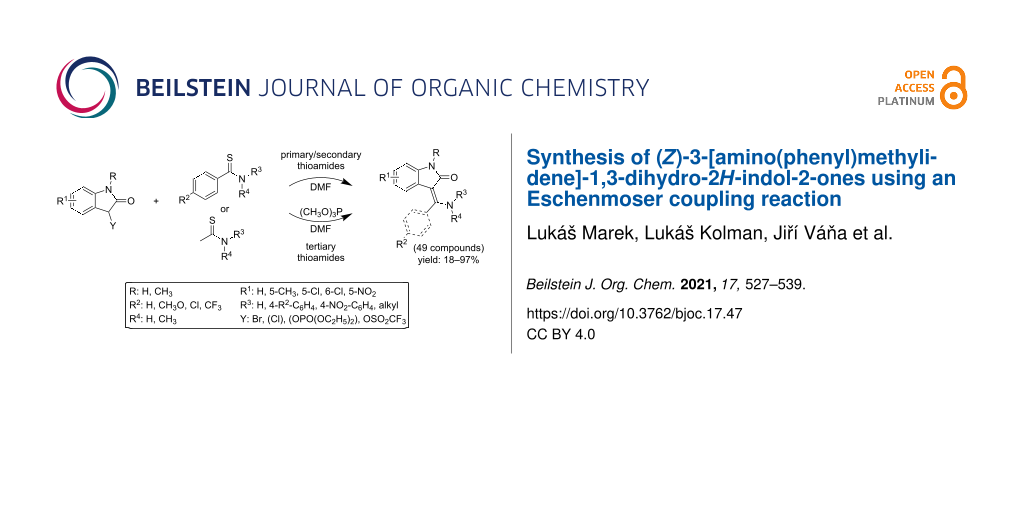
Abstract
A highly modular method for the synthesis of (Z)-3-[amino(phenyl/methyl)methylidene]-1,3-dihydro-2H-indol-2-ones starting from easily available 3-bromooxindoles or (2-oxoindolin-3-yl)triflate and thioacetamides or thiobenzamides is described. A series of 49 compounds, several of which have previously been shown to possess significant tyrosin kinase inhibiting activity, was prepared in yields varying mostly from 70 to 97% and always surpassing those obtained by other published methods. The method includes an Eschenmoser coupling reaction, which is very feasible (even without using a thiophile except tertiary amides) and scalable. The (Z)-configuration of all products was confirmed by NMR techniques.

Graphical Abstract
Introduction
3-(Aminomethylidene)-1,3-dihydro-2H-indol-2-ones (3-(aminomethylidene)oxindoles) belong to a subclass of intensively studied heterocyclic compounds, especially due to their significant pharmacological activity. As early as in the middle of the 1980s researchers at Pfizer Inc. patented [1] these compounds as highly potent gabaergic agents, having possible therapeutic utility as anticonvulsants or anxiolytics [2]. However, the main therapeutic potential of these compounds was discovered ca 15 years later when several patents appeared claiming their inhibiting effect on various kinases [3,4]. Since that time many patents as well as papers have been published [5-11] on that topic but until present only Nintedanib (Figure 1) was approved [12,13] for the treatment of idiopathic pulmonary fibrosis in 2014.
![[1860-5397-17-47-1]](/bjoc/content/figures/1860-5397-17-47-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The title compounds can be prepared using several synthetic approaches (Scheme 1) starting either from 1,3-dihydro-2H-indol-2-ones (oxindoles) and nitriles, amides, amide acetals, and imidoesters [1-3,5,11,14] or from their independently prepared 3-chloromethylidene- [4,9,15-17], 3-hydroxymethylidene- or 3-(alkoxymethylidene)oxindoles [2,5,6,18-20] and the appropriate amines. Recently, original methods involving the construction of the oxindole skeleton were also described starting from 2-alkynylphenyl isocyanates and their precursors [21-23], or from N-phenylpropiolamides [24-26]. The newest method of synthesis, starting from 3-bromo-3-[bromo(phenyl)methyl]oxindole and substituted anilines [27], is similar to a long-known preparation utilizing 3-chloro-3-(nitroalkyl)oxindoles and amines [28].
![[1860-5397-17-47-i1]](/bjoc/content/inline/1860-5397-17-47-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: The known synthetic strategies leading to 3-(aminomethylidene)oxindoles.
Scheme 1: The known synthetic strategies leading to 3-(aminomethylidene)oxindoles.
Except these synthetically valuable methods, several notes concerning the formation of 3-(aminomethylidene)-1,3-dihydro-2H-indol-2-ones under different conditions can be found in the literature [29-31].
In our group we recently discovered [32,33] a new synthetic pathway involving a rearrangement of 2-aryl-5-(2-aminophenyl)-4-hydroxy-1,3-thiazoles (e.g., 8aa–ad in Scheme 2) leading to the 3-[amino(aryl)methylidene]-1,3-dihydro-2H-indol-2-ones containing an unsubstituted amino group and the oxindole nucleus. In the present paper, we want to describe an optimized procedure avoiding the intermediary thiazoles and to demonstrate the usefulness and wide group tolerability of our new synthetic approach for the preparation of a library of substituted phenyl and amino derivatives (Table 1). A number of the compounds that have been prepared using this route (namely 5ac’, 5ca, 5cc’, 5da, 5db, and 5dc’) have significant tyrosin kinase inhibiting properties [11].
Table 1: A survey and labeling of synthesized 3-[amino(phenyl)methylidene]-1,3-dihydro-2H-indol-2-ones.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-47-i5.svg?max-width=637&scale=1.0)
|
|||||||||||
| entry | 1 | R1 | 2 | R2 | R3 = R4 | 3 | R2 = R3 | R4 | 4 | R2 | R3 = R4 |
| 1 | a | H | a | H | H | a | H | C6H5 | a | H | CH3 |
| 2 | b | 5-CH3 | b | OCH3 | H | b | H | 4-OCH3-C6H4 | b | OCH3 | CH3 |
| 3 | c | 5-Cl | c | Cl | H | c | H | 4-Cl-C6H4 | c | Cl | CH3 |
| 4 | d | 6-Cl | c’ | I | H | d | H | 4-CF3-C6H4 | |||
| 5 | e | 5-NO2 | d | CF3 | H | e | H | 4-NO2-C6H4 | |||
| 6 | f | H | CH3 | ||||||||
| 7 | g | H | CH3(CH2)4 | ||||||||
| 8 | h | H | C6H5CH2 | ||||||||
| 9 | i | H | cyclohexyl | ||||||||
aNote: in all shown formulas of products the first letter relates to the substitution of compound 1, whereas the second letter relates to a substitution of 2–4 (e.g., 7ba means combination of 1b + 4a).
Results and Discussion
In our previous paper [33] we reported the formation of 3-[amino(aryl)methylidene]-1,3-dihydro-2H-indol-2-ones (R1, R3, R4: H) through the intermediary (and in most cases isolable) 2-aryl-5-(2-aminophenyl)-4-hydroxy-1,3-thiazoles 8aa–ad formed from 3-bromooxindole (1a) and various substituted primary aromatic thioamides (e.g., thiobenzamides 2a–d) in acetonitrile (Scheme 2). Although the overall yields of such a two-step transformation was acceptable (around 70–80%), later we have found that a further substitution of the 3-bromooxindole (1a), especially with electron-withdrawing groups (R1 is EWG), causes either an exclusive formation of the corresponding 2-aryl-5-(2-aminophenyl)-4-hydroxy-1,3-thiazoles (when starting from 2c,d) or leads to a complex and inseparable mixture of products (when starting from 2a,b). The addition of a base (e.g., triethylamine, N-methylmorpholine, ammonia) which was originally found to be beneficial [33] for the rearrangement of the kinetically formed thiazole to the desired product now caused a complete decomposition giving mainly the isoindigo derivatives 9a–e. Similar issues were observed when the secondary or tertiary thiobenzamides 3a–i or 4a–c were used. The following Scheme 2 summarizes the main possible reaction routes starting from 3-bromooxindoles 1a–e and various primary, secondary, and tertiary thiobenzamides 2a–d, 3a–i, and 4a–c.
![[1860-5397-17-47-i2]](/bjoc/content/inline/1860-5397-17-47-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: The possible intermediates and products occurring in the reactions of 3-bromooxindoles with thiobenzamides.
Scheme 2: The possible intermediates and products occurring in the reactions of 3-bromooxindoles with thioben...
The formation of the different products can be explained as follows. In acetonitrile, that was used in our previous study [33], the C3 hydrogen of the initially formed α-thioiminium salt is not acidic enough to form the carbanion that is necessary for the desired Eschenmoser coupling reaction (route b). Therefore, the nucleophilicity of the conjugated base of the nitrogen (benzenecarbimidothioate or thioimidate) is exerted towards the oxindole carbonyl to give the thiazole. Moreover, if both benzene rings contain electron-withdrawing groups, enhancing either the leaving ability of the oxindole nitrogen (R1: Cl, NO2) or the acidity of the >C=NH2+ group (R2: Cl, CF3) increasing the proportion of the nucleophile >C=NH, then the formation of the thiazole (route a) is further accelerated (Scheme 2). Although the addition of a base generates the required C3 carbanion, such a base also converts the reactive α-thioiminium group to a much less reactive thioimidate at the same time. The C3 carbanion then preferentially attacks (route c) another molecule of the starting material 1a–e and the resulting adducts undergo a subsequent elimination to give the isoindigo derivatives 9a–e. The substituent R4 strongly influences both the equilibrium between >C=N+HR4 and >C=NR4 as well as the nucleophilic attack of the internal C3 carbanion to give the thiirane intermediate in Scheme 2. While the EWG groups decrease the concentration of the α-thioiminium salt (that is much more prone to a nucleophilic addition than the free thioimidate group) they also make the α-thioiminium/thioimidate group more reactive. These two effects are antagonistic and therefore a thorough study of the R4 substituent influence is necessary. On the other hand, mono- as well as disubstitution of the thioamide nitrogen completely prevents the formation of the thiazole (route a).
The rather complicated product isolation procedure of the Eschenmoser coupling reaction (route b) and the long reaction time taken together with problems connected with changing substituents throughout the molecule, encouraged us to find better or even universal reaction conditions compatible with all substituents present in both benzene rings (R1 and R2) as well as at the thioamide nitrogen (R3 and R4).
In order to achieve the desired product of the Eschenmoser coupling reaction (route b), two main prerequisites must be fulfilled – i.e., the substantial acidity of a hydrogen at C3 and a more stable α-thioiminium group necessary for the internal nucleophilic addition giving the thiirane intermediate (route b). It is well known that the acidity of carbon acids in acetonitrile is much lower than in polar aprotic solvents, like DMSO or DMF or in polar protic solvents like alcohols. For carbon acids the ΔpKa is 12.9 between ACN and DMSO but the ΔpKa is only −1.5 between DMF and DMSO [34]. A change of the solvent from acetonitrile to DMF should therefore decrease the pKa by ca. 11 units. In polar protic solvents (e.g., in methanol) such an increase of acidity should be also distinct but it is known that polar protic solvents much better solvate anions and thus decrease their nucleophilicity. Therefore, dimethylformamide has been chosen as the best solvent.
Indeed, when dimethylformamide (DMF) was used as the solvent for the reaction of the substituted 3-bromooxindoles 1a–e with various aromatic primary (2a–d) and secondary (3a–i) thiobenzamides, the corresponding 3-[amino(phenyl)methylidene]oxindoles (series 5 and 6) were obtained in good yields mostly exceeding 70% without the occurrence of intermediary thiazoles (route a). Only with a few secondary thiobenzamides (3c–e) or the tertiary thiobenzamides 4b,c, the yields were lower.
Reaction of 1a–e with primary thiobenzamides 2a–d (R3, R4: H)
All possible combinations of the 3-bromooxindoles 1a–e with the primary thiobenzamides 2a–d were studied in order to test the R1 and R2 substituent tolerance. Moreover, in order to demonstrate the application potential of our method, a few more derivatives containing R2: I instead of Cl (5ac’, 5cc’, 5dc’) which display a significant kinase inhibition activity, have been prepared. It is worth noting that the existing literature synthetic pathway [11] gave only poor yields of these desirable compounds. From the data presented in Table 2, it is clear that the yields obtained in DMF are mostly very good and always much better than the previously reported yields by us in ACN [33] (see Table 2, entries 1–4) or by other authors starting from iminoesters [11] (see Table 2, entries 9, 13, 14, and 21–23). However, the inspection of Table 2 does not show any clear dependence between the electronic properties of the substituent R1 or R2 and the isolated yield. In most cases the yields are higher than 80%, but it seems that the presence of at least one EWG often gives an enhanced yield above 90% (cf. entries 3, 4, 8, 9, 11, 15, 20, and 21 in Table 2). The influence of a single substituent R1 is weaker (e.g., entries 1, 5, 13, and 17 in Table 2) because the electron-withdrawing groups enhance the C3–H acidity and an EDG enhances the C3 carbanion nucleophilicity and these two effects are opposing. The influence of the substituent effects, as quantified using Hammett’s substituent constants [35], on the yields can be represented graphically (Figure 2).
Table 2: The prepared 3-[amino(phenyl)methylidene]-1,3-dihydro-2H-indol-2-ones 5aa–ed with a primary amino group (R3 and R4: H).
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-47-i6.svg?max-width=637&scale=1.0)
|
||||||||
| entry | product | R1 | R2 |
n1a–e/n2a–d
(mmol) |
m1a–e
(mg) |
m2a–d
(mg) |
yield 5aa–ed
(mg)/(%) |
mp
(°C) |
| 1 | 5aa | H | H | 3.3/3 | 700 | 412 | 623/(88, 69a, 84b, 70c) | 239–240 |
| 2 | 5ab | H | OCH3 | 3.3/3 | 700 | 502 | 655/(82, 73a, 10d) | 229.5–231 |
| 3 | 5ac | H | Cl | 3.3/3 | 700 | 515 | 754/(93, 71a) | 237–238.5 |
| 4 | 5ad | H | CF3 | 3.3/3 | 700 | 616 | 884/(97, 77a) | 244–245 |
| 5 | 5ba | 5-CH3 | H | 2.75/2.5 | 622 | 343 | 526/(84) | 206–208 |
| 6 | 5bb | 5-CH3 | OCH3 | 2.75/2.5 | 622 | 418 | 482/(69) | 213–215 |
| 7 | 5bc | 5-CH3 | Cl | 2.75/2.5 | 622 | 429 | 520/(73) | 246–247.5 |
| 8 | 5bd | 5-CH3 | CF3 | 3.3/3 | 746 | 616 | 891/(93) | 234–236 |
| 9 | 5ca | 5-Cl | H | 1.56/1.42 | 350 | 194 | 373/(97, 29d) | 212–214 |
| 10 | 5cb | 5-Cl | OCH3 | 3.3/3 | 813 | 502 | 658/(73, 18d) | 235–237 |
| 11 | 5cc | 5-Cl | Cl | 3.3/3 | 813 | 515 | 824/(90) | 281.5–283 |
| 12 | 5cd | 5-Cl | CF3 | 3.3/3 | 813 | 616 | 770/(76) | 235–238 |
| 13 | 5da | 6-Cl | H | 1.56/1.42 | 350 | 194 | 323/(84, 22d) | 191–193 |
| 14 | 5db | 6-Cl | OCH3 | 2.2/2 | 493 | 334 | 523/(87, 20d) | 229.5–231 |
| 15 | 5dc | 6-Cl | Cl | 3.3/3 | 813 | 515 | 861/(94) | 228–229.5 |
| 16 | 5dd | 6-Cl | CF3 | 3.3/3 | 813 | 616 | 796/(78, 30c) | 292–295 |
| 17 | 5ea | 5-NO2 | H | 2.2/2 | 565 | 274 | 445/(79) | 296–298 |
| 18 | 5eb | 5-NO2 | OCH3 | 2.2/2 | 565 | 334 | 455/(73) | 328–331 |
| 19 | 5ec | 5-NO2 | Cl | 2.2/2 | 565 | 343 | 489/(78) | >372 dec. |
| 20 | 5ed | 5-NO2 | CF3 | 2.2/2 | 565 | 410 | 674/(97) | 327–330 |
| 21 | 5ac’ | H | I | 1.65/1.5 | 350 | 395 | 500/(93, 6d) | 242–245 |
| 22 | 5cc’ | 5-Cl | I | 1.65/1.5 | 407 | 395 | 520/(87, 8d) | 257–258 |
| 23 | 5dc’ | 6-Cl | I | 1.65/1.5 | 407 | 395 | 510/(86, 15d) | 257–260 |
aIn ACN (cf. reference [33]); bprepared from 2-(phenylethynyl)phenylisocyanate (see reference [22]); cprepared from oxindole and the corresponding benzonitrile (see reference [14]); dprepared from oxindole and benzoic acid imidoester (see reference [11]).
![[1860-5397-17-47-2]](/bjoc/content/figures/1860-5397-17-47-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The R1 and R2 substitution influence on the isolated yields of products 5aa–ed.
Figure 2: The R1 and R2 substitution influence on the isolated yields of products 5aa–ed.
Reaction of 1a–e with secondary thiobenzamides 3a–i
Secondary thiobenzamides 3a–i also react well with 3-bromooxindoles 1a–e (Table 3) although the average yields of the corresponding products 6aa–ef are always 10–15% lower when comparing substrates with the same R1 (R2 and R3 are always H). For example, while for the reaction of 1a with 2a the yield is 88% (Table 2, entry 1), a nitrogen substitution (3a: R4: C6H5 or 3f: R4: CH3) lowers the isolated yields to 77% and 76% (Table 3, entries 1 and 6), respectively. The further substitution of the N-phenyl group with an EWG causes a significant decrease in the isolated yield (cf. entries 1, and 3–5 in Table 3) from 77% to only 32% while an EDG substituent (4-OCH3; entry 2 in Table 3) enhances the yield to 88%. From these observations it is clear that the influence of R4 is much stronger than the influence of the substituent R1 (cf. entries 1 and 10–13 or entries 6 and 14–17 in Table 3). The attached EWG substituent R4 lowers the pKa (and thus also the equilibrium concentration) of the intermediary α-thioiminium salt which is much more reactive towards an internal nucleophilic attack than the free thioimidate group. Although an electron-withdrawing substituent R4 also enhances the internal nucleophilic attack to both α-thioiminium and imine groups – such influence is weaker because this addition occurs on a carbon atom more distant from R4. The presumption that the presence of an α-thioiminium group is a necessary prerequisite for the successful reaction progress is further supported by the fact that all N-alkyl-substituted thiobenzamides 3f–i (see entries 6–9 in Table 3) give comparable yields around 70%.
Table 3: The prepared (Z)-3-[amino(phenyl)methylidene]-1,3-dihydro-2H-indol-2-ones 6aa–ef with a secondary amino group (R2 and R3: H).
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-47-i7.svg?max-width=637&scale=1.0)
|
||||||||
| entry | product | R1 | R4 |
n1a–e/n2a–d
(mmol) |
m1a–e
(mg) |
m3a–i
(mg) |
yield 6aa–ee
(mg)/(%) |
mp
(°C) |
| 1 | 6aa | H | C6H5 | 3.3/3 | 700 | 640 | 721/(77%) | 322.5–325 |
| 2 | 6ab | H | 4-CH3O-C6H4 | 3.3/3 | 700 | 682 | 827/(88%, 82%a) | 271–273 |
| 3 | 6ac | H | 4-Cl-C6H4 | 3.3/3 | 700 | 743 | 503/(48%) | 286.5–288 |
| 4 | 6ad | H | 4-CF3-C6H4 | 3.3/3 | 700 | 844 | 488/(43%) | 282–284 |
| 5 | 6ae | H | 4-NO2-C6H4 | 3.3/3 | 700 | 775 | 339/(32%) | 281–283.5 |
| 6 | 6af | H | CH3 | 2.75/2.5 | 556 | 378 | 475/(76%) | 256–258 |
| 7 | 6ag | H | CH3(CH2)4 | 3.3/3 | 700 | 622 | 652/(71%) | 214–216 |
| 8 | 6ah | H | C6H5CH2 | 3.3/3 | 700 | 724 | 696/(71%) | 231–234 |
| 9 | 6ai | H | cyclo-C6H11 | 3.3/3 | 700 | 658 | 613/(64%) | 343–346 |
| 10 | 6ba | 5-CH3 | C6H5 | 3.15/3 | 712 | 640 | 674/(69%) | 258–261 |
| 11 | 6ca | 5-Cl | C6H5 | 1.31/1.25 | 324 | 267 | 380/(89%, 68%b) | 242–243 |
| 12 | 6da | 6-Cl | C6H5 | 3.15/3 | 776 | 640 | 749/(72%) | 319–322 |
| 13 | 6ea | 5-NO2 | C6H5 | 2.1/2 | 540 | 427 | 529/(74%) | 267–269 |
| 14 | 6bf | 5-CH3 | CH3 | 3.15/3 | 712 | 454 | 521/(66%) | 255–257.5 |
| 15 | 6cf | 5-Cl | CH3 | 3.15/3 | 776 | 454 | 605/(71%) | 256–258 |
| 16 | 6df | 6-Cl | CH3 | 3.15/3 | 776 | 454 | 719/(84%) | 274–276.5 |
| 17 | 6ef | 5-NO2 | CH3 | 3.15/3 | 810 | 454 | 656/(74%) | 332.5–335 |
aPrepared from 2-(phenylethynyl)phenylisocyanate (see reference [23]); bthe E-isomer prepared from 3-bromo-3-(bromo(phenyl)methyl)-5-chlorooxindole (see reference [27]).
Reaction of 1a–c with tertiary thiobenzamides 4a–c
The preliminary experiments involving the reaction of 1a with N,N-dimethylthiobenzamide (4a) in DMF have shown that the reaction is much slower (>24 h) and also gives relatively high amounts of the undesirable isoindigo 9a [32] (route c). In order to accelerate the Eschenmoser coupling reaction and to enhance the reaction yield (route b) an addition of a suitable thiophile assisting the sulfur extrusion should be beneficial [36]. Therefore, we tested two P(III) compounds as thiophiles. First, we added triphenylphosphine (1 equiv) but an even more complex mixture of products resulted. Much better results were obtained when trimethyl phosphite was used but this strongly depended on the used molar ratio: the best yield of the product 7aa (83%) was achieved when using 1.5 equiv of trimethyl phosphite. While a lower amount (1 equiv) had only a moderate influence on the reaction progress (yield of 7aa was 50%) a higher amount (3 equiv) caused a significant reduction of 1a to the parent oxindole (yield of 7aa was 44%). Therefore, the optimized amount (1.5 equiv) was used for the synthesis starting from the other thiobenzamides 4b and 4c and 3-bromooxindoles 1b and 1c (Table 4). From the inspection of Table 4 it is obvious that the isolated yields are lower (19–60%) but this is mainly due to the requirement for a more complex separation involving repeated column chromatography and/or crystallization.
Table 4: The prepared (Z)-3-[dimethylamino(phenyl)methylidene]-1,3-dihydro-2H-indol-2-ones 7aa–ca with a tertiary amino group (R3 and R4: CH3).
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-47-i8.svg?max-width=637&scale=1.0)
|
||||||||
| entry | product | R1 | R2 |
n1a–e/n4a–c
(mmol) |
m1a–e
(mg) |
m4a–c
(mg) |
yield 7aa–ca
(mg)/(%) |
mp
(°C) |
| 1 | 7aa | H | H | 1.1/1 | 233 | 165 | 220/(83%) | 239–240 |
| 2 | 7ab | H | CH3O | 1.1/1 | 233 | 195 | 140/(48%) | 272–273 |
| 3 | 7ac | H | Cl | 1.1/1 | 233 | 200 | 179/(60%) | 239.5–241 |
| 4 | 7ba | 5-CH3 | H | 5.5/5 | 1243 | 825 | 740/(53%) | 228–230 |
| 5 | 7ca | 6-Cl | H | 5.5/5 | 1360 | 825 | 280/(19%) | 269–270.5 |
Aliphatic thioamides
Although our concern was mainly dedicated to the synthesis of 3-[amino(aryl)methylidene]oxindoles many of which display a significant tyrosin kinase inhibiting activity, we also verified the versatility of our method for the analogous oxindoles 10a–c carrying a methyl group on the methylidene carbon (Scheme 3).
![[1860-5397-17-47-i3]](/bjoc/content/inline/1860-5397-17-47-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The Eschenmoser coupling reaction of 3-bromooxindole (1a) with thioacetamides.
Scheme 3: The Eschenmoser coupling reaction of 3-bromooxindole (1a) with thioacetamides.
The replacement of thiobenzamide and its N-methyl or N-phenyl derivative with the analogous aliphatic thioacetamides has virtually no influence on the reaction progress and the corresponding oxindoles 10a–c were obtained in 62–89% yields (Table 5).
Table 5: The prepared (Z)-3-[1-aminoethylidene]-1,3-dihydro-2H-indol-2-ones 10a–c.
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-47-i9.svg?max-width=637&scale=1.0)
|
|||||||
| entry | product | R |
n1a/nthioamide
(mmol) |
m1a
(mg) |
mthioamide
(mg) |
yield 10
(mg)/(%) |
mp
(°C) |
| 1 | 10a | H | 2/2 | 424 | 150 | 310/(89%, 65%a) | 261–262.5 |
| 2 | 10b | CH3 | 2/2 | 424 | 178 | 320/(85%) | 281–282 |
| 3 | 10c | C6H5 | 2/2 | 424 | 302 | 312/(62%) | 222–223.5 |
aPrepared from oxindole and the corresponding acetonitrile (see reference [14]).
Alternative electrophilic components for the Eschenmoser coupling reaction
The Eschenmoser coupling reaction [36,37] usually starts from α-substituted ketones, esters, malonates, or nitriles. The group in the α-position must be a good leaving group enabling a facile nucleophilic substitution giving the α-thioiminium salt and the cleaved leaving group should be a poor nucleophile in order to suppress the reverse reaction. In most cases halogens (mainly Br and I) fulfill these criteria but for highly congested thioamides only triflates gave desirable yields [36]. Moreover, several new alternatives to the Eschenmoser coupling reaction can be found in the recent literature [38-40].
In our case, the 3-bromooxindoles 1a–e worked very well but they are accessible from commercially available isatins only through a three-step synthesis in an acceptable overall yield 51–76% (see Table 6 in the Experimental). In order to avoid the relatively long synthesis of the precursors we tried to change Br for another leaving group Y (Scheme 4).
![[1860-5397-17-47-i4]](/bjoc/content/inline/1860-5397-17-47-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: The synthesis of alternative 3-substituted oxindoles and their Eschenmoser coupling reaction with thiobenzamide (2a).
Scheme 4: The synthesis of alternative 3-substituted oxindoles and their Eschenmoser coupling reaction with t...
First, we prepared 3-chlorooxindole (1f) which is available from β-nitrostyrene and acetyl chloride under FeCl3 catalysis [41]. Unfortunately, the Eschenmoser coupling reaction with thiobenzamide (2a) did not proceed at all and only the formation of isoindigo 9a was observed. However, a very easy synthesis of diethyl (2-oxoindolin-3-yl)phosphate (1g) from isatin has recently appeared [42] which encouraged us to submit this reagent to the Eschenmoser reaction. Unfortunately, this reagent reacts with the parent thiobenzamide (2a) very slowly even after heating to 80 °C. A maximum (chromatographic) yield 38% of 5aa was achieved after 32 h when a double molar excess of phosphate 1g was used. A further increase in the excess of 1g had no positive effect on the yield of 5aa. This result is in accordance with the relatively low nucleofugality of diethyl phosphate which is roughly quantified by the pKa = 1.39 of its conjugated acid [43]. We have also examined an addition of 20% of tetrabutylammonium bromide as a nucleophilic catalyst which could transform the phosphate 1g in situ to the more reactive bromo derivative 1a. It was found that such addition has only a small positive effect on the reaction progress, most probably due to only a slightly higher nucleophilicity of the bromide (n = 3.9) [44] in comparison with the thiobenzamide (n = 3.1) [45].
The last possibility for the activation of the oxindole component involves the introduction of the triflate group into position 3. The synthesis of triflates according to the standard protocol involves the reaction of an alcohol with triflic chloride or anhydride. However, these very reactive reagents could react with 3-hydroxyoxindole both on the oxygen as well as on the nitrogen. Therefore, we first examined a protocol starting from N-methylisatin which was reduced to 3-hydroxy-N-methyloxindole [46] (yield 75%; note: the reduction of other isatins gives lower yields) and then treated with triflic anhydride under inert atmosphere at −20 °C. Quite a clean formation of the triflate 1i was observed in an NMR tube but all attempts to isolate the pure compound 1i in a preparative scale failed. Therefore, we decided to carry out an Eschenmoser coupling reaction in a one-pot manner. Into a chilled solution of 3-hydroxy-N-methyloxindole in DMF, one equivalent of triflic anhydride was added first and, after reaction completion, one equivalent of thiobenzamide (2a) was added. By this route, the desired product of the Eschenmoser coupling 11 was isolated in a very good yield of 87% (cf. with 78% in reference [33] and 64% in reference [14]). As a result, a good overall yield of 65% was achieved over three steps starting from N-methylisatin. For comparison, the four-step synthesis starting from N-methylisatin and giving 11 through 3-bromo-N-methyloxindole (1a’), has an overall yield of only 48%. Moreover, no formation of the isoindigo derivative was observed under such conditions – probably due to the presence of triflic acid evolved during the first step. This new route is therefore suitable for the synthesis of N-alkyl-3-(aminomethylidene)oxindoles.
We also tried to apply the same protocol for the parent (i.e. N-unprotected) 3-hydroxyoxindole and thiobenzamide (2a). We partly succeeded because we isolated the coupling product 5aa in 65% yield (48% from isatin) which is still less than when starting from isatin through 1a (57%). For a further two representatives, namely 4’-trifluoromethylthiobenzanilide (3d) and N,N-dimethylthiobenzamide (4a) the same one-pot method gives only 18% of the product 6ad (cf. with 43% from 1a) and 27% (without thiophile) of 7aa (cf. with 86% from 1a). It was obvious that an improved synthetic approach would necessitate the protection of the oxindole nitrogen avoiding the formation of the undesired N-Tf derivative. Such nitrogen protection/deprotection (e.g., acetylation/deacetylation) is often used in the other synthetic strategies described in Scheme 1. Therefore, the starting isatin was acetylated first with acetic anhydride [47] in 95% yield and then reduced with various complex borohydrides (e.g., sodium borohydride, sodium cyanoborohydride, or sodium triacetoxyborohydride) in methanol. Unfortunately, formation of the desired N-acetyl-3-hydroxyoxindole was not observed at all, because a base-catalyzed addition of methanol to the C3=O and subsequent intramolecular rearrangement of the acetyl group occurs to give 3-methoxy-2-oxoindolin-3-yl acetate. Other reducing agents recommended in the literature for the reduction of unprotected isatin (sodium dithionite, zinc, phosphinic acid, etc.) also failed or caused an acetyl group removal. Therefore, we abandoned this strategy.
Configuration of products 5–7
All 3-(aminomethylidene)oxindoles can formally exist as (Z)- or (E)-isomers. Their actual configuration depends on the substitution at the methylidene carbon as well as the nitrogen. While derivatives containing an unsubstituted methylidene carbon and tertiary nitrogen exist as a mixture of rapidly equilibrating (Z)/(E)-isomers [2], the derivatives containing a secondary nitrogen slightly prefer [2,9,48] the (Z)-configuration due to an intramolecular hydrogen bond. A substitution of hydrogen at the methylidene carbon by an aryl group and a secondary nitrogen, enabling an intramolecular hydrogen bond, results in the geometry of a double bond that is locked in a (Z)-configuration [12]. The same (Z)-configuration was observed by us for the primary 3-[amino(aryl)methylidene]oxindoles [33].
The usual method used for the configuration assignment of 3-amino(aryl)methylidene derivatives involves a simple 1H NMR experiment showing either an upfield (δ ≈ 5.7–6.4 ppm for (Z)-configuration [5,12,32,33]) or a downfield shift (δ ≈ 7.7–7.9 ppm for (E)-configuration [27]) of the proton in position 4 of the oxindole moiety. In our previous paper [32] such an assignment was also confirmed in the solid state by X-ray diffraction. The important role of the intramolecular hydrogen bond fixing the (Z)-configuration has also been observed in the crystal structures of other 3-aminomethylidene derivatives containing a primary or secondary nitrogen [14,49-52].
The (Z)-configuration of the newly prepared 3-[amino(aryl)methylidene]oxindoles with primary, secondary, and tertiary nitrogen (series 5–7) has been therefore confirmed by 1H NMR spectroscopy in CDCl3 or DMSO-d6 solution. In all spectra the proton in position 4 of the oxindole moiety shows a typical upfield shift in the range of 5.7–6.4 (series 5), 5.3–5.9 (series 6), and 5.0–5.5 ppm (series 7), except for the nitro derivatives (R1: 5-NO2) where the nitro group always enhances the Ar–H4 chemical shift by about 0.7 ppm. Therefore, a 2D-NOESY experiment was performed for some representative 5-NO2 derivatives (5eb–ed) confirming their (Z)-configuration (see Supporting Information File 1).
Conclusion
We have developed an efficient and versatile synthesis of (Z)-3-[amino(phenyl/methyl)methylidene]-1,3-dihydro-2H-indol-2-ones (series 5, 6, 7, 10, and compound 11) starting from 3-bromooxindoles 1a–e and primary, secondary or tertiary thioacetamides and thiobenzamides 2–4. The superiority of our method in terms of the isolated yield (mostly >70% but often >85%) is clearly seen in all relevant cases (see Table 2 and Table 3) in which a direct comparison with other preparative routes [11,14,22,23] is possible. Although the starting 3-bromooxindoles 1a–e are available from the corresponding isatins by a three-step synthesis (overall yield 51–76%), the total yields related to these commercially available and cheap starting compounds are better or at least competitive to the methods described in Scheme 1 of which several have serious disadvantages. For example, the method starting from N-phenylpropiolamides [24] is not applicable (i.e., 0% yield) for the preparation of 1-unsubstituted 3-(aminomethylidene)oxindoles and the method involving the transformation of 3-bromo-3-[bromo(phenyl)methyl]oxindoles [27] gives the products with inversed configuration of the double bond. Several synthetic strategies starting from isocyanate [22,23] or oxindole [4,12] precursors work well only with protected nitrogen(s) in the starting compounds whose preparation and protection/deprotection also lengthens the total synthesis and decreases the overall isolated yield of the desired 3-(aminomethylidene)oxindoles.
The main advantages of our synthetic approach using an Eschenmoser coupling reaction involve the easy elaboration with no need for an inert atmosphere, extra dry solvents or toxic starting compounds (e.g., triphosgene needed for the synthesis of isocyanates) and (except for tertiary thioamides) addition of a thiophile. Moreover, our method has almost no limitation in terms of substitutions on each part of the skeleton.
Our alternative and shorter synthetic approach also starts from isatin and involves its reduction to 3-hydroxyoxindole and a one-pot triflation/Eschenmoser coupling with thiobenzamides under an inert atmosphere. However, this procedure is only suited for the preparation starting from N-methyl-3-hydroxyoxindole in which excessive N-triflication is blocked and the overall yield of 1-methyl-3-(aminomethylidene)-1,3-dihydro-2H-indol-2-one (11) (to isatin) is better than those obtained through 3-bromo-N-methyloxindole (1a’). In the case of parent isatin such a strategy also gives the desired products 5aa, 6ad, and 7aa but in a lower overall yield than the yield obtained through 1a. Other leaving groups in position 3 of the oxindole are unsuitable because only very slow (phosphate) or even no Eschenmoser coupling reaction (chloride) occurs with them.
Experimental
The starting 3-bromooxindoles 1a–e and 3-bromo-N-methyloxindole (1a’) were synthesized from the corresponding isatins using the procedure described in our previous paper [33] (see also Supporting Information File 1). The following Table 6 summarizes the overall yields after three steps.
Table 6: The yields of the synthesized 3-bromooxindoles 1a–e.a
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-47-i10.svg?max-width=637&scale=1.0)
|
|||
| entry | product 1 | R1 |
overall yield
(%) |
| 1 | 1a | H | 65* |
| 2 | 1b | 5-CH3 | 54 |
| 3 | 1c | 5-Cl | 76 |
| 4 | 1d | 6-Cl | 64 |
| 5 | 1e | 5-NO2 | 55 |
aThe corresponding 3-bromo-N-methyloxindole (1a’) was obtained in 55% yield.
3-Hydroxyoxindole and 3-hydroxy-N-methyloxindole were prepared [46] from the corresponding isatins through the reduction with sodium borohydride in methanol (see also Supporting Information File 1). The primary thiobenzamides 2a–d were prepared by the magnesium chloride-catalyzed thiolysis of commercially available benzonitriles [53] and the secondary thiobenzamides 3a–h were obtained by the thionation of the corresponding N-substituted amides [54] using pyridine–P4S10 as sulfurization agent. The tertiary thiobenzamides 4a–c were synthetized by a one-pot acylation/thionation from the corresponding acid chlorides and dimethylamine [55]. Other chemicals and solvents were purchased from Acros Organics, Sigma-Aldrich, or Fluorochem and were used as received. The melting points are uncorrected. 1H and 13C (APT) NMR spectra were recorded on a Bruker Avance III 400 MHz or on a Bruker Ascend 500 MHz instrument. The chemical shifts δ are referenced to TMS (δ = 0) or solvent residual peaks δ(CDCl3) = 7.24 ppm (1H) and 77.0 ppm (13C), δ(DMSO-d6) = 2.50 ppm (1H) and 39.6 ppm (13C). High-resolution mass spectra were recorded on a MALDI LTQ Orbitrap XL equipped with a nitrogen UV laser (337 nm, 60 Hz, 8–20 μJ) in the positive ion mode. For the CID experiment using the linear trap quadrupole (LTQ) helium was used as the collision gas and 2,5-dihydroxybenzoic acid (DHB) or (2-methylprop-2-en-1-yliden)malononitrile (DCTB) as the MALDI matrix. Elemental analyses were performed on a Flash 2000 Organic Elemental Analyser (Thermofisher). For samples containing chlorine mercurimetric titration was used. IR spectra were recorded on a Nicolet iS50 equipped with an ATR diamond crystal (neat solid samples). Flash chromatography was performed using a Büchi Reveleris® X2 flash chromatography system equipped with a UV–vis/ELSD detector and Reveleris Flash pure cartridges (12–40 g, 35–45 μm, 53–80 Å) or puriFlash® Alumine N 32/63 µm cartridges (12 g).
General procedure for the synthesis of 3-[amino(phenyl)methylidene]-1,3-dihydro-2H-indol-2-ones (5aa–ed and 6aa–ef)
In a 20 mL flask with a magnetic stirrer the solutions of the corresponding 3-bromooxindole 1a–e and substituted thiobenzamide 2a–d or 3a–i (amounts specified in Table 2 and Table 3) in dry DMF (5 + 5 mL for 3 mmol scale) were mixed and the mixture stirred for 5 h (primary thiobenzamides) or 12 h (secondary thiobenzamides) at room temperature. Then, TEA (2 equiv) was added and the mixture stirred for additional 5 min and diluted with water (100 mL). The aqueous solution was extracted with DCM (3 × 60 mL) and the combined organic layers were washed with water (2 × 40 mL) and brine (40 mL), dried with anhydrous Na2SO4 and evaporated. The solid residue was dissolved in methanol and filtered through a plug of Celite. The filtrate was evaporated with silica gel and submitted to preparative flash chromatography (silica gel or alumina cartridge; mobile phase and gradient are specified in Supporting Information File 1). Analytically pure samples were obtained by crystallization from the appropriate solvent.
Compounds 5ea and 5eb were isolated as follows: After the reaction of 1e with 2a,b (4 h) the reaction mixture was poured into cold EtOAc/EtOH 20:1 (80 mL) and stirred for 30 min. The precipitated product was filtered off and washed with EtOAc (2 × 5 mL). The crude 5ea or 5eb was dissolved in boiling CHCl3/CH3OH 1:1 (ca 125 mL) and filtered through a short plug of silica gel which was then washed with another portion (50 mL) of CHCl3/CH3OH 1:1. The collected filtrates were dried and evaporated.
Synthesis of 3-[dimethylamino(phenyl)methylidene]-1,3-dihydro-2H-indol-2-ones (7aa–ca)
The 4-substituted N,N-dimethylthiobenzamide (4a–c, n4a–c) and trimethyl phosphite (1.5 equiv) were dissolved in a minimum amount of dry DMF and a saturated solution of 3-bromooxindole (1a–c, n1a–c) in dry DMF was added. The reaction mixture was stirred for 20 h at room temperature (TLC monitoring; silica gel plates/EtOAc/hexane) and then diluted with an aqueous NaHCO3 solution (5%, 50 mL). The suspension was extracted with EtOAc (3 × 150 mL) and the combined organic layer was washed with water (5 × 100 mL), brine (2 × 100 mL), dried with anhydrous Na2SO4 and evaporated. The residue was repeatedly evaporated with EtOH to remove trimethyl thiophosphate. The crude product was then submitted to preparative flash chromatography (silica gel or alumina cartridge; mobile phase and gradient are specified in the Supporting Information File 1).
Synthesis of 3-[amino(methyl)methylidene]-1,3-dihydro-2H-indol-2-ones (10a–c)
The corresponding thioacetamide (2 mmol) was dissolved in DMF (5 mL) and a solution of 1a (424 mg, 2 mmol) in dry DMF (5 mL) was added. The mixture was stirred overnight at room temperature and then evaporated in vacuum. The residue was dissolved in DCM (40 mL), evaporated with neutral alumina (5 g) and submitted to preparative flash chromatography (silica gel or alumina cartridge; mobile phase and gradient are specified in Supporting Information File 1).
Supporting Information
| Supporting Information File 1: Synthetic procedures, characterization data and copies of spectra. | ||
| Format: PDF | Size: 13.0 MB | Download |
References
-
Howard, H. R., Jr.; Sarges, R. Heteroylidene indolone compounds. U.S. Patent US 4,476,307, Oct 9, 1984.
Return to citation in text: [1] [2] -
Sarges, R.; Howard, H. R.; Koe, B. K.; Weissman, A. J. Med. Chem. 1989, 32, 437–444. doi:10.1021/jm00122a025
Return to citation in text: [1] [2] [3] [4] [5] -
Davis, S. T.; Dickerson, S. H.; Frye, S. V.; Harris, P. A.; Hunter, R. N.; Kuyper, L. F.; Lackney, K. E.; Lackney, M. J.; Veal, J. M.; Walker, D. H. Substituted oxindole derivatives as protein tyrosine kinase and as protein serine/threonine kinase inhibitors. WO Pat. Appl. WO 99/15500 A1, April 1, 1999.
Return to citation in text: [1] [2] -
Grell, W.; Wittneben, H.; van Meel, J. C. A.; Redemann, N.; Walter, R.; Himmelsbach, F.; Haigh, R. Indolinones having kinase-inhibiting activity. U.S. Pat. Appl. US 6,043,254 A, March 28, 2000.
Return to citation in text: [1] [2] [3] -
Bramson, H. N.; Corona, J.; Davis, S. T.; Dickerson, S. H.; Edelstein, M.; Frye, S. V.; Gampe, R. T.; Harris, P. A.; Hassell, A.; Holmes, W. D.; Hunter, R. N.; Lackey, K. E.; Lovejoy, B.; Luzzio, M. J.; Montana, V.; Rocque, W. J.; Rusnak, D.; Shewchuk, L.; Veal, J. M.; Walker, D. H.; Kuyper, L. F. J. Med. Chem. 2001, 44, 4339–4358. doi:10.1021/jm010117d
Return to citation in text: [1] [2] [3] [4] -
Andrews, S. W.; Wurster, J. A.; Wang, E. H.; Malone, T. 3-(Arylamino)methylene-1,3-dihydro-2H-ondol-2-ones as kinase inhibitors. U.S. Pat. Appl. US 2003/0199478 A1, Oct 23, 2003.
Return to citation in text: [1] [2] -
Hauf, S.; Cole, R. W.; LaTerra, S.; Zimmer, C.; Schnapp, G.; Walter, R.; Heckel, A.; van Meel, J.; Rieder, C. L.; Peters, J.-M. J. Cell Biol. 2003, 161, 281–294. doi:10.1083/jcb.200208092
Return to citation in text: [1] -
Wood, E. R.; Kuyper, L.; Petrov, K. G.; Hunter, R. N., III; Harris, P. A.; Lackey, K. Bioorg. Med. Chem. Lett. 2004, 14, 953–957. doi:10.1016/j.bmcl.2003.12.002
Return to citation in text: [1] -
Bouchikhi, F.; Rossignol, E.; Sancelme, M.; Aboab, B.; Anizon, F.; Fabbro, D.; Prudhomme, M.; Moreau, P. Eur. J. Med. Chem. 2008, 43, 2316–2322. doi:10.1016/j.ejmech.2008.01.010
Return to citation in text: [1] [2] [3] -
Jetton, N.; Rothberg, K. G.; Hubbard, J. G.; Wise, J.; Li, Y.; Ball, H. L.; Ruben, L. Mol. Microbiol. 2009, 72, 442–458. doi:10.1111/j.1365-2958.2009.06657.x
Return to citation in text: [1] -
Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006.
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] -
Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466–4480. doi:10.1021/jm900431g
Return to citation in text: [1] [2] [3] [4] -
Roth, G. J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, L.; Park, J.; Pautsch, A.; Kaiser, R. Discovery and Development of Nintedanib: A Novel Antiangiogenic and Antifibrotic Agent. In Successful Drug Discovery; Fischer, J.; Childers, W. E., Eds.; Wiley-VCH: Weinheim, Germany, 2017; Vol. 2, pp 235–266. doi:10.1002/9783527800315.ch11
Return to citation in text: [1] -
Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Wenkert, E.; Udelhofen, J. H.; Bhattacharyya, N. K. J. Am. Chem. Soc. 1959, 81, 3763–3768. doi:10.1021/ja01523a068
Return to citation in text: [1] -
Erba, E.; Pocar, D.; Valle, M. J. Chem. Soc., Perkin Trans. 1 1999, 421–426. doi:10.1039/a809321k
Return to citation in text: [1] -
Pedras, M. S. C.; Sorensen, J. L.; Okanga, F. I.; Zaharia, I. L. Bioorg. Med. Chem. Lett. 1999, 9, 3015–3020. doi:10.1016/s0960-894x(99)00523-5
Return to citation in text: [1] -
Behringer, H.; Weissauer, H. Chem. Ber. 1952, 85, 774–779. doi:10.1002/cber.19520850713
Return to citation in text: [1] -
Wolfbeis, O. S. Monatsh. Chem. 1981, 112, 369–383. doi:10.1007/bf00900767
Return to citation in text: [1] -
Heckel, A.; Roth, J.; Kley, S.; Hoerer, S.; Uphues, I. Alkyl-containing 5-acylindolinones, the preparation thereof and their use as medicaments. U.S. Pat. Appl. US 2005/0209302 A1, Sept 22, 2005.
Return to citation in text: [1] -
Tang, S.; Yu, Q.-F.; Peng; Li, J.-H.; Zhong, P.; Tang, R.-Y. Org. Lett. 2007, 9, 3413–3416. doi:10.1021/ol701450n
Return to citation in text: [1] -
Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Org. Lett. 2009, 11, 2141–2143. doi:10.1021/ol900759f
Return to citation in text: [1] [2] [3] [4] -
Cantagrel, G.; de Carné-Carnavalet, B.; Meyer, C.; Cossy, J. Org. Lett. 2009, 11, 4262–4265. doi:10.1021/ol9014877
Return to citation in text: [1] [2] [3] [4] -
Tang, S.; Peng; Pi, S.-F.; Liang, Y.; Wang, N.-X.; Li, J.-H. Org. Lett. 2008, 10, 1179–1182. doi:10.1021/ol800080w
Return to citation in text: [1] [2] -
Tang, S.; Peng, P.; Wang, Z.-Q.; Tang, B.-X.; Deng, C.-L.; Li, J.-H.; Zhong, P.; Wang, N.-X. Org. Lett. 2008, 10, 1875–1878. doi:10.1021/ol8006315
Return to citation in text: [1] -
Peng, P.; Tang, B.-X.; Pi, S.-F.; Liang, Y.; Li, J.-H. J. Org. Chem. 2009, 74, 3569–3572. doi:10.1021/jo900437p
Return to citation in text: [1] -
Jiang, H.; Feng, Z.; Chen, T.; Li, Z.; Huang, W.; Luo, Y.; Zhao, Y. J. Chem. Res. 2018, 42, 44–49. doi:10.3184/174751918x15166485941737
Return to citation in text: [1] [2] [3] [4] -
Otomasu, H.; Yoshida, K.; Natori, K. Chem. Pharm. Bull. 1975, 23, 1436–1439. doi:10.1248/cpb.23.1436
Return to citation in text: [1] -
Aurich, H. G. Justus Liebigs Ann. Chem. 1970, 732, 195–198. doi:10.1002/jlac.19707320117
Return to citation in text: [1] -
Reisch, J.; Ossenkop, W. F. Chem. Ber. 1973, 106, 2070–2071. doi:10.1002/cber.19731060635
Return to citation in text: [1] -
Stauss, U.; Härter, H. P.; Neuenschwander, M.; Schindler, O. Helv. Chim. Acta 1972, 55, 771–780. doi:10.1002/hlca.19720550305
Return to citation in text: [1] -
Kammel, R.; Tarabová, D.; Růžičková, Z.; Hanusek, J. Tetrahedron Lett. 2015, 56, 2548–2550. doi:10.1016/j.tetlet.2015.03.052
Return to citation in text: [1] [2] [3] [4] -
Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] -
Cox, B. G. Acids and Bases. Solvent Effects on Acid-Base Strength; Oxford University Press: Oxford, UK, 2013; pp 133–137. doi:10.1093/acprof:oso/9780199670512.001.0001
Return to citation in text: [1] -
Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004
Return to citation in text: [1] -
Shiosaki, K. The Eschenmoser Coupling Reaction. In Comprehensive Organic Synthesis – Selectivity, Strategy and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1993; Vol. 2, pp 865–892. doi:10.1016/b978-0-08-052349-1.00051-2
Return to citation in text: [1] [2] [3] -
Hussaini, S. R.; Chamala, R. R.; Wang, Z. Tetrahedron 2015, 71, 6017–6086. doi:10.1016/j.tet.2015.06.026
Return to citation in text: [1] -
Huang, P.-Q.; Ou, W.; Ye, J.-L. Org. Chem. Front. 2015, 2, 1094–1106. doi:10.1039/c5qo00191a
Return to citation in text: [1] -
Huang, P.-Q.; Ou, W. Eur. J. Org. Chem. 2017, 582–592. doi:10.1002/ejoc.201601326
Return to citation in text: [1] -
Pal, A.; Hussaini, S. R. ACS Omega 2019, 4, 269–280. doi:10.1021/acsomega.8b02633
Return to citation in text: [1] -
Guillaumel, J.; Demerseman, P.; Clavel, J.-M.; Royer, R.; Platzer, N.; Brevard, C. Tetrahedron 1980, 36, 2459–2465. doi:10.1016/0040-4020(80)80224-9
Return to citation in text: [1] -
Rokade, B. V.; Guiry, P. J. J. Org. Chem. 2020, 85, 6172–6180. doi:10.1021/acs.joc.0c00370
Return to citation in text: [1] -
Kumler, W. D.; Eiler, J. J. J. Am. Chem. Soc. 1943, 65, 2355–2361. doi:10.1021/ja01252a028
Return to citation in text: [1] -
Swain, C. G.; Scott, C. B. J. Am. Chem. Soc. 1953, 75, 141–147. doi:10.1021/ja01097a041
Return to citation in text: [1] -
Manikyamba, P.; Sundaram, E. V. Int. J. Chem. Kinet. 1990, 22, 1153–1165. doi:10.1002/kin.550221105
Return to citation in text: [1] -
Trost, B. M.; Hirano, K. Org. Lett. 2012, 14, 2446–2449. doi:10.1021/ol300577y
Return to citation in text: [1] [2] -
El-Faham, A.; Al Marhoon, Z.; Abdel-Megeed, A.; Albericio, F. Molecules 2013, 18, 14747–14759. doi:10.3390/molecules181214747
Return to citation in text: [1] -
Chen, G.; Liu, B.; Tang, Y.; Deng, Q.; Hao, X.-j. Heterocycl. Commun. 2010, 16, 25–32. doi:10.1515/hc.2010.16.1.25
Return to citation in text: [1] -
Li, C.; Go, Y.; Mao, Z.; Koyano, K.; Kai, Y.; Kanehisa, N.; Zhu, Q.; Zhou, Z.; Wu, S. Bull. Chem. Soc. Jpn. 1996, 69, 1621–1627. doi:10.1246/bcsj.69.1621
Return to citation in text: [1] -
Periyaraja, S.; Mandal, A. B.; Shanmugam, P. Org. Lett. 2011, 13, 4980–4983. doi:10.1021/ol2022164
Return to citation in text: [1] -
Liu, Y.-Y.; Duan, S.-W.; Zhang, R.; Liu, Y.-H.; Chen, J.-R.; Xiao, W.-J. Org. Biomol. Chem. 2016, 14, 5224–5228. doi:10.1039/c6ob00891g
Return to citation in text: [1] -
Hassan, A. A.; El-Shaieb, K. M. A.; Abd El-Aal, A. S.; Bräse, S.; Nieger, M. J. Heterocycl. Chem. 2016, 53, 2025–2029. doi:10.1002/jhet.2524
Return to citation in text: [1] -
Manaka, A.; Sato, M. Synth. Commun. 2005, 35, 761–764. doi:10.1081/scc-200050393
Return to citation in text: [1] -
Bergman, J.; Pettersson, B.; Hasimbegovic, V.; Svensson, P. H. J. Org. Chem. 2011, 76, 1546–1553. doi:10.1021/jo101865y
Return to citation in text: [1] -
Voß, J.; Walter, W. Justus Liebigs Ann. Chem. 1968, 716, 209–211. doi:10.1002/jlac.19687160131
Return to citation in text: [1]
| 23. | Cantagrel, G.; de Carné-Carnavalet, B.; Meyer, C.; Cossy, J. Org. Lett. 2009, 11, 4262–4265. doi:10.1021/ol9014877 |
| 27. | Jiang, H.; Feng, Z.; Chen, T.; Li, Z.; Huang, W.; Luo, Y.; Zhao, Y. J. Chem. Res. 2018, 42, 44–49. doi:10.3184/174751918x15166485941737 |
| 32. | Kammel, R.; Tarabová, D.; Růžičková, Z.; Hanusek, J. Tetrahedron Lett. 2015, 56, 2548–2550. doi:10.1016/j.tetlet.2015.03.052 |
| 42. | Rokade, B. V.; Guiry, P. J. J. Org. Chem. 2020, 85, 6172–6180. doi:10.1021/acs.joc.0c00370 |
| 43. | Kumler, W. D.; Eiler, J. J. J. Am. Chem. Soc. 1943, 65, 2355–2361. doi:10.1021/ja01252a028 |
| 38. | Huang, P.-Q.; Ou, W.; Ye, J.-L. Org. Chem. Front. 2015, 2, 1094–1106. doi:10.1039/c5qo00191a |
| 39. | Huang, P.-Q.; Ou, W. Eur. J. Org. Chem. 2017, 582–592. doi:10.1002/ejoc.201601326 |
| 40. | Pal, A.; Hussaini, S. R. ACS Omega 2019, 4, 269–280. doi:10.1021/acsomega.8b02633 |
| 41. | Guillaumel, J.; Demerseman, P.; Clavel, J.-M.; Royer, R.; Platzer, N.; Brevard, C. Tetrahedron 1980, 36, 2459–2465. doi:10.1016/0040-4020(80)80224-9 |
| 36. | Shiosaki, K. The Eschenmoser Coupling Reaction. In Comprehensive Organic Synthesis – Selectivity, Strategy and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1993; Vol. 2, pp 865–892. doi:10.1016/b978-0-08-052349-1.00051-2 |
| 37. | Hussaini, S. R.; Chamala, R. R.; Wang, Z. Tetrahedron 2015, 71, 6017–6086. doi:10.1016/j.tet.2015.06.026 |
| 36. | Shiosaki, K. The Eschenmoser Coupling Reaction. In Comprehensive Organic Synthesis – Selectivity, Strategy and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1993; Vol. 2, pp 865–892. doi:10.1016/b978-0-08-052349-1.00051-2 |
| 36. | Shiosaki, K. The Eschenmoser Coupling Reaction. In Comprehensive Organic Synthesis – Selectivity, Strategy and Efficiency in Modern Organic Chemistry; Trost, B. M.; Fleming, I., Eds.; Pergamon Press: Oxford, UK, 1993; Vol. 2, pp 865–892. doi:10.1016/b978-0-08-052349-1.00051-2 |
| 14. | Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771 |
| 44. | Swain, C. G.; Scott, C. B. J. Am. Chem. Soc. 1953, 75, 141–147. doi:10.1021/ja01097a041 |
| 45. | Manikyamba, P.; Sundaram, E. V. Int. J. Chem. Kinet. 1990, 22, 1153–1165. doi:10.1002/kin.550221105 |
| 46. | Trost, B. M.; Hirano, K. Org. Lett. 2012, 14, 2446–2449. doi:10.1021/ol300577y |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 5. | Bramson, H. N.; Corona, J.; Davis, S. T.; Dickerson, S. H.; Edelstein, M.; Frye, S. V.; Gampe, R. T.; Harris, P. A.; Hassell, A.; Holmes, W. D.; Hunter, R. N.; Lackey, K. E.; Lovejoy, B.; Luzzio, M. J.; Montana, V.; Rocque, W. J.; Rusnak, D.; Shewchuk, L.; Veal, J. M.; Walker, D. H.; Kuyper, L. F. J. Med. Chem. 2001, 44, 4339–4358. doi:10.1021/jm010117d |
| 12. | Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466–4480. doi:10.1021/jm900431g |
| 32. | Kammel, R.; Tarabová, D.; Růžičková, Z.; Hanusek, J. Tetrahedron Lett. 2015, 56, 2548–2550. doi:10.1016/j.tetlet.2015.03.052 |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 2. | Sarges, R.; Howard, H. R.; Koe, B. K.; Weissman, A. J. Med. Chem. 1989, 32, 437–444. doi:10.1021/jm00122a025 |
| 9. | Bouchikhi, F.; Rossignol, E.; Sancelme, M.; Aboab, B.; Anizon, F.; Fabbro, D.; Prudhomme, M.; Moreau, P. Eur. J. Med. Chem. 2008, 43, 2316–2322. doi:10.1016/j.ejmech.2008.01.010 |
| 48. | Chen, G.; Liu, B.; Tang, Y.; Deng, Q.; Hao, X.-j. Heterocycl. Commun. 2010, 16, 25–32. doi:10.1515/hc.2010.16.1.25 |
| 12. | Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466–4480. doi:10.1021/jm900431g |
| 47. | El-Faham, A.; Al Marhoon, Z.; Abdel-Megeed, A.; Albericio, F. Molecules 2013, 18, 14747–14759. doi:10.3390/molecules181214747 |
| 2. | Sarges, R.; Howard, H. R.; Koe, B. K.; Weissman, A. J. Med. Chem. 1989, 32, 437–444. doi:10.1021/jm00122a025 |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 14. | Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771 |
| 32. | Kammel, R.; Tarabová, D.; Růžičková, Z.; Hanusek, J. Tetrahedron Lett. 2015, 56, 2548–2550. doi:10.1016/j.tetlet.2015.03.052 |
| 14. | Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771 |
| 49. | Li, C.; Go, Y.; Mao, Z.; Koyano, K.; Kai, Y.; Kanehisa, N.; Zhu, Q.; Zhou, Z.; Wu, S. Bull. Chem. Soc. Jpn. 1996, 69, 1621–1627. doi:10.1246/bcsj.69.1621 |
| 50. | Periyaraja, S.; Mandal, A. B.; Shanmugam, P. Org. Lett. 2011, 13, 4980–4983. doi:10.1021/ol2022164 |
| 51. | Liu, Y.-Y.; Duan, S.-W.; Zhang, R.; Liu, Y.-H.; Chen, J.-R.; Xiao, W.-J. Org. Biomol. Chem. 2016, 14, 5224–5228. doi:10.1039/c6ob00891g |
| 52. | Hassan, A. A.; El-Shaieb, K. M. A.; Abd El-Aal, A. S.; Bräse, S.; Nieger, M. J. Heterocycl. Chem. 2016, 53, 2025–2029. doi:10.1002/jhet.2524 |
| 27. | Jiang, H.; Feng, Z.; Chen, T.; Li, Z.; Huang, W.; Luo, Y.; Zhao, Y. J. Chem. Res. 2018, 42, 44–49. doi:10.3184/174751918x15166485941737 |
| 1. | Howard, H. R., Jr.; Sarges, R. Heteroylidene indolone compounds. U.S. Patent US 4,476,307, Oct 9, 1984. |
| 12. | Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466–4480. doi:10.1021/jm900431g |
| 13. | Roth, G. J.; Binder, R.; Colbatzky, F.; Dallinger, C.; Schlenker-Herceg, R.; Hilberg, F.; Wollin, L.; Park, J.; Pautsch, A.; Kaiser, R. Discovery and Development of Nintedanib: A Novel Antiangiogenic and Antifibrotic Agent. In Successful Drug Discovery; Fischer, J.; Childers, W. E., Eds.; Wiley-VCH: Weinheim, Germany, 2017; Vol. 2, pp 235–266. doi:10.1002/9783527800315.ch11 |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 46. | Trost, B. M.; Hirano, K. Org. Lett. 2012, 14, 2446–2449. doi:10.1021/ol300577y |
| 5. | Bramson, H. N.; Corona, J.; Davis, S. T.; Dickerson, S. H.; Edelstein, M.; Frye, S. V.; Gampe, R. T.; Harris, P. A.; Hassell, A.; Holmes, W. D.; Hunter, R. N.; Lackey, K. E.; Lovejoy, B.; Luzzio, M. J.; Montana, V.; Rocque, W. J.; Rusnak, D.; Shewchuk, L.; Veal, J. M.; Walker, D. H.; Kuyper, L. F. J. Med. Chem. 2001, 44, 4339–4358. doi:10.1021/jm010117d |
| 6. | Andrews, S. W.; Wurster, J. A.; Wang, E. H.; Malone, T. 3-(Arylamino)methylene-1,3-dihydro-2H-ondol-2-ones as kinase inhibitors. U.S. Pat. Appl. US 2003/0199478 A1, Oct 23, 2003. |
| 7. | Hauf, S.; Cole, R. W.; LaTerra, S.; Zimmer, C.; Schnapp, G.; Walter, R.; Heckel, A.; van Meel, J.; Rieder, C. L.; Peters, J.-M. J. Cell Biol. 2003, 161, 281–294. doi:10.1083/jcb.200208092 |
| 8. | Wood, E. R.; Kuyper, L.; Petrov, K. G.; Hunter, R. N., III; Harris, P. A.; Lackey, K. Bioorg. Med. Chem. Lett. 2004, 14, 953–957. doi:10.1016/j.bmcl.2003.12.002 |
| 9. | Bouchikhi, F.; Rossignol, E.; Sancelme, M.; Aboab, B.; Anizon, F.; Fabbro, D.; Prudhomme, M.; Moreau, P. Eur. J. Med. Chem. 2008, 43, 2316–2322. doi:10.1016/j.ejmech.2008.01.010 |
| 10. | Jetton, N.; Rothberg, K. G.; Hubbard, J. G.; Wise, J.; Li, Y.; Ball, H. L.; Ruben, L. Mol. Microbiol. 2009, 72, 442–458. doi:10.1111/j.1365-2958.2009.06657.x |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 3. | Davis, S. T.; Dickerson, S. H.; Frye, S. V.; Harris, P. A.; Hunter, R. N.; Kuyper, L. F.; Lackney, K. E.; Lackney, M. J.; Veal, J. M.; Walker, D. H. Substituted oxindole derivatives as protein tyrosine kinase and as protein serine/threonine kinase inhibitors. WO Pat. Appl. WO 99/15500 A1, April 1, 1999. |
| 4. | Grell, W.; Wittneben, H.; van Meel, J. C. A.; Redemann, N.; Walter, R.; Himmelsbach, F.; Haigh, R. Indolinones having kinase-inhibiting activity. U.S. Pat. Appl. US 6,043,254 A, March 28, 2000. |
| 29. | Aurich, H. G. Justus Liebigs Ann. Chem. 1970, 732, 195–198. doi:10.1002/jlac.19707320117 |
| 30. | Reisch, J.; Ossenkop, W. F. Chem. Ber. 1973, 106, 2070–2071. doi:10.1002/cber.19731060635 |
| 31. | Stauss, U.; Härter, H. P.; Neuenschwander, M.; Schindler, O. Helv. Chim. Acta 1972, 55, 771–780. doi:10.1002/hlca.19720550305 |
| 4. | Grell, W.; Wittneben, H.; van Meel, J. C. A.; Redemann, N.; Walter, R.; Himmelsbach, F.; Haigh, R. Indolinones having kinase-inhibiting activity. U.S. Pat. Appl. US 6,043,254 A, March 28, 2000. |
| 12. | Roth, G. J.; Heckel, A.; Colbatzky, F.; Handschuh, S.; Kley, J.; Lehmann-Lintz, T.; Lotz, R.; Tontsch-Grunt, U.; Walter, R.; Hilberg, F. J. Med. Chem. 2009, 52, 4466–4480. doi:10.1021/jm900431g |
| 2. | Sarges, R.; Howard, H. R.; Koe, B. K.; Weissman, A. J. Med. Chem. 1989, 32, 437–444. doi:10.1021/jm00122a025 |
| 32. | Kammel, R.; Tarabová, D.; Růžičková, Z.; Hanusek, J. Tetrahedron Lett. 2015, 56, 2548–2550. doi:10.1016/j.tetlet.2015.03.052 |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 21. | Tang, S.; Yu, Q.-F.; Peng; Li, J.-H.; Zhong, P.; Tang, R.-Y. Org. Lett. 2007, 9, 3413–3416. doi:10.1021/ol701450n |
| 22. | Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Org. Lett. 2009, 11, 2141–2143. doi:10.1021/ol900759f |
| 23. | Cantagrel, G.; de Carné-Carnavalet, B.; Meyer, C.; Cossy, J. Org. Lett. 2009, 11, 4262–4265. doi:10.1021/ol9014877 |
| 27. | Jiang, H.; Feng, Z.; Chen, T.; Li, Z.; Huang, W.; Luo, Y.; Zhao, Y. J. Chem. Res. 2018, 42, 44–49. doi:10.3184/174751918x15166485941737 |
| 27. | Jiang, H.; Feng, Z.; Chen, T.; Li, Z.; Huang, W.; Luo, Y.; Zhao, Y. J. Chem. Res. 2018, 42, 44–49. doi:10.3184/174751918x15166485941737 |
| 2. | Sarges, R.; Howard, H. R.; Koe, B. K.; Weissman, A. J. Med. Chem. 1989, 32, 437–444. doi:10.1021/jm00122a025 |
| 5. | Bramson, H. N.; Corona, J.; Davis, S. T.; Dickerson, S. H.; Edelstein, M.; Frye, S. V.; Gampe, R. T.; Harris, P. A.; Hassell, A.; Holmes, W. D.; Hunter, R. N.; Lackey, K. E.; Lovejoy, B.; Luzzio, M. J.; Montana, V.; Rocque, W. J.; Rusnak, D.; Shewchuk, L.; Veal, J. M.; Walker, D. H.; Kuyper, L. F. J. Med. Chem. 2001, 44, 4339–4358. doi:10.1021/jm010117d |
| 6. | Andrews, S. W.; Wurster, J. A.; Wang, E. H.; Malone, T. 3-(Arylamino)methylene-1,3-dihydro-2H-ondol-2-ones as kinase inhibitors. U.S. Pat. Appl. US 2003/0199478 A1, Oct 23, 2003. |
| 18. | Behringer, H.; Weissauer, H. Chem. Ber. 1952, 85, 774–779. doi:10.1002/cber.19520850713 |
| 19. | Wolfbeis, O. S. Monatsh. Chem. 1981, 112, 369–383. doi:10.1007/bf00900767 |
| 20. | Heckel, A.; Roth, J.; Kley, S.; Hoerer, S.; Uphues, I. Alkyl-containing 5-acylindolinones, the preparation thereof and their use as medicaments. U.S. Pat. Appl. US 2005/0209302 A1, Sept 22, 2005. |
| 28. | Otomasu, H.; Yoshida, K.; Natori, K. Chem. Pharm. Bull. 1975, 23, 1436–1439. doi:10.1248/cpb.23.1436 |
| 22. | Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Org. Lett. 2009, 11, 2141–2143. doi:10.1021/ol900759f |
| 23. | Cantagrel, G.; de Carné-Carnavalet, B.; Meyer, C.; Cossy, J. Org. Lett. 2009, 11, 4262–4265. doi:10.1021/ol9014877 |
| 4. | Grell, W.; Wittneben, H.; van Meel, J. C. A.; Redemann, N.; Walter, R.; Himmelsbach, F.; Haigh, R. Indolinones having kinase-inhibiting activity. U.S. Pat. Appl. US 6,043,254 A, March 28, 2000. |
| 9. | Bouchikhi, F.; Rossignol, E.; Sancelme, M.; Aboab, B.; Anizon, F.; Fabbro, D.; Prudhomme, M.; Moreau, P. Eur. J. Med. Chem. 2008, 43, 2316–2322. doi:10.1016/j.ejmech.2008.01.010 |
| 15. | Wenkert, E.; Udelhofen, J. H.; Bhattacharyya, N. K. J. Am. Chem. Soc. 1959, 81, 3763–3768. doi:10.1021/ja01523a068 |
| 16. | Erba, E.; Pocar, D.; Valle, M. J. Chem. Soc., Perkin Trans. 1 1999, 421–426. doi:10.1039/a809321k |
| 17. | Pedras, M. S. C.; Sorensen, J. L.; Okanga, F. I.; Zaharia, I. L. Bioorg. Med. Chem. Lett. 1999, 9, 3015–3020. doi:10.1016/s0960-894x(99)00523-5 |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 14. | Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771 |
| 22. | Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Org. Lett. 2009, 11, 2141–2143. doi:10.1021/ol900759f |
| 23. | Cantagrel, G.; de Carné-Carnavalet, B.; Meyer, C.; Cossy, J. Org. Lett. 2009, 11, 4262–4265. doi:10.1021/ol9014877 |
| 1. | Howard, H. R., Jr.; Sarges, R. Heteroylidene indolone compounds. U.S. Patent US 4,476,307, Oct 9, 1984. |
| 2. | Sarges, R.; Howard, H. R.; Koe, B. K.; Weissman, A. J. Med. Chem. 1989, 32, 437–444. doi:10.1021/jm00122a025 |
| 3. | Davis, S. T.; Dickerson, S. H.; Frye, S. V.; Harris, P. A.; Hunter, R. N.; Kuyper, L. F.; Lackney, K. E.; Lackney, M. J.; Veal, J. M.; Walker, D. H. Substituted oxindole derivatives as protein tyrosine kinase and as protein serine/threonine kinase inhibitors. WO Pat. Appl. WO 99/15500 A1, April 1, 1999. |
| 5. | Bramson, H. N.; Corona, J.; Davis, S. T.; Dickerson, S. H.; Edelstein, M.; Frye, S. V.; Gampe, R. T.; Harris, P. A.; Hassell, A.; Holmes, W. D.; Hunter, R. N.; Lackey, K. E.; Lovejoy, B.; Luzzio, M. J.; Montana, V.; Rocque, W. J.; Rusnak, D.; Shewchuk, L.; Veal, J. M.; Walker, D. H.; Kuyper, L. F. J. Med. Chem. 2001, 44, 4339–4358. doi:10.1021/jm010117d |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 14. | Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771 |
| 24. | Tang, S.; Peng; Pi, S.-F.; Liang, Y.; Wang, N.-X.; Li, J.-H. Org. Lett. 2008, 10, 1179–1182. doi:10.1021/ol800080w |
| 25. | Tang, S.; Peng, P.; Wang, Z.-Q.; Tang, B.-X.; Deng, C.-L.; Li, J.-H.; Zhong, P.; Wang, N.-X. Org. Lett. 2008, 10, 1875–1878. doi:10.1021/ol8006315 |
| 26. | Peng, P.; Tang, B.-X.; Pi, S.-F.; Liang, Y.; Li, J.-H. J. Org. Chem. 2009, 74, 3569–3572. doi:10.1021/jo900437p |
| 24. | Tang, S.; Peng; Pi, S.-F.; Liang, Y.; Wang, N.-X.; Li, J.-H. Org. Lett. 2008, 10, 1179–1182. doi:10.1021/ol800080w |
| 34. | Cox, B. G. Acids and Bases. Solvent Effects on Acid-Base Strength; Oxford University Press: Oxford, UK, 2013; pp 133–137. doi:10.1093/acprof:oso/9780199670512.001.0001 |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 55. | Voß, J.; Walter, W. Justus Liebigs Ann. Chem. 1968, 716, 209–211. doi:10.1002/jlac.19687160131 |
| 53. | Manaka, A.; Sato, M. Synth. Commun. 2005, 35, 761–764. doi:10.1081/scc-200050393 |
| 54. | Bergman, J.; Pettersson, B.; Hasimbegovic, V.; Svensson, P. H. J. Org. Chem. 2011, 76, 1546–1553. doi:10.1021/jo101865y |
| 14. | Bisht, G. S.; Gnanaprakasam, B. J. Org. Chem. 2019, 84, 13516–13527. doi:10.1021/acs.joc.9b01771 |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
| 22. | Miura, T.; Toyoshima, T.; Takahashi, Y.; Murakami, M. Org. Lett. 2009, 11, 2141–2143. doi:10.1021/ol900759f |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 35. | Hansch, C.; Leo, A.; Taft, R. W. Chem. Rev. 1991, 91, 165–195. doi:10.1021/cr00002a004 |
| 11. | Burgdorf, L. T.; Bruge, D.; Greiner, H.; Kordowicz, M.; Sirrenberg, C.; Zenke, F. Oxindoles as kinase inhibitors. WO Pat. Appl. WO 2006/131186 A1, Dec 14, 2006. |
| 33. | Kammel, R.; Tarabová, D.; Brož, B.; Hladíková, V.; Hanusek, J. Tetrahedron 2017, 73, 1861–1866. doi:10.1016/j.tet.2017.02.045 |
© 2021 Marek et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)