1Department of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, United Kingdom
2Vertex Pharmaceuticals (Europe) Ltd., 86–88 Jubilee Avenue, Milton Park, Abingdon, OX14 4RW, United Kingdom
3current address: MSD UK Discovery Centre, Francis Crick Institute, 1 Midland Road, London, NW1 1AT, United Kingdom
Corresponding author email
Associate Editor: T. J. J. Müller Beilstein J. Org. Chem.2021,17, 334–342.https://doi.org/10.3762/bjoc.17.31 Received 10 Nov 2020,
Accepted 19 Jan 2021,
Published 02 Feb 2021
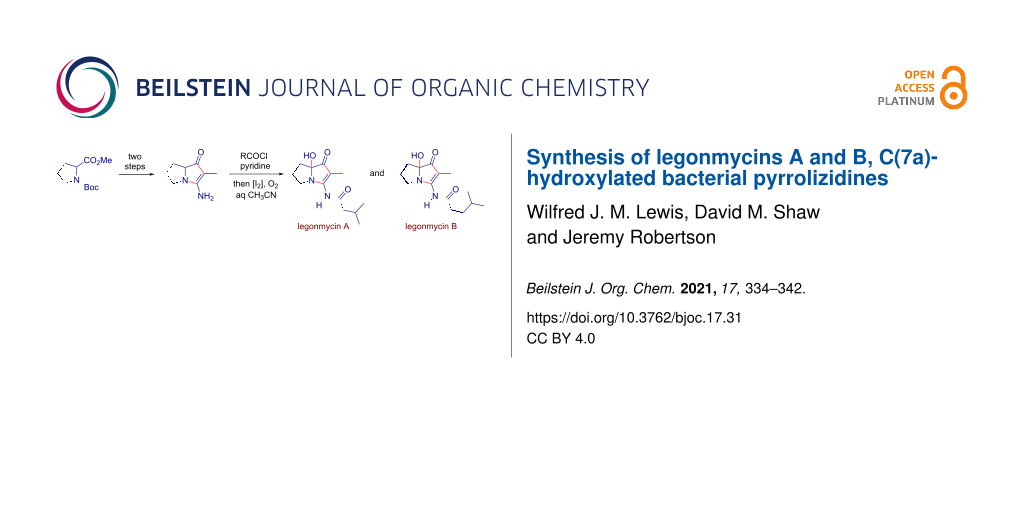
A one-flask, two-step procedure from 3-amino-2-methyl-5,6,7,7a-tetrahydro-1H-pyrrolizin-1-one affords the Streptomyces secondary metabolites legonmycins A and B – three operations overall from methyl N-Boc-prolinate. The key step proceeds in each case via N,O-diacylation, then selective oxidative hydrolysis of the intermediate bicyclic pyrrole and establishes a precedent for the synthesis of related C(7a)-hydroxylated pyrrolizidines.
At least 40 members of the large class of pyrrolizidine alkaloids [1-4] have so far been characterized from bacterial cultures. Of these ‘bacterial pyrrolizidines’, those of the vinylogous urea type are particularly intriguing from a chemical and biosynthetic perspective. Their emergence dates back to 1977 with a patent filing from The Upjohn Company describing an unidentified ‘antibiotic 354’ isolated as a fermentation product of Streptomyces puniceus subsp. doliceus; spectroscopic characterization, including 1H NMR data, was provided, but no structure [5]. In 1980, the same group reported that antibiotic 354 is equivalent to clazamycin B (Figure 1) [6], one of two 7a-hydroxylated iminopyrrolizinones that had been described a year earlier by a group at The Institute of Microbial Chemistry in Tokyo [7,8]. The two clazamycins, A and B, are related as 7a-epimers that interconvert in aqueous solution to form a pH-dependent equilibrium ratio of the two [9]; they are weakly antibacterial and were shown to prolong “the survival period of mice inoculated with leukaemia L-120 cells” [7,8]. Although the clazamycins are not vinylogous ureas, since they lack the C(1) carbonyl group, their structural relationship with bohemamine, the first genuine member of the group, qualifies them as honorary members. The structure of bohemamine, named by its discoverers at Bristol–Myers Company and Cornell University after the opera La Bohème, was also reported in 1980 [10]. Following a 23-year hiatus, two papers submitted within two weeks of each other reported, respectively: (1) the isolation from Streptomyces sp. UMA-044 and characterization of NP25302, that differs from bohemamine in lacking the 6,7-epoxide functionality [11] and (2) the jenamidines A–C, from another Streptomyces sp. strain [12], although these molecules were not recognized as pyrrolizidines until Snider’s subsequent synthetic work revised the structural assignment [13,14] and, separately, established the absolute configuration of natural (+)-NP25302 [15]. Contemporaneously, three further bohemamines were described [16]; five more plus a related 7a-hydroxylated variant [17], various dimeric bohemamines [18,19], and spithioneines A and B [20] extended the range more recently [21].
Figure 1:
The clazamycins, and selected bacterial pyrrolizidines of the vinylogous urea type. For consistency, the standard numbering convention used for the plant pyrrolizidines, as depicted for the clazamycins, is used throughout.
Figure 1:
The clazamycins, and selected bacterial pyrrolizidines of the vinylogous urea type. For consistency...
Almost 40 years after the bacterial pyrrolizidines were first recognized, two 2015 papers addressed their biosynthesis. Thus, the investigation of the metabolites of Streptomyces sp. MA37 (from a soil sample obtained in Legon, Ghana) revealed the production of legonmycins A (3) and B (4) (Figure 2) and found that just four genes (lgnA–D) were necessary for their biosynthesis [22]. Proteins coded by three of these genes (LgnA, LgnB, and LgnD) were shown to assemble legonindolizidines A (1) and B (2) – from proline, threonine, and a fatty acid component – which are then converted by LgnC, a flavin-dependent monooxygenase, into the corresponding legonmycins (3 and 4) via a sequence of Baeyer–Villiger-type ring expansion, hydrolysis and decarboxylation, cyclization and dehydration, and finally hydroxylation at C(7a). Just one month later, Bode reported the identification of an unknown gene cluster in the symbiotic bacterium Xenorhabdus stockiae[23]. Cloning and expression of this pxaAB gene cluster in E. coli, and analysis of the metabolites by differential 2D NMR spectroscopy, led to the isolation and characterization of pyrrolizixenamide A (9) and, subsequently, pyrrolizixenamides B–D (10–12). Ultimately, an analogous biosynthetic pathway to that proposed for the legonmycins was established with, in this case, PxaB achieving the oxidative steps from indolizidine intermediates 5–8 produced by PxaA. An important aspect of this work was the finding that at least 90 different bacterial strains, spanning 23 species, contain gene sequences encoding proteins related to PxaB. These species include both Gram-positive and Gram-negative bacteria, indicating that pyrrolizidines are potential secondary metabolites of a variety of bacterial genera. The most recent addition to the bacterial pyrrolizidine literature also concerns their biosynthesis and addresses the origin of the methyl groups in the bohemamines and NP25302 [24].
Figure 2:
Key species in the biosynthesis of legonmycins A (3) and B (4), and the pyrrolizixenamides A–D (9–12).
Figure 2:
Key species in the biosynthesis of legonmycins A (3) and B (4), and the pyrrolizixenamides A–D (9–12...
Our interest in this area originated in reports of significant biological activity for those bacterial pyrrolizidines for which such effects have been assessed. The clazamycins, jenamidine A, dibohemamines D–F, and quinohemamine are all reported to be cytotoxic against a variety of cancer cell lines, and both bohemamine and NP25302 inhibit HL-60 cell adhesion to Chinese hamster ovary cells expressing intercellular adhesion molecule ICAM-1 (CD-54). This interest led us to develop a total synthesis of NP25302 [25] and its 5-normethyl analog and, as described in this paper, concise syntheses of the legonmycins.
Results and Discussion
The legonmycins are unique among the bacterial pyrrolizidines in bearing a C(2) methyl group. The biosynthetic study [22] demonstrates that this methyl group originates from the terminal carbon in threonine and not, as might be thought to be theoretically possible [18,19], via a late-stage addition to an intact pyrrolizidine core. The molecules are isolated as their racemates and, by analogy to the clazamycins, even if the LgnC-mediated hydroxylation is fully stereoselective, the C(7a) center is expected to be configurationally unstable in aqueous solution. Chiral HPLC analysis of samples of pyrrolizixenamide A isolated from culture clearly indicated racemic specimens but also revealed the presence of the two C(7a)-hydroxylated derivatives whose presence in the mixture became enriched during purification. Taken together, it seems reasonable to envisage that ready equilibration of the pyrrolizixenamide enantiomers (and those of related molecules such as jenamidine A and the prelegonmycins) via the hydroxypyrrole tautomer, and aerial oxidation of this electron-rich intermediate, would offer a non-enzymatic route to the various C(7a)-hydroxylated bacterial pyrrolizidines including the legonmycins, jenamidines B and C, and bohemamine D (cf. [26-30]).
With all this in mind, we set out to develop a simplification of Snider’s route [14,15] to the vinylogous urea core for application to the synthesis of legonmycins A and B, expecting to facilitate the C(7a) hydroxylation by exploiting the tendency to C(1–7a) enolization. Since Snider was unsuccessful in adapting his route to encompass the synthesis of jenamidines B and C [15], a successful route to the legonmycins would establish conditions for the synthesis of further C(7a)-hydroxylated bacterial pyrrolizidines and related molecules.
Pyrrolizidine 14 (Scheme 1), the key intermediate in Snider’s improved route to jenamidine A and Bode’s preparation of pyrrolizixenamides A (9) and D (12), is formed by N-cyclization onto the nitrile group in cyano-β-ketoester 13. The appended ester functionality in 14 has, at some point, to be removed, as its presence complicates a potential application to the (2-methyl-substituted) legonmycins, and it exerts a potentially stabilizing electronic effect on pyrrolic intermediates that could be unhelpful in downstream oxidation chemistry. Accordingly, our route began with the addition [31-33] of 2-lithiopropionitrile to methyl N-Boc-prolinate (15) to give α-cyanoketone 16 which was used in crude form or purified by chromatography to a colorless solid, albeit with significant loss in yield. The removal of the Boc protecting group under acidic conditions was accompanied by cyclization in situ [14,15,34,35] and pyrrolizinone derivative 17 was obtained efficiently on a multigram scale over two steps.
Originally, it was expected that adapting the conditions (NaH, RCOCl, THF) used for the acylation step in the synthesis of NP25302 [15,25] would deliver both prelegonmycins by the appropriate choice of the acylating agent. In the event, no monoacylated product was observed and attempts to isolate the individual components from the crude mixtures by chromatography provided material of insufficient purity to allow the definitive assignment of the structures. Diacylation is a feature of reactions of this type, with both Snider and Bode observing such side-products, although the extent of diacylation in those cases appears to be reduced, perhaps by virtue of the 2-carboxyl group present in their work. Recognizing that the ester and amide carbonyl groups in the diacylation product (18, Scheme 2) should be easily differentiated, and that the ester represents an enol(ate) equivalent, improved conditions for the diacylation were developed. Ultimately, the reaction with isovaleryl chloride in acetonitrile, with pyridine as a scavenger for the liberated HCl, gave solutions of crude diacylated product 18 in acceptable purity after simple filtration as work-up. It was reasoned that the activation of the electron-rich pyrrole (with generic electrophile X2 as shown) would hasten cleavage of the ester and that, irrespective of the site of activation, the subsequent ejection of the activating reagent would generate an extended iminium ion whose capture by adventitious water would produce legonmycin A directly. For context, the treatment of enol esters with oxidizing agents to give α-hydroxyketones, or halogens to give α-haloketones, is well known [36,37]. The halogen-mediated oxidation of 3-acyloxypyrroles and related compounds, to give α-hydroxy-3-ketopyrroles, is not reported but oxidations that represent equivalent reactivity have been reported [38-41].
Scheme 2:
Hypothesis for the oxidative hydrolysis of diacylated pyrrolizidinone 18 (R = iBu).
Scheme 2:
Hypothesis for the oxidative hydrolysis of diacylated pyrrolizidinone 18 (R = iBu).
The concept was tested in an NMR experiment in which a −78 °C solution of the crude diacylated species 18 in methanol-d4 was treated sequentially with a solution of I2 (1.0 equiv) in methanol-d4 and pyridine-d5 (≈4.5 equiv). The 1H NMR spectrum acquired after 20 min showed loss of the triplet at 3.76 ppm arising from the enantiotopic CH2N protons in pyrrole 18, and the appearance of new resonances at 3.21 (ddd, J = 11.5, 9.0, 2.5 Hz, 1H) and 3.64 (dt, J = 11.5, 8.5 Hz, 1H) ppm that correspond visually with those reported for the diastereotopic CH2N protons in legonmycin A. HRMS (ESI+) analysis of material isolated from this NMR experiment showed m/z = 270.1892, suggesting that in methanol-d4 the reported [22] spectrum is not that of legonmycin A itself but that of the C(7a)-OCD3 derivative (21, Scheme 3; m/z calcd for C14H20D3N2O [M + H]+, 270.1892). A sample of the corresponding OCH3 derivative 22, containing a small amount of residual toluene from the chromatography solvent, was obtained by an analogous reaction in (non-deuterated) methanol. The 13C NMR spectrum for this derivative corresponded closely to that obtained by us and reported for legonmycin A in methanol-d4, with an additional resonance at 51.7 ppm for the OCH3 carbon (3.12 ppm in the 1H NMR spectrum) and a shift in the C(7a) resonance.
Scheme 3:
Preparation of C(7a)-functionalized pyrrolizinone derivatives and synthesis of legonmycins A and B.
Scheme 3:
Preparation of C(7a)-functionalized pyrrolizinone derivatives and synthesis of legonmycins A and B.
The complication of solvent incorporation was avoided by running both the acylation and oxidative hydrolysis steps in acetonitrile, with water added in the second step. The oxidation step could be carried out either with a stoichiometric amount of I2, or just 10 mol % when O2 was bubbled gently through the reaction mixture. Additionally, since both the optimized diacylation and oxidative hydrolysis steps were carried out in acetonitrile, the transformation of the intermediate 17 into legonmycin A was conveniently performed as a one-flask process, delivering the natural product in 56% yield (corrected for the presence of ≈10 wt % toluene) on a ≈2.0 mmol scale. The repetition of the process, using isocaproyl chloride instead of isovaleryl chloride, afforded legonmycin B in moderate yield from ≈0.5 mmol of intermediate 17. In a larger-scale reaction (≈1.1 mmol), carried out with no added I2, an approximately 50:50 ratio of the diacylated intermediate 23 and legonmycin B was obtained.
Conclusion
This work demonstrates that, under carefully chosen conditions, the diacylation of pyrrolizinone derivatives such as compound 17 can be combined with the electrophilic activation and hydrolysis of the resulting electron-rich pyrroles in an overall N-acylation/C(7a) hydroxylation. This transformation is central to a synthesis of legonmycins A and B that requires just three laboratory operations from commercially available proline derivative 15.
It is noteworthy that Snider’s attempt to oxidize, by epoxidation, the N,O-diacetyl derivative of compound 14 was not successful. Indeed, in our own work, the use of either NBS, MCPBA, or O2 with a transition-metal catalyst also gave unpromising results in attempts to oxidize compound 17. Future work will explore whether or not the nature of the C(2)-substituent (CO2t-Bu in 15; Me in 17) has a significant influence on the oxidation chemistry, and how effectively these results can be translated to syntheses of further C(7a)-hydroxylated bacteral pyrrolizidines and their analogs.
Experimental
General experimental details
Dry solvents were obtained from an MBraun SPS-800 solvent purification system, except that THF was freshly distilled from Na/benzophenone. All other reagents or solvents were used as received from commercial suppliers, unless indicated otherwise. Column chromatography was performed using Merck Geduran Si 60 silica gel as the stationary phase unless otherwise specified. “Brine” refers to sat. aq NaCl solution. Thin-layer chromatography (TLC) was performed with Merck TLC 60 Silica gel F254 plates as the stationary phase and spots were visualized with KMnO4, vanillin, anisaldehyde, or ultraviolet light (254 nm). Retention factors (Rf) are quoted to the nearest 0.05. NMR spectra were recorded on Bruker AVIII HD 500, AVII 500, or AVIII HD 400 spectrometers, 500/125 MHz spectra being recorded by the NMR Service at the Chemistry Research Laboratory, University of Oxford. Chemical shifts are reported in ppm downfield of tetramethysilane, internally referenced (in MestReNova) to the appropriate solvent peak: CDCl3, 7.26/77.16; DMSO-d6, 2.50/39.52; CD3OD, 3.31/49.00. Coupling constants (J) are rounded to the nearest 0.5 Hz. IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer on a diamond ATR module, and νmax values are expressed in cm−1. HRMS measurements were performed by the Mass Spectrometry Service at the Chemistry Research Laboratory, University of Oxford, using a Bruker microTOF spectrometer. Values of m/z are calculated to 0.0001 Daltons from the chemical formula, and all reported values are within 5 ppm of the calculated theoretical values. Melting points were recorded on a Griffin mp apparatus and are uncorrected.
To a solution of LHMDS (100 mL, 1.0 M in THF, 100 mmol), cooled to −78 °C and under a N2 atmosphere, was added dropwise propionitrile (7.33 mL, 103 mmol). The mixture was stirred vigorously at −78 °C for 40 min, after which time a solution of proline derivative 15 (11.2 g, 48.8 mmol) in THF (60 mL) was added dropwise via syringe. The mixture was warmed to rt and stirred for 42 h. The mixture was then diluted with aq acetic acid (20 mL, H2O/AcOH 3:1 (v/v)), concentrated, and extracted with ethyl acetate (3 × 100 mL). The combined organic extracts were washed sequentially with water (40 mL) and brine (10 mL), then dried (Na2SO4) and concentrated to afford a pale yellow solid which could be used in the next step without further purification (14.1 g, containing ≈5% w/w propionitrile and ≈10% w/w trimethylsilyl acetate). An analytical sample, a white solid, was obtained by column chromatography (pentane → pentane/ethyl acetate 4:1) as a 1:1 mixture of diastereomers, each an ≈2:1 mixture of rotamers (in CDCl3). Rf 0.50 (pentane/ethyl acetate 1:1); mp 92–102 °C; IR νmax: 2244m, 1738m, 1688s, 1394s, 1163s, 1119m; 1H NMR (400 MHz, CDCl3) δ 1.40 (s, ≈1.5H), 1.41 (s, ≈1.5H), 1.43 (s, ≈3H), 1.47 (s, ≈3H), 1.49 (d, J = 7.0 Hz, ≈2H), 1.51 (2 × d, J = 7.0 Hz, 2 × ≈0.5H), 1.83–2.43 (m, 4H), 3.39–3.62 (m, ≈2.17H), 3.63 (q, J = 7.0 Hz, ≈0.17H), 3.79 (2 × q, J = 7.0 Hz, 2 × ≈0.33H), 4.47 (dd, J = 8.5, 5.5 Hz, ≈0.33H), 4.55–4.60 (m, ≈0.33H), 4.62 (dd, J = 8.0, 5.0 Hz, ≈0.33H); 13C NMR (100 MHz, CDCl3) δ 13.5, 14.2, 14.3, 23.9, 24.8, 24.9, 28.5 (two peaks), 29.7, 29.9, 30.7, 31.0, 33.3, 33.8, 35.2, 36.2, 47.0 (three peaks), 63.3, 63.8, 64.5, 64.7, 80.5, 80.9, 81.0, 81.1, 117.8, 118.0, 118.1, 154.7, 155.4, 200.6 (two peaks), 201.0, 201.6; HRMS–ESI+ (m/z): [M + Na]+ calcd for C13H20N2O3Na, 275.1366; found, 275.1366.
Acetyl chloride (300 μL, 4.22 mmol) was added dropwise to ethanol (2 mL) stirring at −78 °C under N2. The mixture was briefly warmed to rt (5 min) then re-cooled to −78 °C. A solution of crude cyanoketone 16 (1.0 g, 85% purity, 3.37 mmol) in ethanol (1 mL) was added, the cooling bath replaced with a rt water bath, and stirring continued for 3.5 h. The mixture was concentrated, with azeotropic removal of ethanol and other volatiles with toluene, to afford the title compound as a caramel-colored solid (561 mg) that was used directly in the subsequent acylation and oxidation steps [the NMR spectra reproduced on page S3 of Supporting Information File 1 are for the crude material]. IR νmax: 3283br, 3140br, 1661m, 1607s, 1522s, 1483s, 1446s, 1382s; 1H NMR (400 MHz, CD3OD) δ 1.35–1.45 (m, 1H), 1.76 (d, J = 0.5 Hz, 3H), 2.23–2.32 (m, 1H), 2.32–2.46 (m, 2H), 3.24 (td, J = 10.0, 7.5 Hz, 1H), 3.49 (ddd, J = 10.0, 8.5, 2.5 Hz, 1H), 4.44 (dd, J = 10.5, 5.5 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 5.8, 28.4, 30.2, 46.5, 70.0, 101.3 (from a separate spectrum of a stronger but impure sample), 174.4, 176.7; HRMS–ESI+ (m/z): [M]+ calcd for C8H13N2O, 153.1022; found, 153.1022. The free-base was also prepared in a separate large-scale experiment [from crude 16 (17.6 g, 69.8 mmol) giving hydrochloride 17 (13.1 g, 69.4 mmol)]. Thus, a 6.4 g sample of the crude salt 17 was dissolved in methanol/triethylamine 9:1, concentrated, then re-dissolved in the minimum amount of methanol to allow precipitation of triethylamine hydrochloride by the addition of THF. The solid was filtered off and the filtrate concentrated. The residue was purified by two sequential rounds of column chromatography (ethyl acetate/triethylamine 99:1 → ethyl acetate/methanol/triethylamine 89:10:1) to produce the pyrrolizinone free-base (2.28 g, 15.0 mmol, equating to 44% overall yield from proline derivative 16). Rf 0.10 (ethyl acetate/methanol 9:1); 1H NMR (400 MHz, CDCl3) δ [the resonances vary with sample concentration and source of CDCl3] 1.56–1.65 (m, 1H) overlaying 1.60 (s, 3H), 1.94–2.04 (m, 2H), 2.10–2.29 (m, 1H), 3.08–3.15 (m, 1H), 3.17–3.23 (m, 1H), 3.86 (t, J = 8.0 Hz, 1H), 4.67 (br s, 2H); 13C NMR (100 MHz, CD3OD) δ 5.6, 28.1, 29.0, 47.9, 71.1, 93.0, 177.6, 194.3.
N-(7a-Methoxy-2-methyl-1-oxo-5,6,7,7a-tetrahydro-1H-pyrrolizin-3-yl)-3-methylbutanamide, O-methyl-legonmycin A (22)
This experiment was performed as described in order to trial various work-up and chromatography options. To a mixture of 3-aminopyrrolizine hydrochloride derivative 17 (2.0 g, 10.6 mmol), distilled pyridine (3.42 mL, 42.4 mmol), and acetonitrile (21 mL) at rt was added isovaleryl chloride (2.58 mL, 21.2 mmol). The mixture was stirred for 1 h, then half of the reaction mixture was removed via syringe to a second flask and concentrated (the remaining half was used for a separate reaction). The residue was dissolved in methanol (53 mL) then the stirred mixture was cooled to −78 °C and a pre-cooled (−78 °C) solution of I2 (1.35 g, 5.32 mmol) in methanol (53 mL) was added via cannula over 15 min. The reaction mixture was allowed to warm to rt over 14 h, the volatiles were removed, then pyridine (2 mL) and activated charcoal (5 g) were added. The resulting slurry was part-purified by column chromatography (dichloromethane → methanol) and the product-containing fractions were combined and recolumned (dichloromethane → dichloromethane/methanol 9:1). The product-containing fractions were combined and diluted with CuSO4·5H2O solution (20 mL; saturated, in methanol), shaken with activated carbon (5 g), filtered, and concentrated to afford a brown oil (1.33 g). This residue was subjected to a third round of chromatography (toluene/ethyl acetate 3:2 → toluene/ethyl acetate/acetone 2:2:1) to afford the title compound as an oil (661 mg, 47%). Rf 0.35 (dichloromethane/methanol 9:1); 1H NMR (400 MHz, CD3OD) δ 1.02 (d, J = 6.5 Hz, 3H), 1.03 (d, J = 6.5 Hz, 3H), 1.59 (s, 3H), 1.64 (ddd, J = 13.0, 10.5, 7.5 Hz, 1H), 1.94 (ddd, J = 13.0, 6.5, 3.0 Hz, 1H), 1.98–2.09 (m, 1H), 2.16 (≈ nonet, apparent J = 7.0 Hz, 1H), 2.32–2.34 (m, partially obscured by residual PhCH3, 1H), 2.34 (dd, J = 14.5, 7.0 Hz, 1H), 2.40 (dd, J = 14.5, 7.0 Hz, 1H), 3.10 (≈dt, J = 11.0, 8.0 Hz, 1H) overlays 3.12 (s, 3H), 3.37 (ddd, J = 11.0, 8.0, 3.5 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 6.7, 22.6, 22.7, 27.2, 27.7, 33.4, 46.6, 49.3, 51.7, 101.6, 102.0, 173.1, 199.9 (one resonance not resolved); LRMS–ESI+ (m/z): 289.1 ([M + Na]+, 26%), 235.2 ([M + H – MeOH]+, 100%), 151.0 (24%); HRMS–ESI+ (m/z): [M − OMe]+ calcd for C13H19N2O2, 235.1441; found, 235.1443.
N-(7a-Hydroxy-2-methyl-1-oxo-5,6,7,7a-tetrahydro-1H-pyrrolizin-3-yl)-3-methylbutanamide, legonmycin A (3)
To a mixture of 3-aminopyrrolizine hydrochloride derivative 17 (412 mg, 2.18 mmol), distilled pyridine (882 µL, 10.9 mmol), and acetonitrile (4.36 mL) at rt was added isovaleryl chloride (532 µL, 4.36 mmol). The mixture was stirred for 1 h, then a solution of I2 (54.5 mg, 0.215 mmol) in water (2.2 mL) and acetonitrile (15.2 mL) was added. O2 was bubbled gently through the solution for 15 h, and the mixture was concentrated with azeotropic removal of water with toluene (2 × 50 mL). The residue was purified by column chromatography (toluene/ethyl acetate/acetone 3:2:0–12:8:5) to afford the title compound as a yellow amorphous solid (345 mg, 56% corrected for the presence of ≈10 wt % toluene). The data are consistent with literature values [22]. Rf 0.10 (toluene/ethyl acetate/acetone 2:2:1); mp 128 °C [lit. mp not given]; IR νmax 3234br, 2958m, 1667m, 1571s, 1517s, 1437s, 1197m, 730m; 1H NMR (400 MHz, CD3OD) δ 1.01 (d, J = 6.5 Hz, 3H), 1.02 (d, J = 6.5 Hz, 3H), 1.58 (s, 3H), 1.65 (ddd, J = 13.0, 10.5, 7.5 Hz, 1H), 1.93 (ddd, J = 13.0, 6.5, 3.5 Hz, 1H), 1.99–2.11 (m, 1H), 2.15 (nonet, apparent J = 6.5 Hz, 1H), 2.32 (dd, J = 14.5, 7.0 Hz, 1H) overlaying 2.28–2.39 (m, 1H), 2.38 (dd, J = 14.5, 7.0 Hz, 1H), 3.04 (ddd, J = 11.0, 8.5, 7.0 Hz, 1H), 3.43 (ddd, J = 11.0, 8.0, 4.0 Hz, 1H); 13C NMR (100 MHz, CD3OD) δ 7.0, 22.6, 22.7, 27.2, 27.8, 33.8, 46.6, 49.3, 97.7, 99.4, 169.4, 172.9 (carbonyl resonance at ≈201–202 not seen); HRMS–ESI+ (m/z): [M – OH]+ calcd for C13H19N2O2, 235.1441; found, 235.1439.
N-(7a-Hydroxy-2-methyl-1-oxo-5,6,7,7a-tetrahydro-1H-pyrrolizin-3-yl)-3-methylpentanamide, legonmycin B (4)
Two separate small-scale diacylation reactions were run in parallel and then combined for oxidative hydrolysis. The quantities for each reaction are separated by a forward slash. To a mixture of 3-aminopyrrolizine hydrochloride 17 (22.3/43.6 mg, 0.118/0.231 mmol) and a solution of distilled pyridine in acetonitrile (1.20/2.30 mL, 0.5 M, 0.590/1.15 mmol) was added isocaproyl chloride (32/63 µL, 0.237/0.468 mmol). Both reaction mixtures were stirred for 18 h and then combined. Water (350 µL) and I2 (8.4 mg, 0.0331 mmol) were added sequentially. O2 was bubbled gently through the solution for 21 h and the mixture was concentrated. The residue was purified by column chromatography (toluene/ethyl acetate/acetone 3:2:0–0:1:1) to afford the title compound as a yellow glass (34.7 mg, 37%). The data are consistent with literature values [22]. Rf 0.40 (acetone/ethyl acetate/toluene 2:1:1); mp 49 °C [lit. mp not given]; IR νmax 3234br, 2957m, 1698m, 1668m, 1571s, 1454m, 1195m; 1H NMR (400 MHz, DMSO-d6) δ 0.88 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.5 Hz, 3H), 1.42–1.60 (m, 4H) overlaying 1.44 (s, 3H), 1.71 (ddd, J = 13.0, 6.5, 4.0 Hz, 1H), 1.77–1.87 (m, 1H), 2.05–2.17 (m, 1H), 2.37 (dd, J = 15.0, 7.5 Hz, 1H), 2.44 (dd, J = 15.0, 7.5 Hz, 1H), 2.91 (dt, J = 11.0, 7.5 Hz, 1H), 3.20 (ddd, J = 11.0, 7.5, 4.5 Hz, 1H), 6.04 (s, 1H), 10.16 (s, 1H); 13C NMR (100 MHz, DMSO-d6) δ 7.3, 22.2 (two peaks), 26.1, 27.2, 32.9, 33.7, 34.0, 47.6, 95.6, 97.3, 166.1, 170.9, 198.8; HRMS–ESI+ (m/z): [M + H]+ calcd for C14H23N2O3, 267.1703; found, 267.1703.
Supporting Information
Supporting Information File 1:
Copies of the NMR spectra for compounds 16, 17 (crude HCl salt and purified free-base), 22, 3 (legonmycin A), and 4 (legonmycin B).
WJML was supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1).
References
Robertson, J.; Stevens, K. Nat. Prod. Rep.2014,31, 1721–1788. doi:10.1039/c4np00055b
Return to citation in text:
[1]
Robertson, J.; Stevens, K. Nat. Prod. Rep.2017,34, 62–89. doi:10.1039/c5np00076a
Return to citation in text:
[1]
Tamariz, J.; Burgueño-Tapia, E.; Vázquez, M. A.; Delgado, F. Pyrrolizidine alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press, 2018; Vol. 80, pp 1–314. doi:10.1016/bs.alkal.2018.03.001
Return to citation in text:
[1]
Ratmanova, N. K.; Andreev, I. A.; Leontiev, A. V.; Momotova, D.; Novoselov, A. M.; Ivanova, O. A.; Trushkov, I. V. Tetrahedron2020,76, 131031. doi:10.1016/j.tet.2020.131031
Return to citation in text:
[1]
DeBoer, C.; Dolak, L. A.; Peterson, D. H. Composition of matter and process. Novel antibiotic formulations of antibiotic 354 (U-54,703). U.S. Patent 4,113,855, Sept 12, 1978.
Return to citation in text:
[1]
Horiuchi, Y.; Kondo, S.; Ikeda, T.; Ikeda, D.; Miura, K.; Hamada, M.; Takeuchi, T.; Umezawa, H. J. Antibiot.1979,32, 762–764. doi:10.7164/antibiotics.32.762
Return to citation in text:
[1]
[2]
Nakamura, H.; Iitaka, Y.; Umezawa, H. J. Antibiot.1979,32, 765–767. doi:10.7164/antibiotics.32.765
Return to citation in text:
[1]
[2]
Buechter, D. D.; Thurston, D. E. J. Nat. Prod.1987,50, 360–367. doi:10.1021/np50051a004
Return to citation in text:
[1]
Doyle, T. W.; Nettleton, D. E.; Balitz, D. M.; Moseley, J. E.; Grulich, R. E.; McCabe, T.; Clardy, J. J. Org. Chem.1980,45, 1324–1326. doi:10.1021/jo01295a037
Return to citation in text:
[1]
Zhang, Q.; Schrader, K. K.; ElSohly, H. N.; Takamatsu, S. J. Antibiot.2003,56, 673–681. doi:10.7164/antibiotics.56.673
Return to citation in text:
[1]
Hu, J.-F.; Wunderlich, D.; Thiericke, R.; Dahse, H.-M.; Grabley, S.; Feng, X.-Z.; Sattler, I. J. Antibiot.2003,56, 747–754. doi:10.7164/antibiotics.56.747
Return to citation in text:
[1]
Snider, B. B.; Duvall, J. R.; Sattler, I.; Huang, X. Tetrahedron Lett.2004,45, 6725–6727. doi:10.1016/j.tetlet.2004.07.055
Return to citation in text:
[1]
Snider, B. B.; Duvall, J. R. Org. Lett.2005,7, 4519–4522. doi:10.1021/ol0518784
Return to citation in text:
[1]
[2]
[3]
Duvall, J. R.; Wu, F.; Snider, B. B. J. Org. Chem.2006,71, 8579–8590. doi:10.1021/jo061650+
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Bugni, T. S.; Woolery, M.; Kauffman, C. A.; Jensen, P. R.; Fenical, W. J. Nat. Prod.2006,69, 1626–1628. doi:10.1021/np0602721
Return to citation in text:
[1]
Jiang, B.; Zhao, W.; Li, S.; Liu, H.; Yu, L.; Niu, W.; He, H.; Wu, L. J. Antibiot.2018,71, 965–967. doi:10.1038/s41429-018-0084-5
Return to citation in text:
[1]
Huang, S.; Tabudravu, J.; Elsayed, S. S.; Travert, J.; Peace, D.; Tong, M. H.; Kyeremeh, K.; Kelly, S. M.; Trembleau, L.; Ebel, R.; Jaspars, M.; Yu, Y.; Deng, H. Angew. Chem., Int. Ed.2015,54, 12697–12701. doi:10.1002/anie.201502902
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
Schimming, O.; Challinor, V. L.; Tobias, N. J.; Adihou, H.; Grün, P.; Pöschel, L.; Richter, C.; Schwalbe, H.; Bode, H. B. Angew. Chem., Int. Ed.2015,54, 12702–12705. doi:10.1002/anie.201504877
Return to citation in text:
[1]
Liu, L.; Li, S.; Sun, R.; Qin, X.; Ju, J.; Zhang, C.; Duan, Y.; Huang, Y. Org. Lett.2020,22, 4614–4619. doi:10.1021/acs.orglett.0c01224
Return to citation in text:
[1]
Stevens, K.; Tyrrell, A. J.; Skerratt, S.; Robertson, J. Org. Lett.2011,13, 5964–5967. doi:10.1021/ol202381m
Return to citation in text:
[1]
[2]
McNab, H.; Monahan, L. C.; Walton, J. C. J. Chem. Soc., Perkin Trans. 21988, 759–764. doi:10.1039/p29880000759
Return to citation in text:
[1]
Gaber, A. E.-A. M.; Hunter, G. A.; McNab, H. J. Chem. Soc., Perkin Trans. 12002, 548–554. doi:10.1039/b109788a
Return to citation in text:
[1]
McNab, H.; Morrow, M.; Parsons, S.; Shannon, D. A.; Withell, K. Org. Biomol. Chem.2009,7, 4936–4942. doi:10.1039/b911951e
Return to citation in text:
[1]
Kondakal, V. V. R.; Qamar, M. I.; Hemming, K. Tetrahedron Lett.2012,53, 4100–4103. doi:10.1016/j.tetlet.2012.05.117
Return to citation in text:
[1]
Jamshaid, F.; Kondakal, V. V. R.; Newman, C. D.; Dobson, R.; João, H.; Rice, C. R.; Mwansa, J. M.; Thapa, B.; Hemming, K. Tetrahedron2020,76, 131570. doi:10.1016/j.tet.2020.131570
Return to citation in text:
[1]
Volovenko, Y. M.; Shokol, T. V.; Babichev, F. S. Dopov. Nats. Akad. Nauk Ukr.1986, 34–36.
Return to citation in text:
[1]
Wittenberger, S. J. J. Org. Chem.1996,61, 356–358. doi:10.1021/jo9509723
Return to citation in text:
[1]
Haight, A. R.; Stuk, T. L.; Menzia, J. A.; Robbins, T. A. Tetrahedron Lett.1997,38, 4191–4194. doi:10.1016/s0040-4039(97)00866-6
Return to citation in text:
[1]
Gola, A.; Samartzi, E.; Bardakos, V.; Petroliagi, M.; Igglessi-Markopoulou, O.; Markopoulos, J.; Barkley, J. V. J. Heterocycl. Chem.2000,37, 681–686. doi:10.1002/jhet.5570370402
Return to citation in text:
[1]
Petroliagi, M.; Igglessi-Markopoulou, O. J. Heterocycl. Chem.2001,38, 917–922. doi:10.1002/jhet.5570380416
Return to citation in text:
[1]
Gardner, P. D. J. Am. Chem. Soc.1956,78, 3421–3424. doi:10.1021/ja01595a043
Return to citation in text:
[1]
Tamariz, J.; Burgueño-Tapia, E.; Vázquez, M. A.; Delgado, F. Pyrrolizidine alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press, 2018; Vol. 80, pp 1–314. doi:10.1016/bs.alkal.2018.03.001
4.
Ratmanova, N. K.; Andreev, I. A.; Leontiev, A. V.; Momotova, D.; Novoselov, A. M.; Ivanova, O. A.; Trushkov, I. V. Tetrahedron2020,76, 131031. doi:10.1016/j.tet.2020.131031
DeBoer, C.; Dolak, L. A.; Peterson, D. H. Composition of matter and process. Novel antibiotic formulations of antibiotic 354 (U-54,703). U.S. Patent 4,113,855, Sept 12, 1978.
Doyle, T. W.; Nettleton, D. E.; Balitz, D. M.; Moseley, J. E.; Grulich, R. E.; McCabe, T.; Clardy, J. J. Org. Chem.1980,45, 1324–1326. doi:10.1021/jo01295a037
Duvall, J. R.; Wu, F.; Snider, B. B. J. Org. Chem.2006,71, 8579–8590. doi:10.1021/jo061650+
34.
Gola, A.; Samartzi, E.; Bardakos, V.; Petroliagi, M.; Igglessi-Markopoulou, O.; Markopoulos, J.; Barkley, J. V. J. Heterocycl. Chem.2000,37, 681–686. doi:10.1002/jhet.5570370402
Jamshaid, F.; Kondakal, V. V. R.; Newman, C. D.; Dobson, R.; João, H.; Rice, C. R.; Mwansa, J. M.; Thapa, B.; Hemming, K. Tetrahedron2020,76, 131570. doi:10.1016/j.tet.2020.131570



![[1860-5397-17-31-1]](/bjoc/content/figures/1860-5397-17-31-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-31-2]](/bjoc/content/figures/1860-5397-17-31-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-31-i1]](/bjoc/content/inline/1860-5397-17-31-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-31-i2]](/bjoc/content/inline/1860-5397-17-31-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-31-i3]](/bjoc/content/inline/1860-5397-17-31-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)