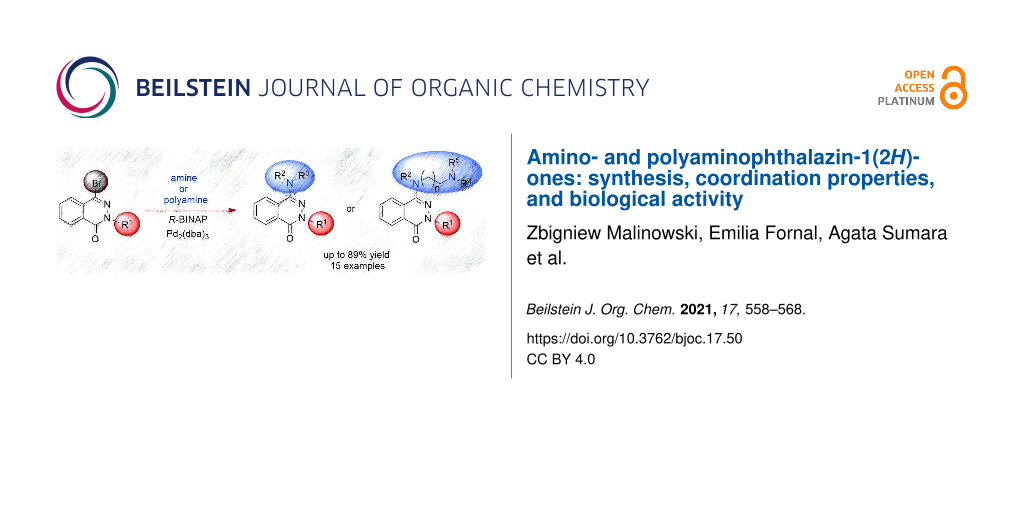
Abstract
Amino- and polyaminophthalazinones were synthesized by the palladium‐catalyzed amination (alkyl- and arylamines, polyamines) of 4-bromophthalazinones in good yields. The coordinating properties of selected aminophthalazinones towards Cu(II) ions were investigated and the participation of the nitrogen atoms in the complexation of the metal ion was shown. A biological screening of the potential cytotoxicity of selected synthesized compounds on HT-29 and PC-3 cell lines, as well as on the L-929 cell line, proved that some amino derivatives of phthalazinone show interesting anticancer activities. The detailed synthesis, spectroscopic data, and biological assays are reported.

Graphical Abstract
Introduction
Phthalazine and its analogs are an interesting group of pharmacologically active heterocycles [1,2], many of which possess, e.g., antimicrobial [3,4], antifungal [5], antidepressant [6,7], and antihistaminic [8-10] properties. Amino- and amidophthalazine derivatives have been examined, e.g., as inhibitors of PGE2 production [11] that plays an important role in the growth of various cancerous tissues (colon, lung, and breast cancer) and as antagonists of the human A3 adenosine receptor [12].
Aminophthalazines have been also evaluated for their inhibitory activity toward phosphodiesterases such as PDE-5 (Figure 1) [13,14] and PDE-10 [15] for a potential use in the treatment of chronic pain and neurodegenerative or psychiatric disorders. Some of these derivatives are known to possess anti-inflammatory (p38 MAP kinase inhibitors, Figure 1) [16], cardiotonic [17], and anticancer (Aurora-A kinase inhibitors) properties [18-20].
![[1860-5397-17-50-1]](/bjoc/content/figures/1860-5397-17-50-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structure of biologically active phthalazine derivatives.
Figure 1: Structure of biologically active phthalazine derivatives.
The aminophthalazine and also hydrazinylphthalazine moiety can be also found in the core of many commercial drugs such as hydralazine [21-23], budralazine [24], and carbazeran [25] that are used for the treatment of heart failure as well as in the structure of the effective anticancer drug vatalanib [26-28] (Figure 1). On the other hand, aminophthalazinones can be prospective candidates as N- and O-donor ligands to form complexes with biological significant metal ions, such as copper or zinc [29].
Recently, we have demonstrated a strategy for the synthesis of phthalazinone and phthalazine derivatives of type 4 containing an alkylsulfanyl functional group at the 4 position, that is based on the Pd-catalyzed coupling reaction between mercaptanes and 4-bromolactams (Scheme 1, route A) [30]. In continuing our efforts on the functionalization of phthalazinones and quinazolinones [30,31] and taking into consideration the biological importance of aminophthalazine derivatives, we decided to apply the methodology based on the palladium-catalyzed C–N-bond formation (Buchwald–Hartwig-type reaction) as a convenient and effective approach for the synthesis of the new phthalazinone derivatives 5 and 6 (Scheme 1, route B).
![[1860-5397-17-50-i1]](/bjoc/content/inline/1860-5397-17-50-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route to aminophthalazinones 5 and 6.
Scheme 1: Synthetic route to aminophthalazinones 5 and 6.
In the literature, the number of reported synthetic methods for 4-aminophthalazin-1(2H)-ones is limited to a few examples and they usually involve two main approaches: 1) the application of multicomponent reactions starting from, e.g., the available o-bromobenzoate via palladium-catalyzed isocyanide insertion [32,33] (a method that is limited to tertiary-substituted isocyanides) or 2) the palladium or copper-catalyzed coupling of bromolactams with amines (a method that requires the usually lengthy synthesis of the bromoprecursors) [19,34].
Therefore, the need to develop new and improve the existing methods for the synthesis of aminophthalazinones is important from the point of view of their properties and pharmaceutical industry interest.
In the present work, we report results of our research on the synthesis, application as ligands in complexes, and cytotoxic activity of amino- and polyaminophthalazinone derivatives.
Results and Discussion
Chemistry
Synthesis of aminophthalazinones
The synthetic route toward the aminophthalazinones 5 and 6 is shown in Scheme 1 (route B). A current literature review [19,34] and our experience [35] proved, that the direct Pd-coupling of bromophthalazinones of type 2 and also bromoquinazolinones, having an N–H moiety (acidic hydrogen atom) with amines does not yield satisfying results without an N-protection of the amide function.
Therefore, in the initial stage of our studies, the N-substituted phthalazin-1(2H)-ones 3a–d were prepared in two steps starting from phthalazin-1(2H)-one (1). The 4-bromo-derivative 2 was synthesized directly from lactam 1, which underwent the selective bromination at the 4-position using the combination of Br2 and KBr (KBr3) in acetate buffer, following the method previously reported by us [30]. It is known that the alkylation of phthalazinones depends on the reaction conditions and can proceed in two ways involving either the nitrogen or the oxygen atom (lactam–lactim tautomerism). It has been proven that potassium salts of phthalazinones or the bromo derivatives are selectively alkylated on the nitrogen atom [30,36]. For our purposes, the simple alkyl halides (MeI, iPrI), and 2-chloro-N,N-dimethylethylamine hydrochloride and 4-(2-chloroethyl)morpholine hydrochloride, were chosen as the alkylating agents. Thus, the desired N-methyl and N-isopropyl lactams 3a,b (3a R1 = Me, 85%; 3b R1 = iPr, 84%) were obtained by the direct alkylation of bromophthalazinone 2 with methyl or isopropyl iodide in the presence of K2CO3 in dry acetone as the solvent (conventional heating). In the similar way also the 2-aminoethyl lactams 3c (R1 = CH2CH2NMe2) and 3d (R1 = CH2CH2(morpholin-4-yl) were synthesized with 53% and 61% yields, respectively. The methodology turned out to be the most efficient out of the tested ones, especially for the products 3c and 3d. The formation of the N-alkylated products 3 was confirmed on the basis of their spectral analysis (see Supporting Information File 1).
In the next step, the bromolactams 3 were subjected to a thermal palladium cross-coupling reaction with various amines and polyamines. To optimize the reaction conditions, we investigated the reaction of 4-bromo-2-methylphthalazin-1(2H)-one (3a) with morpholine as the model substrates. Previously, we have observed that the coupling system involving Xantphos/Pd(OAc)2 (used in the ratio of 15 mol %/15 mol % or 30 mol %/30 mol %) and t-BuOK or DIPEA in 1,4-dioxane as the solvent was effective for the C–N or C–S bond formation [30,31,35].
Unfortunately, it turned out, that the application of Xantphos/Pd(OAc)2/t-BuOK and our standard procedure [30,35], in which the amine is added after the lactam 3a, for the initial experiments ended with failure. In most cases, regardless of the used catalytic system (Pd source: Pd(OAc)2, Pd2(dba)3, ligand: DPEPhos, DavePhos, BINAP), solvent (1,4-dioxane, toluene) and base (t-BuOK, DIPEA, Cs2CO3), the 1H NMR spectra of the post-reaction mixtures indicated the presence of unchanged substrate 3a. Also, the variation of the reactant quantities (ligand/Pd source = 15:15, 23:15, 23:7.5, 20:20 mol %) and the reaction time did not have a positive effect on the course of the reaction and in several cases resulted in the formation of 2-methylphthalazin-1(2H)-one, i.e., the debromination product of bromophthalazinone 3a.
The commonly adopted view on the mechanism of the Pd-mediated C–N-bond formation (Buchwald–Hartwig-type coupling) [37-40] assumes that the coordination of the amine takes place after the oxidative addition of the organic halide to the palladium–ligand complex. Based on the analysis of our results we have concluded that in the case of systems 3, the order of reagent addition could have a significant impact on a proper course of the cross-coupling reaction. To confirm this idea, we carried out an experiment in which to the in situ-generated (BINAP)Pd complex (Pd2(dba)3/rac-BINAP or (R)-BINAP; 15:15 mol %) morpholine was added prior to the addition of lactam 3a. As a result, we obtained the target 4-(morpholin-4-yl) derivative 5a in 77% yield. Moreover, it turned out that this amination reaction also proceeded with a reduced amount of Pd2(dba)3 from 15 mol % to 2 mol % without loss of the product yield.
These results showed that the coordination of the amine to the (BINAP)Pd complex probably leads to the formation of a (BINAP)Pd–amine system (Figure 2), facilitating the oxidative addition of the bromolactam 3a and is the crucial step in the amination procedure of 4-bromophthalazinones [40].
![[1860-5397-17-50-2]](/bjoc/content/figures/1860-5397-17-50-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Proposed catalytic cycles for the amination of 4-bromophthalazinones of type 3 (Phthal: phthalazinone, PhthalBr: 4-bromophthalazinone, PhthalNR1R2: 4-aminophthalazinone).
Figure 2: Proposed catalytic cycles for the amination of 4-bromophthalazinones of type 3 (Phthal: phthalazino...
Based on the results of our initial studies, we next examined the reaction scope of the bromolactams 3 with various amines and polyamines. As can be seen from Scheme 2 the yields of the amination products 5 and 6 were dependent on the nature of the amine as well as on the used phthalazinone. Efficient results of coupling were received using, inter alia, cyclic, aromatic or benzylamines. The reaction of 3a with piperidine gave a higher yield of the target product 5c (85%) compared to the use of morpholine (5a, 77%), and especially thiomorpholine (5b, 62%). In turn, the use of 1-aminohexane (NH2Hex) in the reaction with 3b resulted in a significant decrease in the yield of product 5e (50%) in comparison with 2-(thiophen-2-yl)ethan-1-amine (5f, 80%), cyclohexylamine (5d, 85%), and also with the 4-CF3 and 4-OMe-benzylamines (5g, 85%; 5h, 65%). Additionally, when using primary amines in the reaction the formation of polysubstituted products was not observed.
![[1860-5397-17-50-i2]](/bjoc/content/inline/1860-5397-17-50-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of 4-amino- and 4-polyaminophthalazinones 5 and 6 (the yields refer to the isolated compounds).
Scheme 2: Synthesis of 4-amino- and 4-polyaminophthalazinones 5 and 6 (the yields refer to the isolated compo...
On the other hand, N-phenylpyridin-2-amine afforded the appropriate compound 5i in a lower yield as compared with aliphatic amines. Moreover, it turned out that an aminoalkyl substituent attached to the lactam scaffold did not disturb its functionalization at the 4 position by a simple amine. The corresponding product 5j was obtained in 89% yield.
It was found that also polyamines are suitable reagents in this Pd-coupling reaction. Only the reaction of 3c with N1,N1-diethylpentane-1,4-diamine proceeded in a lower yield and the target derivative 6e was isolated with 44% yield. In the case of N,N′-dimethylethylenediamine the substitution of both nitrogen atoms was not observed. Instead, the partial demethylation of the amine occurred. As a result, a mixture of the products 6c and 6d (molar ratio 6c/6d = 1:4) was obtained. With the increase in the amount of palladium (15 mol %) the phthalazin-1(2H)-one derivative 6d was obtained as the main product (70%).
Complexation behavior of aminophthalazinones with Cu(II) ions
To conclude the synthetic research, we looked at the potential of aminophthalazinones as polydentate ligands for the synthesis of complexes with metal ions. Examples of the use of phthalazinone and its derivatives in the synthesis of coordination compounds with La(III), Co(II), Cu(II), Mn(II) were extremely interesting [41,42]. Despite the fact that the phthalazinone molecule is potentially a tridentate NNO ligand, so far, in the examples described in the literature the coordination takes place through the oxygen atom [41,42]. Having synthesized 4-aminophthalazinones, we decided to investigate how these compounds behave towards Cu(II) ions. For testing, we chose the derivatives of ethylenediamine 6d, N-phenylpyridin-2-amine 5i, and additionally the pyridin-2-yl derivative 7 (Figure 3). Compound 7 was synthesized according to the method described by us [43].
![[1860-5397-17-50-3]](/bjoc/content/figures/1860-5397-17-50-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The phthalazinone derivatives that were used to test the complexation of Cu(II) ions.
Figure 3: The phthalazinone derivatives that were used to test the complexation of Cu(II) ions.
All selected compounds (Figure 3) contain some specific structural elements (e.g., an ethylenediamine moiety, similarity to bipyridyl or even to isocyclam skeleton) allowing them to act as ligands and form complexes. In our tests, we hoped that putting in the 4-position of the skeleton a substituent with a donor nitrogen atom could result in the formation of complexes following a different way of coordination with the metal ion than described in the literature [41].
The complexing properties of compounds 5i, 6d, and 7 were investigated by mass spectrometry (ESIMS). The spectra were recorded after time intervals of 0 h, 1 h, and 24 h starting from mixing the solutions containing equimolar amounts of the ligand and CuCl2. Unexpectedly, compound 6d (L1) did not show any complexing properties. In all spectra a signal of the same ligand, despite long reaction times, was only observed (ESIMS, negative ion mode, m/z = 231.1 Da; ESIMS, positive ion mode, m/z = 233.1 Da (100%)).
In the case of compound 5i (L2), the formation of two types of complexes was detected: 1) with m/z = 426.2 Da [(L2)63Cu(II)Cl]+, 428.2 Da [(L2)65Cu(II)Cl]+ and 2) with m/z = 719.5 Da and 721.5 Da, which corresponded to the 63Cu2+/65Cu2+ ion complex containing two ligands L2. However, a significant amount of unchanged ligand was also visible. For the ion at m/z = 719.5 Da, tandem spectra were recorded. The first fragmentation gave ions at m/z = 329.3 Da (L2 + H+) and 391.4 Da (ligand L2 and copper). The further fragmentation of the ion at m/z = 391.4 Da gave rise to an ion series: 362.4 Da (probably after elimination of HCO), 311.4 Da, 286.3 Da (100%), and 235.4 Da. We did not observe in the tandem spectra the signal after the elimination of copper alone.
Similarly, the ESIMS spectrum of an equimolar mixture of compound 7 (L3) and CuCl2 showed the presence of two types of complexes containing one and two phthalazinone ligands: [(L3)Cu(II)Cl]+ and [(L3)2Cu2(II)Cl3]+. The most abundant peak at m/z = 422.3 Da corresponded to the complex [(L3)Cu(II)Cl]+. The MS/MS fragmentation of the ions at m/z = 422.3 Da for 63Cu and 424.3 Da for 65Cu followed the same fragmentation pattern for both ions. The proposal of the fragmentation pathway, based on the X-ray crystal structure of the Cu(II) complex with 7 (L3) (Figure 4, vide infra), is shown in Scheme 3. The MS/MS fragmentation analysis of the [(L3)Cu(II)Cl]+ complex 8 (m/z = 422.3 and 424.3 Da) showed at the first step the loss of an aminoalkyl fragment (C2H3NMe2 = 71.1 Da) to form the ions 9 ⇌ 10 (m/z = 351.2 and 353.2 Da). Because of the lactam–lactim tautomerism the further complex decomposition can proceed through two fragmentation routes: 1) with the loss of HCl/CO or 2) with the loss of HCl/N2 (≈64 Da). In both cases, the pyridazinone moiety undergoes degradation to different ions 11, 12 with the same m/z = 287.3 and 289.3 Da. In the next stage, the copper cation is detached to form ions 13 and 14 (224.3 Da) indicating that the copper is well fitted into compound moiety.
![[1860-5397-17-50-i3]](/bjoc/content/inline/1860-5397-17-50-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: The proposal of the fragmentation pathway of the Cu(II) complex with compound 7.
Scheme 3: The proposal of the fragmentation pathway of the Cu(II) complex with compound 7.
Based on the results of X-ray structural analysis of the Cu(II) complex with 7, it can be assumed, that also in the case of ligand 5i (L2) the nitrogen atoms of the pyridin-2-yl and azomethin moiety participate in the coordination with Cu(II) ions.
Crystallography of complex 17
The copper(II) complex 17 [(L3)Cu(II)Cl2] was synthesized and characterized by X-ray analysis, FTIR and vis–NIR spectroscopy (for details see Supporting Information File 2). The molecular structure of the complex 17 is shown in Figure 4 and Figure 5.
![[1860-5397-17-50-4]](/bjoc/content/figures/1860-5397-17-50-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-50-5]](/bjoc/content/figures/1860-5397-17-50-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: Molecular structure of complex 17 with atom numbering scheme. The anisotropic displacement parameters are shown at the 50% probability level.
Figure 5: Molecular structure of complex 17 with atom numbering scheme. The anisotropic displacement paramete...
The basic experimental details and selected crystallographic data are summarized in Table 1. For full details on the comparison of experimental and calculated bond lengths and bond angles of complex 17 are provided in Supporting Information File 2, Table S1. The complex 17 crystallizes in the monoclinic Pc space group with two molecules A and B in one asymmetric unit. Their geometry was fully optimized in vacuum using the DFT method with the crystal structure coordinates as the input geometry (optimized at the CAM-B3LYP/6–311++G(d,p)/LanL2DZ(Cu) level of theory). However, due to the convergence failure during the geometry optimization of the dimer of molecules A and B, the geometry optimization was performed for the isolated molecules A and B.
Table 1: Crystallographic data for complex 17.
| crystal data | complex 17 |
|---|---|
| empirical formula | C18H20Cl2CuN4O2 |
| formula weight | 458.83 |
| crystal system | monoclinic |
| space group | Pc |
| unit cell dimensions | |
| a (Å) | 11.5529(4) |
| b (Å) | 11.4432(4) |
| c (Å) | 14.3523(5) |
| α (°) | |
| β (°) | 97.337(3) |
| γ (°) | |
| V (Å3) | 1881.87(11) |
| Z | 4 |
| T (K) | 100(2) |
| F(000) | 940 |
| Dx (g cm−3) | 1.620 |
| μ (mm−1) | 1.466 |
| scan method | Ω-scan |
| θ range (°) | 2.9, 26.5 |
| measured reflections | 13642 |
| unique reflections | 5023 |
| observed reflections [I>2σ(I)] | 4548 |
| completeness to θmax (%) | 99.7 |
| R [I>2σ(I)] | 0.0408 |
| wR (all data) | 0.0943 |
| S | 1.04 |
| largest diff. peak, hole (e Å−3) | −0.45, 0.93 |
In a solid phase the Cu(II) central ion is five-coordinated by three nitrogen atoms of 7 and two chloride anions. The CuN3Cl2 coordination center adopts the strongly distorted square pyramidal geometry with the five-coordinate geometry index (τ5) of 0.45 and 0.42 for molecules A and B, respectively. In particular, in molecules A and B, the pyridin-2-yl nitrogen atoms (N3 and N5), the azomethine nitrogen atoms (N1 and N7), and the tertiary nitrogen atom of the (2-(dimethylamino)ethyl group (N4 and N6) act as donors of coordination bonds. The coordination sphere of the Cu1 and Cu2 central ions is completed with Cl1, Cl2 and Cl3, Cl4 chloride anions, respectively. In the molecules A and B the pyridin-2-yl substituents are twisted in opposite directions relative to the mean plane of the 1,2-diazine moiety with the dihedral angles equal to 27.61 and 28.71°, respectively, and moreover, the methyl groups of the methoxy substituents are directed opposite and towards to the pyridin-2-yl substituents, respectively. The crystal structure of complex 17 is stabilized with a 3D intermolecular hydrogen bond network (Figure S1a, Supporting Information File 2). Additionally, the complex 17 in a solid state is also stabilized with π–π stacking interactions between the 1,2-diazine moieties and the pyridin-2-yl substituents of the ligands (Figure S1b, Supporting Information File 2).
The FTIR spectrum recorded for complex 17 confirmed the coordination of copper(II) through nitrogen atoms of the pyridin-2-yl substituent, the pyridazin-3-one moiety, and the 2-(dimethylamino)ethyl group. In the vis–NIR spectrum recorded in methanol, based on TD-DFT calculations, the d–d, metal–ligand charge transfer (MLCT), ligand–metal charge transfer (MLCT), and intraligand charge transfer (ILCT) transitions were identified. The vis–NIR spectroscopy revealed that in complex 17 the metal-to-ligand stoichiometry is equal to 1:1. In turn, cyclic voltammetry was used to investigate the electrochemical behavior of complex 17. The measurements on a platinum disc electrode suggested that the [(L3)Cu(II)Cl2]/[(L3)Cu(I)Cl2] redox couple was formed. For more details, see Supporting Information File 2.
Bioactivity
Cytotoxicity analysis – MTT assay
We used the MTT assay to evaluate compounds 5c, 5d, 5f, 5g, 6b, and 6d for their potential activity on cytotoxicity, proliferation and growth of HT-29 (human colon adenocarcinoma cell line), PC-3 (human prostate cancer cell line) and L-929 cells (mouse fibroblast cell line). The results obtained from the MTT assay revealed that the tested phthalazinone derivatives were cytotoxic towards the tested cell lines in the concentration range from 20.1 μM to 92.93 μM (Figure 6 and Figure 7).
![[1860-5397-17-50-6]](/bjoc/content/figures/1860-5397-17-50-6.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 6: Determination of relative cell viability (% of control) in different cell lines (HT-29; PC-3 and L-929) treated with compounds 6d (1), 6b (2), 5d (3), 5c (4), 5g (5), 5f (6) using the MTT assay. The SEM value is shown for each tested concentration value.
Figure 6: Determination of relative cell viability (% of control) in different cell lines (HT-29; PC-3 and L-...
![[1860-5397-17-50-7]](/bjoc/content/figures/1860-5397-17-50-7.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 7: Cytotoxic properties of the phthalazinone derivatives expressed as IC50 after 72 h of cell treatment. Values significantly differing from the control cell line (L-929) by ANOVA and Dunnett’s test: * increase and # decrease.
Figure 7: Cytotoxic properties of the phthalazinone derivatives expressed as IC50 after 72 h of cell treatmen...
The compounds 5d, 5g and 5f exhibited the strongest cytotoxic effects as compared to the other examined compounds. The IC50 values for the tested cell lines were respectively: HT-29 (IC50 = 29.79 µM; 31.45 µM; 27.41 µM), PC-3 (IC50 = 20.12 µM; 22.4 µM; 27.27 µM) and L-929 (IC50 = 20.44 µM; 21.32 µM; 20.1µM). However, derivatives 6d and 5c appeared to be less cytotoxic against all tested cell lines. The IC50 values for the tested cell lines were respectively: HT-29 (IC50 = 75.58 µM; 36.67 µM), PC-3 (IC50 = 82.95 µM; 45.98 µM) and L-929 (IC50 = 52.69 µM; 27.35 µM). Despite the fact that the compounds were cytotoxic to tumor cell lines, they were also similarly or even more cytotoxic to the control line (L-929), which is not a desirable feature for any kind of a new potential pharmaceutical drug.
Among the analyzed compounds only compound 6b significantly influenced the viability of the tumor cell lines HT-29 (IC50 = 51.65 μM) and PC-3 (IC50 = 66.27 μM) compared to normal cells L-929 (IC50 = 92.93 μM). However, in order to determine further biological utility of compound 6b, additional research has to be done,
Conclusion
In conclusion, we have demonstrated an efficient synthesis of 2-substituted (alkyl, aminoalkyl) 4-aminophthalazinones 5 and 6 via the direct bromination of phthalazin-1(2H)-one (1) with potassium tribromide, followed by the alkylation of 4-bromophthalazinone 2 with methyl iodide, isopropyl iodide or 2-chloro-N,N-dimethylethylamine and 4-(2-chloroethyl)morpholine and further palladium-catalyzed amination of lactams 3 with aliphatic, aromatic, benzylic, cyclic amines and polyamines. Furthermore, we have demonstrated that some of the phthalazinone derivatives act as ligands and form stable coordination compounds with Cu(II) ions. The results of biological tests showed that the compounds containing an amino- or polyamino-substituent at the 4-position of the phthalazinone moiety could have potential applications as new anticancer agents.
Supporting Information
| Supporting Information File 1: Experimental details, synthetic procedures, and characterization data of new compounds including copies of spectra. | ||
| Format: PDF | Size: 3.4 MB | Download |
| Supporting Information File 2: Experimental details, computational details, X-ray crystallographic data, FTIR and vis–NIR spectra, and cyclic voltammetry for compounds 7 and 17. | ||
| Format: PDF | Size: 706.8 KB | Download |
References
-
Vila, N.; Besada, P.; Costas, T.; Costas-Lago, M. C.; Terán, C. Eur. J. Med. Chem. 2015, 97, 462–482. doi:10.1016/j.ejmech.2014.11.043
Return to citation in text: [1] -
Asif, M. Chem. Int. 2019, 5 (1), 97–108.
https://bosaljournals.com/chemint/images/pdffiles/19-4.pdf
Return to citation in text: [1] -
Sridhara, A. M.; Reddy, K. R. V.; Keshavayya, J.; Vadiraj, S. G.; Bose, P.; Ambika, D. S.; Raju, C. K.; Shashidhara, S.; Raju, N. H. J. J. Pharm. Res. (Gurgaon, India) 2011, 4, 496–500.
http://jprsolutions.info/newfiles/journal-file-56cd4abbe72b30.45259996.pdf
Return to citation in text: [1] -
Moustafa, A. H.; El-Sayed, H. A.; Abd El-Hady, R. A.; Haikal, A. Z.; El-Hashash, M. J. Heterocycl. Chem. 2016, 53, 789–799. doi:10.1002/jhet.2316
Return to citation in text: [1] -
El-Wahab, A. H. F. A.; Mohamed, H. M.; El-Agrody, A. M.; El-Nassag, M. A.; Bedair, A. H. Pharmaceuticals 2011, 4, 1158–1170. doi:10.3390/ph4081158
Return to citation in text: [1] -
Cashman, J. R.; Voelker, T.; Johnson, R.; Janowsky, A. Bioorg. Med. Chem. 2009, 17, 337–343. doi:10.1016/j.bmc.2008.10.065
Return to citation in text: [1] -
Cashman, J. R.; Voelker, T.; Zhang, H.-T.; O’Donnell, J. M. J. Med. Chem. 2009, 52, 1530–1539. doi:10.1021/jm8010993
Return to citation in text: [1] -
Yamaguchi, M.; Kamei, K.; Koga, T.; Akima, M.; Kuroki, T.; Ohi, N. J. Med. Chem. 1993, 36, 4052–4060. doi:10.1021/jm00077a008
Return to citation in text: [1] -
Procopiou, P. A.; Browning, C.; Gore, P. M.; Lynn, S. M.; Richards, S. A.; Slack, R. J.; Sollis, S. L. Bioorg. Med. Chem. 2012, 20, 6097–6108. doi:10.1016/j.bmc.2012.08.032
Return to citation in text: [1] -
Procopiou, P. A.; Ford, A. J.; Gore, P. M.; Looker, B. E.; Hodgson, S. T.; Holmes, D. S.; Vile, S.; Clark, K. L.; Saunders, K. A.; Slack, R. J.; Rowedder, J. E.; Watts, C. J. ACS Med. Chem. Lett. 2017, 8, 577–581. doi:10.1021/acsmedchemlett.7b00112
Return to citation in text: [1] -
Medda, F.; Sells, E.; Chang, H.-H.; Dietrich, J.; Chappeta, S.; Smith, B.; Gokhale, V.; Meuillet, E. J.; Hulme, C. Bioorg. Med. Chem. Lett. 2013, 23, 528–531. doi:10.1016/j.bmcl.2012.11.030
Return to citation in text: [1] -
Poli, D.; Catarzi, D.; Colotta, V.; Varano, F.; Filacchioni, G.; Daniele, S.; Trincavelli, L.; Martini, C.; Paoletta, S.; Moro, S. J. Med. Chem. 2011, 54, 2102–2113. doi:10.1021/jm101328n
Return to citation in text: [1] -
Watanabe, N.; Adachi, H.; Takase, Y.; Ozaki, H.; Matsukura, M.; Miyazaki, K.; Ishibashi, K.; Ishihara, H.; Kodama, K.; Nishino, M.; Kakiki, M.; Kabasawa, Y. J. Med. Chem. 2000, 43, 2523–2529. doi:10.1021/jm9905054
Return to citation in text: [1] -
Bollenbach, M.; Lugnier, C.; Kremer, M.; Salvat, E.; Megat, S.; Bihel, F.; Bourguignon, J.-J.; Barrot, M.; Schmitt, M. Eur. J. Med. Chem. 2019, 177, 269–290. doi:10.1016/j.ejmech.2019.05.026
Return to citation in text: [1] -
Humphrey, J. M. Aminophthalazine derivative compounds. WO Pat. Appl. WO2007/085954 A2, Aug 2, 2007.
Return to citation in text: [1] -
Herberich, B.; Cao, G.-Q.; Chakrabarti, P. P.; Falsey, J. R.; Pettus, L.; Rzasa, R. M.; Reed, A. B.; Reichelt, A.; Sham, K.; Thaman, M.; Wurz, R. P.; Xu, S.; Zhang, D.; Hsieh, F.; Lee, M. R.; Syed, R.; Li, V.; Grosfeld, D.; Plant, M. H.; Henkle, B.; Sherman, L.; Middleton, S.; Wong, L. M.; Tasker, A. S. J. Med. Chem. 2008, 51, 6271–6279. doi:10.1021/jm8005417
Return to citation in text: [1] -
Nomoto, Y.; Obase, H.; Takai, H.; Teranishi, M.; Nakamura, J.; Kubo, K. Chem. Pharm. Bull. 1990, 38, 2179–2183. doi:10.1248/cpb.38.2179
Return to citation in text: [1] -
Li, J.; Zhao, Y.-F.; Yuan, X.-Y.; Xu, J.-X.; Gong, P. Molecules 2006, 11, 574–582. doi:10.3390/11070574
Return to citation in text: [1] -
Prime, M. E.; Courtney, S. M.; Brookfield, F. A.; Marston, R. W.; Walker, V.; Warne, J.; Boyd, A. E.; Kairies, N. A.; von der Saal, W.; Limberg, A.; Georges, G.; Engh, R. A.; Goller, B.; Rueger, P.; Rueth, M. J. Med. Chem. 2011, 54, 312–319. doi:10.1021/jm101346r
Return to citation in text: [1] [2] [3] -
Wang, W.; Feng, X.; Liu, H.-X.; Chen, S.-W.; Hui, L. Bioorg. Med. Chem. 2018, 26, 3217–3226. doi:10.1016/j.bmc.2018.04.048
Return to citation in text: [1] -
Reece, P. A. Med. Res. Rev. 1981, 1, 73–96. doi:10.1002/med.2610010105
Return to citation in text: [1] -
Leiro, J. M.; Álvarez, E.; Arranz, J. A.; Cano, E.; Orallo, F. Int. Immunopharmacol. 2004, 4, 163–177. doi:10.1016/j.intimp.2003.10.004
Return to citation in text: [1] -
Graça, I.; Sousa, E. J.; Costa-Pinheiro, P.; Vieira, F. Q.; Torres-Ferreira, J.; Martins, M. G.; Henrique, R.; Jerónimo, C. Oncotarget 2014, 5, 5950–5964. doi:10.18632/oncotarget.1909
Return to citation in text: [1] -
Tanaka, S.; Tanaka, M.; Akashi, A. Stroke 1989, 20, 1724–1729. doi:10.1161/01.str.20.12.1724
Return to citation in text: [1] -
Moroi, R.; Ono, K.; Saito, T.; Akimoto, T.; Sano, M. Chem. Pharm. Bull. 1977, 25, 830–835. doi:10.1248/cpb.25.830
Return to citation in text: [1] -
Jost, L. M.; Gschwind, H.-P.; Jalava, T.; Wang, Y.; Guenther, C.; Souppart, C.; Rottmann, A.; Denner, K.; Waldmeier, F.; Gross, G.; Masson, E.; Laurent, D. Drug Metab. Dispos. 2006, 34, 1817–1828. doi:10.1124/dmd.106.009944
Return to citation in text: [1] -
Dragovich, T.; Laheru, D.; Dayyani, F.; Bolejack, V.; Smith, L.; Seng, J.; Burris, H.; Rosen, P.; Hidalgo, M.; Ritch, P.; Baker, A. F.; Raghunand, N.; Crowley, J.; Von Hoff, D. D. Cancer Chemother. Pharmacol. 2014, 74, 379–387. doi:10.1007/s00280-014-2499-4
Return to citation in text: [1] -
Wang, F.; Molina, J.; Satele, D.; Yin, J.; Lim, V.-S.; Adjei, A. A. Invest. New Drugs 2019, 37, 658–665. doi:10.1007/s10637-018-0690-x
Return to citation in text: [1] -
Holló, B.; Magyari, J.; Živković-Radovanović, V.; Vučković, G.; Tomić, Z. D.; Szilágyi, I. M.; Pokol, G.; Mészáros Szécsényi, K. Polyhedron 2014, 80, 142–150. doi:10.1016/j.poly.2014.03.007
Return to citation in text: [1] -
Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Malinowski, Z.; Fornal, E.; Nowak, M.; Kontek, R.; Gajek, G.; Borek, B. Monatsh. Chem. 2015, 146, 1723–1731. doi:10.1007/s00706-015-1508-6
Return to citation in text: [1] [2] -
Vlaar, T.; Ruijter, E.; Znabet, A.; Janssen, E.; de Kanter, F. J. J.; Maes, B. U. W.; Orru, R. V. A. Org. Lett. 2011, 13, 6496–6499. doi:10.1021/ol202784d
Return to citation in text: [1] -
Vlaar, T.; Mampuys, P.; Helliwell, M.; Maes, B. U. W.; Orru, R. V. A.; Ruijter, E. J. Org. Chem. 2013, 78, 6735–6745. doi:10.1021/jo401131p
Return to citation in text: [1] -
Krishnananthan, S.; Smith, D.; Wu, D.-R.; Yip, S.; Gunaga, P.; Mathur, A.; Li, J. J. Org. Chem. 2016, 81, 1520–1526. doi:10.1021/acs.joc.5b02652
Return to citation in text: [1] [2] -
Nowak, M.; Malinowski, Z.; Jóźwiak, A.; Fornal, E.; Błaszczyk, A.; Kontek, R. Tetrahedron 2014, 70, 5153–5160. doi:10.1016/j.tet.2014.05.117
Return to citation in text: [1] [2] [3] -
Patel, N. R. Phthalazines. In Condensed Pyridazines Including Cinnolines and Phthalazines; Castle, R. N., Ed.; John Wiley & Sons: New York, NY, USA, 1973; Vol. 27, pp 375–392.
Return to citation in text: [1] -
Heravi, M. M.; Kheilkordi, Z.; Zadsirjan, V.; Heydari, M.; Malmir, M. J. Organomet. Chem. 2018, 861, 17–104. doi:10.1016/j.jorganchem.2018.02.023
Return to citation in text: [1] -
Schlummer, B.; Scholz, U. Adv. Synth. Catal. 2004, 346, 1599–1626. doi:10.1002/adsc.200404216
Return to citation in text: [1] -
Shekhar, S.; Ryberg, P.; Hartwig, J. F.; Mathew, J. S.; Blackmond, D. G.; Strieter, E. R.; Buchwald, S. L. J. Am. Chem. Soc. 2006, 128, 3584–3591. doi:10.1021/ja045533c
Return to citation in text: [1] -
Singh, U. K.; Strieter, E. R.; Blackmond, D. G.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 14104–14114. doi:10.1021/ja026885r
Return to citation in text: [1] [2] -
Wang, Y.; Yang, Z.-Y.; Wang, Q.; Cai, Q.-K.; Yu, K.-B. J. Organomet. Chem. 2005, 690, 4557–4563. doi:10.1016/j.jorganchem.2005.07.046
Return to citation in text: [1] [2] [3] -
Öztürk, N.; Bahçeli, S. Suleyman Demirel Univ. Fen Bilimleri Enst. Derg. 2012, 7, 1–8.
Return to citation in text: [1] [2] -
Pakulska, W.; Malinowski, Z.; Szcześniak, A. K.; Czarnecka, E.; Epsztajn, J. Arch. Pharm. (Weinheim, Ger.) 2009, 342, 41–47. doi:10.1002/ardp.200800016
Return to citation in text: [1]
| 37. | Heravi, M. M.; Kheilkordi, Z.; Zadsirjan, V.; Heydari, M.; Malmir, M. J. Organomet. Chem. 2018, 861, 17–104. doi:10.1016/j.jorganchem.2018.02.023 |
| 38. | Schlummer, B.; Scholz, U. Adv. Synth. Catal. 2004, 346, 1599–1626. doi:10.1002/adsc.200404216 |
| 39. | Shekhar, S.; Ryberg, P.; Hartwig, J. F.; Mathew, J. S.; Blackmond, D. G.; Strieter, E. R.; Buchwald, S. L. J. Am. Chem. Soc. 2006, 128, 3584–3591. doi:10.1021/ja045533c |
| 40. | Singh, U. K.; Strieter, E. R.; Blackmond, D. G.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 14104–14114. doi:10.1021/ja026885r |
| 40. | Singh, U. K.; Strieter, E. R.; Blackmond, D. G.; Buchwald, S. L. J. Am. Chem. Soc. 2002, 124, 14104–14114. doi:10.1021/ja026885r |
| 41. | Wang, Y.; Yang, Z.-Y.; Wang, Q.; Cai, Q.-K.; Yu, K.-B. J. Organomet. Chem. 2005, 690, 4557–4563. doi:10.1016/j.jorganchem.2005.07.046 |
| 42. | Öztürk, N.; Bahçeli, S. Suleyman Demirel Univ. Fen Bilimleri Enst. Derg. 2012, 7, 1–8. |
| 1. | Vila, N.; Besada, P.; Costas, T.; Costas-Lago, M. C.; Terán, C. Eur. J. Med. Chem. 2015, 97, 462–482. doi:10.1016/j.ejmech.2014.11.043 |
| 2. |
Asif, M. Chem. Int. 2019, 5 (1), 97–108.
https://bosaljournals.com/chemint/images/pdffiles/19-4.pdf |
| 8. | Yamaguchi, M.; Kamei, K.; Koga, T.; Akima, M.; Kuroki, T.; Ohi, N. J. Med. Chem. 1993, 36, 4052–4060. doi:10.1021/jm00077a008 |
| 9. | Procopiou, P. A.; Browning, C.; Gore, P. M.; Lynn, S. M.; Richards, S. A.; Slack, R. J.; Sollis, S. L. Bioorg. Med. Chem. 2012, 20, 6097–6108. doi:10.1016/j.bmc.2012.08.032 |
| 10. | Procopiou, P. A.; Ford, A. J.; Gore, P. M.; Looker, B. E.; Hodgson, S. T.; Holmes, D. S.; Vile, S.; Clark, K. L.; Saunders, K. A.; Slack, R. J.; Rowedder, J. E.; Watts, C. J. ACS Med. Chem. Lett. 2017, 8, 577–581. doi:10.1021/acsmedchemlett.7b00112 |
| 25. | Moroi, R.; Ono, K.; Saito, T.; Akimoto, T.; Sano, M. Chem. Pharm. Bull. 1977, 25, 830–835. doi:10.1248/cpb.25.830 |
| 6. | Cashman, J. R.; Voelker, T.; Johnson, R.; Janowsky, A. Bioorg. Med. Chem. 2009, 17, 337–343. doi:10.1016/j.bmc.2008.10.065 |
| 7. | Cashman, J. R.; Voelker, T.; Zhang, H.-T.; O’Donnell, J. M. J. Med. Chem. 2009, 52, 1530–1539. doi:10.1021/jm8010993 |
| 26. | Jost, L. M.; Gschwind, H.-P.; Jalava, T.; Wang, Y.; Guenther, C.; Souppart, C.; Rottmann, A.; Denner, K.; Waldmeier, F.; Gross, G.; Masson, E.; Laurent, D. Drug Metab. Dispos. 2006, 34, 1817–1828. doi:10.1124/dmd.106.009944 |
| 27. | Dragovich, T.; Laheru, D.; Dayyani, F.; Bolejack, V.; Smith, L.; Seng, J.; Burris, H.; Rosen, P.; Hidalgo, M.; Ritch, P.; Baker, A. F.; Raghunand, N.; Crowley, J.; Von Hoff, D. D. Cancer Chemother. Pharmacol. 2014, 74, 379–387. doi:10.1007/s00280-014-2499-4 |
| 28. | Wang, F.; Molina, J.; Satele, D.; Yin, J.; Lim, V.-S.; Adjei, A. A. Invest. New Drugs 2019, 37, 658–665. doi:10.1007/s10637-018-0690-x |
| 5. | El-Wahab, A. H. F. A.; Mohamed, H. M.; El-Agrody, A. M.; El-Nassag, M. A.; Bedair, A. H. Pharmaceuticals 2011, 4, 1158–1170. doi:10.3390/ph4081158 |
| 21. | Reece, P. A. Med. Res. Rev. 1981, 1, 73–96. doi:10.1002/med.2610010105 |
| 22. | Leiro, J. M.; Álvarez, E.; Arranz, J. A.; Cano, E.; Orallo, F. Int. Immunopharmacol. 2004, 4, 163–177. doi:10.1016/j.intimp.2003.10.004 |
| 23. | Graça, I.; Sousa, E. J.; Costa-Pinheiro, P.; Vieira, F. Q.; Torres-Ferreira, J.; Martins, M. G.; Henrique, R.; Jerónimo, C. Oncotarget 2014, 5, 5950–5964. doi:10.18632/oncotarget.1909 |
| 3. |
Sridhara, A. M.; Reddy, K. R. V.; Keshavayya, J.; Vadiraj, S. G.; Bose, P.; Ambika, D. S.; Raju, C. K.; Shashidhara, S.; Raju, N. H. J. J. Pharm. Res. (Gurgaon, India) 2011, 4, 496–500.
http://jprsolutions.info/newfiles/journal-file-56cd4abbe72b30.45259996.pdf |
| 4. | Moustafa, A. H.; El-Sayed, H. A.; Abd El-Hady, R. A.; Haikal, A. Z.; El-Hashash, M. J. Heterocycl. Chem. 2016, 53, 789–799. doi:10.1002/jhet.2316 |
| 24. | Tanaka, S.; Tanaka, M.; Akashi, A. Stroke 1989, 20, 1724–1729. doi:10.1161/01.str.20.12.1724 |
| 15. | Humphrey, J. M. Aminophthalazine derivative compounds. WO Pat. Appl. WO2007/085954 A2, Aug 2, 2007. |
| 17. | Nomoto, Y.; Obase, H.; Takai, H.; Teranishi, M.; Nakamura, J.; Kubo, K. Chem. Pharm. Bull. 1990, 38, 2179–2183. doi:10.1248/cpb.38.2179 |
| 41. | Wang, Y.; Yang, Z.-Y.; Wang, Q.; Cai, Q.-K.; Yu, K.-B. J. Organomet. Chem. 2005, 690, 4557–4563. doi:10.1016/j.jorganchem.2005.07.046 |
| 13. | Watanabe, N.; Adachi, H.; Takase, Y.; Ozaki, H.; Matsukura, M.; Miyazaki, K.; Ishibashi, K.; Ishihara, H.; Kodama, K.; Nishino, M.; Kakiki, M.; Kabasawa, Y. J. Med. Chem. 2000, 43, 2523–2529. doi:10.1021/jm9905054 |
| 14. | Bollenbach, M.; Lugnier, C.; Kremer, M.; Salvat, E.; Megat, S.; Bihel, F.; Bourguignon, J.-J.; Barrot, M.; Schmitt, M. Eur. J. Med. Chem. 2019, 177, 269–290. doi:10.1016/j.ejmech.2019.05.026 |
| 18. | Li, J.; Zhao, Y.-F.; Yuan, X.-Y.; Xu, J.-X.; Gong, P. Molecules 2006, 11, 574–582. doi:10.3390/11070574 |
| 19. | Prime, M. E.; Courtney, S. M.; Brookfield, F. A.; Marston, R. W.; Walker, V.; Warne, J.; Boyd, A. E.; Kairies, N. A.; von der Saal, W.; Limberg, A.; Georges, G.; Engh, R. A.; Goller, B.; Rueger, P.; Rueth, M. J. Med. Chem. 2011, 54, 312–319. doi:10.1021/jm101346r |
| 20. | Wang, W.; Feng, X.; Liu, H.-X.; Chen, S.-W.; Hui, L. Bioorg. Med. Chem. 2018, 26, 3217–3226. doi:10.1016/j.bmc.2018.04.048 |
| 12. | Poli, D.; Catarzi, D.; Colotta, V.; Varano, F.; Filacchioni, G.; Daniele, S.; Trincavelli, L.; Martini, C.; Paoletta, S.; Moro, S. J. Med. Chem. 2011, 54, 2102–2113. doi:10.1021/jm101328n |
| 41. | Wang, Y.; Yang, Z.-Y.; Wang, Q.; Cai, Q.-K.; Yu, K.-B. J. Organomet. Chem. 2005, 690, 4557–4563. doi:10.1016/j.jorganchem.2005.07.046 |
| 42. | Öztürk, N.; Bahçeli, S. Suleyman Demirel Univ. Fen Bilimleri Enst. Derg. 2012, 7, 1–8. |
| 11. | Medda, F.; Sells, E.; Chang, H.-H.; Dietrich, J.; Chappeta, S.; Smith, B.; Gokhale, V.; Meuillet, E. J.; Hulme, C. Bioorg. Med. Chem. Lett. 2013, 23, 528–531. doi:10.1016/j.bmcl.2012.11.030 |
| 16. | Herberich, B.; Cao, G.-Q.; Chakrabarti, P. P.; Falsey, J. R.; Pettus, L.; Rzasa, R. M.; Reed, A. B.; Reichelt, A.; Sham, K.; Thaman, M.; Wurz, R. P.; Xu, S.; Zhang, D.; Hsieh, F.; Lee, M. R.; Syed, R.; Li, V.; Grosfeld, D.; Plant, M. H.; Henkle, B.; Sherman, L.; Middleton, S.; Wong, L. M.; Tasker, A. S. J. Med. Chem. 2008, 51, 6271–6279. doi:10.1021/jm8005417 |
| 43. | Pakulska, W.; Malinowski, Z.; Szcześniak, A. K.; Czarnecka, E.; Epsztajn, J. Arch. Pharm. (Weinheim, Ger.) 2009, 342, 41–47. doi:10.1002/ardp.200800016 |
| 30. | Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022 |
| 31. | Malinowski, Z.; Fornal, E.; Nowak, M.; Kontek, R.; Gajek, G.; Borek, B. Monatsh. Chem. 2015, 146, 1723–1731. doi:10.1007/s00706-015-1508-6 |
| 29. | Holló, B.; Magyari, J.; Živković-Radovanović, V.; Vučković, G.; Tomić, Z. D.; Szilágyi, I. M.; Pokol, G.; Mészáros Szécsényi, K. Polyhedron 2014, 80, 142–150. doi:10.1016/j.poly.2014.03.007 |
| 30. | Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022 |
| 30. | Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022 |
| 31. | Malinowski, Z.; Fornal, E.; Nowak, M.; Kontek, R.; Gajek, G.; Borek, B. Monatsh. Chem. 2015, 146, 1723–1731. doi:10.1007/s00706-015-1508-6 |
| 35. | Nowak, M.; Malinowski, Z.; Jóźwiak, A.; Fornal, E.; Błaszczyk, A.; Kontek, R. Tetrahedron 2014, 70, 5153–5160. doi:10.1016/j.tet.2014.05.117 |
| 30. | Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022 |
| 35. | Nowak, M.; Malinowski, Z.; Jóźwiak, A.; Fornal, E.; Błaszczyk, A.; Kontek, R. Tetrahedron 2014, 70, 5153–5160. doi:10.1016/j.tet.2014.05.117 |
| 30. | Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022 |
| 30. | Malinowski, Z.; Fornal, E.; Sierocińska, B.; Czeczko, R.; Nowak, M. Tetrahedron 2016, 72, 7942–7951. doi:10.1016/j.tet.2016.10.022 |
| 36. | Patel, N. R. Phthalazines. In Condensed Pyridazines Including Cinnolines and Phthalazines; Castle, R. N., Ed.; John Wiley & Sons: New York, NY, USA, 1973; Vol. 27, pp 375–392. |
| 19. | Prime, M. E.; Courtney, S. M.; Brookfield, F. A.; Marston, R. W.; Walker, V.; Warne, J.; Boyd, A. E.; Kairies, N. A.; von der Saal, W.; Limberg, A.; Georges, G.; Engh, R. A.; Goller, B.; Rueger, P.; Rueth, M. J. Med. Chem. 2011, 54, 312–319. doi:10.1021/jm101346r |
| 34. | Krishnananthan, S.; Smith, D.; Wu, D.-R.; Yip, S.; Gunaga, P.; Mathur, A.; Li, J. J. Org. Chem. 2016, 81, 1520–1526. doi:10.1021/acs.joc.5b02652 |
| 35. | Nowak, M.; Malinowski, Z.; Jóźwiak, A.; Fornal, E.; Błaszczyk, A.; Kontek, R. Tetrahedron 2014, 70, 5153–5160. doi:10.1016/j.tet.2014.05.117 |
| 32. | Vlaar, T.; Ruijter, E.; Znabet, A.; Janssen, E.; de Kanter, F. J. J.; Maes, B. U. W.; Orru, R. V. A. Org. Lett. 2011, 13, 6496–6499. doi:10.1021/ol202784d |
| 33. | Vlaar, T.; Mampuys, P.; Helliwell, M.; Maes, B. U. W.; Orru, R. V. A.; Ruijter, E. J. Org. Chem. 2013, 78, 6735–6745. doi:10.1021/jo401131p |
| 19. | Prime, M. E.; Courtney, S. M.; Brookfield, F. A.; Marston, R. W.; Walker, V.; Warne, J.; Boyd, A. E.; Kairies, N. A.; von der Saal, W.; Limberg, A.; Georges, G.; Engh, R. A.; Goller, B.; Rueger, P.; Rueth, M. J. Med. Chem. 2011, 54, 312–319. doi:10.1021/jm101346r |
| 34. | Krishnananthan, S.; Smith, D.; Wu, D.-R.; Yip, S.; Gunaga, P.; Mathur, A.; Li, J. J. Org. Chem. 2016, 81, 1520–1526. doi:10.1021/acs.joc.5b02652 |
© 2021 Malinowski et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)