized LiAlH4 reduction by AlCl3 activation of the polyamides 1 and 2.
Successful Au(III) coordination of the open tetraamine ligand 5a and the new cyclam derivatives 6a,b gave the corresponding tetracoordinated N,N,N,N-Au(III) cyclam 5a and 6a,b complexes (50–96%) with alternating five- and six-membered chelate rings. Verification of cyclam tetraamino-coordination was obtained by 1H,15N-HMBC NMR. The polyamides (1, 2) failed to undergo Au(III) coordination, which confirmed the previously observed resistance of amides to coordinate to Au(III).
The catalytic ability of the new Au(III) complexes were screened in selected test reactions. A high catalytic ability was shown for novel N,N,N,N-Au(III) complexes 5a and 6a in alkyne carboalkoxylation and propargyl ester cyclopropanation (full conversion in 1–24 h, 62–97% product yields). No enantioselectivity was observed in the test reactions.
The activity and stability of the Au(III) complexes were strongly depending on the structure of the tetraamine ligands, demonstrating the importance of ligand design. Hence, the present study on cyclam based Au(III) complexes represents the first study on such chiral cyclam metal complexes and contributes to a better knowledge of the tetraamine ligand preparation and Au(III) coordination, as well as an increased understanding of Au(III) ligand design for optimal reaction outcomes.
Supporting Information
| Supporting Information File 1: Experimental procedures and NMR data for new ligands and gold(III) complexes, as well as a method for testing of catalytic activity. | ||
| Format: PDF | Size: 2.7 MB | Download |
References
-
Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u
Return to citation in text: [1] -
Hashmi, A. S. K.; Buehrle, M. Aldrichimica Acta 2010, 43, 27–33.
Return to citation in text: [1] -
Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c
Return to citation in text: [1] -
Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l
Return to citation in text: [1] -
Nevado, C. Chimia 2010, 64, 247–251. doi:10.2533/chimia.2010.247
Return to citation in text: [1] -
Sengupta, S.; Shi, X. ChemCatChem 2010, 2, 609–619. doi:10.1002/cctc.201000070
Return to citation in text: [1] -
Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g
Return to citation in text: [1] -
Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d
Return to citation in text: [1] -
Mascareñas, J. L.; Varela, I.; López, F. Acc. Chem. Res. 2019, 52, 465–479. doi:10.1021/acs.accounts.8b00567
Return to citation in text: [1] -
Mato, M.; García‐Morales, C.; Echavarren, A. M. ChemCatChem 2019, 11, 53–72. doi:10.1002/cctc.201801201
Return to citation in text: [1] -
García-Morales, C.; Echavarren, A. M. Synlett 2018, 29, 2225–2237. doi:10.1055/s-0037-1610203
Return to citation in text: [1] -
Wang, Y.-M.; Lackner, A. D.; Toste, F. D. Acc. Chem. Res. 2014, 47, 889–901. doi:10.1021/ar400188g
Return to citation in text: [1] -
Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/c3cc45518a
Return to citation in text: [1] -
Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k
Return to citation in text: [1] -
Zi, W.; Dean Toste, F. Chem. Soc. Rev. 2016, 45, 4567–4589. doi:10.1039/c5cs00929d
Return to citation in text: [1] -
Kimura, E.; Kurogi, Y.; Takahashi, T. Inorg. Chem. 1991, 30, 4117–4121. doi:10.1021/ic00022a007
Return to citation in text: [1] [2] -
Kavaklı, C.; Özvatan, N.; Tuncel, S. A.; Salih, B. Anal. Chim. Acta 2002, 464, 313–322. doi:10.1016/s0003-2670(02)00484-1
Return to citation in text: [1] -
Yau, J.; Mingos, D. M. P.; Powell, H. R. Polyhedron 1996, 15, 367–369. doi:10.1016/0277-5387(95)00278-z
Return to citation in text: [1] -
Kimura, E.; Kurogi, Y.; Koike, T.; Shionoya, M.; Iitaka, Y. J. Coord. Chem. 1993, 28, 33–49. doi:10.1080/00958979308035142
Return to citation in text: [1] [2] -
Nardin, G.; Randaccio, L.; Annibale, G.; Natile, G.; Pitteri, B. J. Chem. Soc., Dalton Trans. 1980, 220–223. doi:10.1039/dt9800000220
Return to citation in text: [1] -
Cinellu, M. A.; Minghetti, G.; Pinna, M. V.; Stoccoro, S.; Zucca, A.; Manassero, M. J. Chem. Soc., Dalton Trans. 2000, 1261–1265. doi:10.1039/a910188h
Return to citation in text: [1] -
Carotti, S.; Guerri, A.; Mazzei, T.; Messori, L.; Mini, E.; Orioli, P. Inorg. Chim. Acta 1998, 281, 90–94. doi:10.1016/s0020-1693(98)00148-0
Return to citation in text: [1] [2] -
Navarro, M. Coord. Chem. Rev. 2009, 253, 1619–1626. doi:10.1016/j.ccr.2008.12.003
Return to citation in text: [1] -
Gabbiani, C.; Casini, A.; Messori, L. Gold Bull. 2007, 40, 73–81. doi:10.1007/bf03215296
Return to citation in text: [1] [2] -
Messori, L.; Orioli, P.; Tempi, C.; Marcon, G. Biochem. Biophys. Res. Commun. 2001, 281, 352–360. doi:10.1006/bbrc.2001.4358
Return to citation in text: [1] [2] -
Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P. J.; Keppler, B. K.; Messori, L. J. Inorg. Biochem. 2008, 102, 564–575. doi:10.1016/j.jinorgbio.2007.11.003
Return to citation in text: [1] [2] -
Marcon, G.; Messori, L.; Orioli, P.; Cinellu, M. A.; Minghetti, G. Eur. J. Biochem. 2003, 270, 4655–4661. doi:10.1046/j.1432-1033.2003.03862.x
Return to citation in text: [1] [2] -
Sun, R. W.-Y.; Che, C.-M. Coord. Chem. Rev. 2009, 253, 1682–1691. doi:10.1016/j.ccr.2009.02.017
Return to citation in text: [1] -
Messori, L.; Abbate, F.; Marcon, G.; Orioli, P.; Fontani, M.; Mini, E.; Mazzei, T.; Carotti, S.; O'Connell, T.; Zanello, P. J. Med. Chem. 2000, 43, 3541–3548. doi:10.1021/jm990492u
Return to citation in text: [1] [2] [3] -
Schneider, C. R.; Lewis, L. C.; Shafaat, H. S. Dalton Trans. 2019, 48, 15810–15821. doi:10.1039/c9dt03114f
Return to citation in text: [1] -
Mash, B. L.; Raghavan, A.; Ren, T. Eur. J. Inorg. Chem. 2019, 2065–2070. doi:10.1002/ejic.201801198
Return to citation in text: [1] -
Nichols, E. M.; Chang, C. J. Organometallics 2019, 38, 1213–1218. doi:10.1021/acs.organomet.8b00308
Return to citation in text: [1] -
Burrows, C. J.; Muller, J. G.; Poulter, G. T.; Rokita, S. E. Acta Chem. Scand. 1996, 50, 337–344. doi:10.3891/acta.chem.scand.50-0337
Return to citation in text: [1] -
Boiocchi, M.; Ciarrocchi, C.; Fabbrizzi, L.; Licchelli, M.; Mangano, C.; Poggi, A.; Vázquez López, M. Inorg. Chem. 2015, 54, 10197–10207. doi:10.1021/acs.inorgchem.5b01273
Return to citation in text: [1] -
Shircliff, A. D.; Wilson, K. R.; Cannon-Smith, D. J.; Jones, D. G.; Zhang, Z.; Chen, Z.; Yin, G.; Prior, T. J.; Hubin, T. J. Inorg. Chem. Commun. 2015, 59, 71–75. doi:10.1016/j.inoche.2015.07.002
Return to citation in text: [1] -
De, C. K.; Paul, A.; Emge, T. J.; Seidel, D. Supramol. Chem. 2016, 28, 168–175. doi:10.1080/10610278.2015.1119276
Return to citation in text: [1] [2] [3] -
Dogan, A.; Schwederski, B.; Schleid, T.; Lissner, F.; Fiedler, J.; Kaim, W. Inorg. Chem. Commun. 2004, 7, 220–223. doi:10.1016/j.inoche.2003.11.006
Return to citation in text: [1] -
Yang, T.; Tu, C.; Zhang, J.; Lin, L.; Zhang, X.; Liu, Q.; Ding, J.; Xu, Q.; Guo, Z. Dalton Trans. 2003, 3419–3424. doi:10.1039/b305109a
Return to citation in text: [1] -
Fan, D.; Yang, C.-T.; Ranford, J. D.; Vittal, J. J. Dalton Trans. 2003, 4749–4753. doi:10.1039/b309310g
Return to citation in text: [1] -
Kilpin, K. J.; Henderson, W.; Nicholson, B. K. Dalton Trans. 2008, 3899–3906. doi:10.1039/b803835j
Return to citation in text: [1] -
Hill, D. T.; Burns, K.; Titus, D. D.; Girard, G. R.; Reiff, W. M.; Mascavage, L. M. Inorg. Chim. Acta 2003, 346, 1–6. doi:10.1016/s0020-1693(02)01425-1
Return to citation in text: [1] -
Cheung, T.-C.; Lai, T.-F.; Che, C.-M. Polyhedron 1994, 13, 2073–2077. doi:10.1016/s0277-5387(00)83492-0
Return to citation in text: [1] -
Reiersølmoen, A. C.; Csókás, D.; Pápai, I.; Fiksdahl, A.; Erdélyi, M. J. Am. Chem. Soc. 2019, 141, 18221–18229. doi:10.1021/jacs.9b09108
Return to citation in text: [1] [2] -
Johnson, M. W.; DiPasquale, A. G.; Bergman, R. G.; Toste, F. D. Organometallics 2014, 33, 4169–4172. doi:10.1021/om500663m
Return to citation in text: [1] -
Reiersølmoen, A. C.; Csókás, D.; Øien-Ødegaard, S.; Vanderkooy, A.; Gupta, A. K.; Carlsson, A.-C. C.; Orthaber, A.; Fiksdahl, A.; Pápai, I.; Erdélyi, M. J. Am. Chem. Soc. 2020, 142, 6439–6446. doi:10.1021/jacs.0c01941
Return to citation in text: [1] -
Reiersølmoen, A. C.; Fiksdahl, A. Eur. J. Org. Chem. 2020, 2867–2877. doi:10.1002/ejoc.202000139
Return to citation in text: [1] -
Dubé, P.; Toste, F. D. J. Am. Chem. Soc. 2006, 128, 12062–12063. doi:10.1021/ja064209+
Return to citation in text: [1] -
Zi, W.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 12600–12603. doi:10.1021/ja407150h
Return to citation in text: [1] -
Sperger, C. A.; Tungen, J. E.; Fiksdahl, A. Eur. J. Org. Chem. 2011, 3719–3722. doi:10.1002/ejoc.201100291
Return to citation in text: [1] -
Iqbal, N.; Sperger, C. A.; Fiksdahl, A. Eur. J. Org. Chem. 2013, 907–914. doi:10.1002/ejoc.201201328
Return to citation in text: [1] -
Reiersølmoen, A. C.; Østrem, E.; Fiksdahl, A. Eur. J. Org. Chem. 2018, 3317–3325. doi:10.1002/ejoc.201800419
Return to citation in text: [1] [2] -
Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 18002–18003. doi:10.1021/ja0552500
Return to citation in text: [1]
| 43. | Reiersølmoen, A. C.; Csókás, D.; Pápai, I.; Fiksdahl, A.; Erdélyi, M. J. Am. Chem. Soc. 2019, 141, 18221–18229. doi:10.1021/jacs.9b09108 |
| 45. | Reiersølmoen, A. C.; Csókás, D.; Øien-Ødegaard, S.; Vanderkooy, A.; Gupta, A. K.; Carlsson, A.-C. C.; Orthaber, A.; Fiksdahl, A.; Pápai, I.; Erdélyi, M. J. Am. Chem. Soc. 2020, 142, 6439–6446. doi:10.1021/jacs.0c01941 |
| 46. | Reiersølmoen, A. C.; Fiksdahl, A. Eur. J. Org. Chem. 2020, 2867–2877. doi:10.1002/ejoc.202000139 |
| 37. | Dogan, A.; Schwederski, B.; Schleid, T.; Lissner, F.; Fiedler, J.; Kaim, W. Inorg. Chem. Commun. 2004, 7, 220–223. doi:10.1016/j.inoche.2003.11.006 |
| 38. | Yang, T.; Tu, C.; Zhang, J.; Lin, L.; Zhang, X.; Liu, Q.; Ding, J.; Xu, Q.; Guo, Z. Dalton Trans. 2003, 3419–3424. doi:10.1039/b305109a |
| 39. | Fan, D.; Yang, C.-T.; Ranford, J. D.; Vittal, J. J. Dalton Trans. 2003, 4749–4753. doi:10.1039/b309310g |
| 40. | Kilpin, K. J.; Henderson, W.; Nicholson, B. K. Dalton Trans. 2008, 3899–3906. doi:10.1039/b803835j |
| 41. | Hill, D. T.; Burns, K.; Titus, D. D.; Girard, G. R.; Reiff, W. M.; Mascavage, L. M. Inorg. Chim. Acta 2003, 346, 1–6. doi:10.1016/s0020-1693(02)01425-1 |
| 42. | Cheung, T.-C.; Lai, T.-F.; Che, C.-M. Polyhedron 1994, 13, 2073–2077. doi:10.1016/s0277-5387(00)83492-0 |
| 43. | Reiersølmoen, A. C.; Csókás, D.; Pápai, I.; Fiksdahl, A.; Erdélyi, M. J. Am. Chem. Soc. 2019, 141, 18221–18229. doi:10.1021/jacs.9b09108 |
| 44. | Johnson, M. W.; DiPasquale, A. G.; Bergman, R. G.; Toste, F. D. Organometallics 2014, 33, 4169–4172. doi:10.1021/om500663m |
| 16. | Kimura, E.; Kurogi, Y.; Takahashi, T. Inorg. Chem. 1991, 30, 4117–4121. doi:10.1021/ic00022a007 |
| 19. | Kimura, E.; Kurogi, Y.; Koike, T.; Shionoya, M.; Iitaka, Y. J. Coord. Chem. 1993, 28, 33–49. doi:10.1080/00958979308035142 |
| 29. | Messori, L.; Abbate, F.; Marcon, G.; Orioli, P.; Fontani, M.; Mini, E.; Mazzei, T.; Carotti, S.; O'Connell, T.; Zanello, P. J. Med. Chem. 2000, 43, 3541–3548. doi:10.1021/jm990492u |
| 1. | Corma, A.; Leyva-Pérez, A.; Sabater, M. J. Chem. Rev. 2011, 111, 1657–1712. doi:10.1021/cr100414u |
| 2. | Hashmi, A. S. K.; Buehrle, M. Aldrichimica Acta 2010, 43, 27–33. |
| 3. | Jiménez-Núñez, E.; Echavarren, A. M. Chem. Commun. 2007, 333–346. doi:10.1039/b612008c |
| 4. | Li, Z.; Brouwer, C.; He, C. Chem. Rev. 2008, 108, 3239–3265. doi:10.1021/cr068434l |
| 5. | Nevado, C. Chimia 2010, 64, 247–251. doi:10.2533/chimia.2010.247 |
| 6. | Sengupta, S.; Shi, X. ChemCatChem 2010, 2, 609–619. doi:10.1002/cctc.201000070 |
| 7. | Gorin, D. J.; Sherry, B. D.; Toste, F. D. Chem. Rev. 2008, 108, 3351–3378. doi:10.1021/cr068430g |
| 8. | Arcadi, A. Chem. Rev. 2008, 108, 3266–3325. doi:10.1021/cr068435d |
| 18. | Yau, J.; Mingos, D. M. P.; Powell, H. R. Polyhedron 1996, 15, 367–369. doi:10.1016/0277-5387(95)00278-z |
| 19. | Kimura, E.; Kurogi, Y.; Koike, T.; Shionoya, M.; Iitaka, Y. J. Coord. Chem. 1993, 28, 33–49. doi:10.1080/00958979308035142 |
| 20. | Nardin, G.; Randaccio, L.; Annibale, G.; Natile, G.; Pitteri, B. J. Chem. Soc., Dalton Trans. 1980, 220–223. doi:10.1039/dt9800000220 |
| 21. | Cinellu, M. A.; Minghetti, G.; Pinna, M. V.; Stoccoro, S.; Zucca, A.; Manassero, M. J. Chem. Soc., Dalton Trans. 2000, 1261–1265. doi:10.1039/a910188h |
| 36. | De, C. K.; Paul, A.; Emge, T. J.; Seidel, D. Supramol. Chem. 2016, 28, 168–175. doi:10.1080/10610278.2015.1119276 |
| 17. | Kavaklı, C.; Özvatan, N.; Tuncel, S. A.; Salih, B. Anal. Chim. Acta 2002, 464, 313–322. doi:10.1016/s0003-2670(02)00484-1 |
| 36. | De, C. K.; Paul, A.; Emge, T. J.; Seidel, D. Supramol. Chem. 2016, 28, 168–175. doi:10.1080/10610278.2015.1119276 |
| 16. | Kimura, E.; Kurogi, Y.; Takahashi, T. Inorg. Chem. 1991, 30, 4117–4121. doi:10.1021/ic00022a007 |
| 35. | Shircliff, A. D.; Wilson, K. R.; Cannon-Smith, D. J.; Jones, D. G.; Zhang, Z.; Chen, Z.; Yin, G.; Prior, T. J.; Hubin, T. J. Inorg. Chem. Commun. 2015, 59, 71–75. doi:10.1016/j.inoche.2015.07.002 |
| 9. | Mascareñas, J. L.; Varela, I.; López, F. Acc. Chem. Res. 2019, 52, 465–479. doi:10.1021/acs.accounts.8b00567 |
| 10. | Mato, M.; García‐Morales, C.; Echavarren, A. M. ChemCatChem 2019, 11, 53–72. doi:10.1002/cctc.201801201 |
| 11. | García-Morales, C.; Echavarren, A. M. Synlett 2018, 29, 2225–2237. doi:10.1055/s-0037-1610203 |
| 12. | Wang, Y.-M.; Lackner, A. D.; Toste, F. D. Acc. Chem. Res. 2014, 47, 889–901. doi:10.1021/ar400188g |
| 13. | Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16–28. doi:10.1039/c3cc45518a |
| 14. | Dorel, R.; Echavarren, A. M. Chem. Rev. 2015, 115, 9028–9072. doi:10.1021/cr500691k |
| 15. | Zi, W.; Dean Toste, F. Chem. Soc. Rev. 2016, 45, 4567–4589. doi:10.1039/c5cs00929d |
| 36. | De, C. K.; Paul, A.; Emge, T. J.; Seidel, D. Supramol. Chem. 2016, 28, 168–175. doi:10.1080/10610278.2015.1119276 |
| 25. | Messori, L.; Orioli, P.; Tempi, C.; Marcon, G. Biochem. Biophys. Res. Commun. 2001, 281, 352–360. doi:10.1006/bbrc.2001.4358 |
| 30. | Schneider, C. R.; Lewis, L. C.; Shafaat, H. S. Dalton Trans. 2019, 48, 15810–15821. doi:10.1039/c9dt03114f |
| 31. | Mash, B. L.; Raghavan, A.; Ren, T. Eur. J. Inorg. Chem. 2019, 2065–2070. doi:10.1002/ejic.201801198 |
| 32. | Nichols, E. M.; Chang, C. J. Organometallics 2019, 38, 1213–1218. doi:10.1021/acs.organomet.8b00308 |
| 33. | Burrows, C. J.; Muller, J. G.; Poulter, G. T.; Rokita, S. E. Acta Chem. Scand. 1996, 50, 337–344. doi:10.3891/acta.chem.scand.50-0337 |
| 51. | Reiersølmoen, A. C.; Østrem, E.; Fiksdahl, A. Eur. J. Org. Chem. 2018, 3317–3325. doi:10.1002/ejoc.201800419 |
| 22. | Carotti, S.; Guerri, A.; Mazzei, T.; Messori, L.; Mini, E.; Orioli, P. Inorg. Chim. Acta 1998, 281, 90–94. doi:10.1016/s0020-1693(98)00148-0 |
| 34. | Boiocchi, M.; Ciarrocchi, C.; Fabbrizzi, L.; Licchelli, M.; Mangano, C.; Poggi, A.; Vázquez López, M. Inorg. Chem. 2015, 54, 10197–10207. doi:10.1021/acs.inorgchem.5b01273 |
| 24. | Gabbiani, C.; Casini, A.; Messori, L. Gold Bull. 2007, 40, 73–81. doi:10.1007/bf03215296 |
| 26. | Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P. J.; Keppler, B. K.; Messori, L. J. Inorg. Biochem. 2008, 102, 564–575. doi:10.1016/j.jinorgbio.2007.11.003 |
| 29. | Messori, L.; Abbate, F.; Marcon, G.; Orioli, P.; Fontani, M.; Mini, E.; Mazzei, T.; Carotti, S.; O'Connell, T.; Zanello, P. J. Med. Chem. 2000, 43, 3541–3548. doi:10.1021/jm990492u |
| 47. | Dubé, P.; Toste, F. D. J. Am. Chem. Soc. 2006, 128, 12062–12063. doi:10.1021/ja064209+ |
| 48. | Zi, W.; Toste, F. D. J. Am. Chem. Soc. 2013, 135, 12600–12603. doi:10.1021/ja407150h |
| 22. | Carotti, S.; Guerri, A.; Mazzei, T.; Messori, L.; Mini, E.; Orioli, P. Inorg. Chim. Acta 1998, 281, 90–94. doi:10.1016/s0020-1693(98)00148-0 |
| 23. | Navarro, M. Coord. Chem. Rev. 2009, 253, 1619–1626. doi:10.1016/j.ccr.2008.12.003 |
| 24. | Gabbiani, C.; Casini, A.; Messori, L. Gold Bull. 2007, 40, 73–81. doi:10.1007/bf03215296 |
| 25. | Messori, L.; Orioli, P.; Tempi, C.; Marcon, G. Biochem. Biophys. Res. Commun. 2001, 281, 352–360. doi:10.1006/bbrc.2001.4358 |
| 26. | Casini, A.; Hartinger, C.; Gabbiani, C.; Mini, E.; Dyson, P. J.; Keppler, B. K.; Messori, L. J. Inorg. Biochem. 2008, 102, 564–575. doi:10.1016/j.jinorgbio.2007.11.003 |
| 27. | Marcon, G.; Messori, L.; Orioli, P.; Cinellu, M. A.; Minghetti, G. Eur. J. Biochem. 2003, 270, 4655–4661. doi:10.1046/j.1432-1033.2003.03862.x |
| 28. | Sun, R. W.-Y.; Che, C.-M. Coord. Chem. Rev. 2009, 253, 1682–1691. doi:10.1016/j.ccr.2009.02.017 |
| 29. | Messori, L.; Abbate, F.; Marcon, G.; Orioli, P.; Fontani, M.; Mini, E.; Mazzei, T.; Carotti, S.; O'Connell, T.; Zanello, P. J. Med. Chem. 2000, 43, 3541–3548. doi:10.1021/jm990492u |
| 27. | Marcon, G.; Messori, L.; Orioli, P.; Cinellu, M. A.; Minghetti, G. Eur. J. Biochem. 2003, 270, 4655–4661. doi:10.1046/j.1432-1033.2003.03862.x |
| 49. | Sperger, C. A.; Tungen, J. E.; Fiksdahl, A. Eur. J. Org. Chem. 2011, 3719–3722. doi:10.1002/ejoc.201100291 |
| 50. | Iqbal, N.; Sperger, C. A.; Fiksdahl, A. Eur. J. Org. Chem. 2013, 907–914. doi:10.1002/ejoc.201201328 |
| 51. | Reiersølmoen, A. C.; Østrem, E.; Fiksdahl, A. Eur. J. Org. Chem. 2018, 3317–3325. doi:10.1002/ejoc.201800419 |
| 52. | Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, F. D. J. Am. Chem. Soc. 2005, 127, 18002–18003. doi:10.1021/ja0552500 |
© 2021 Reiersølmoen et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)