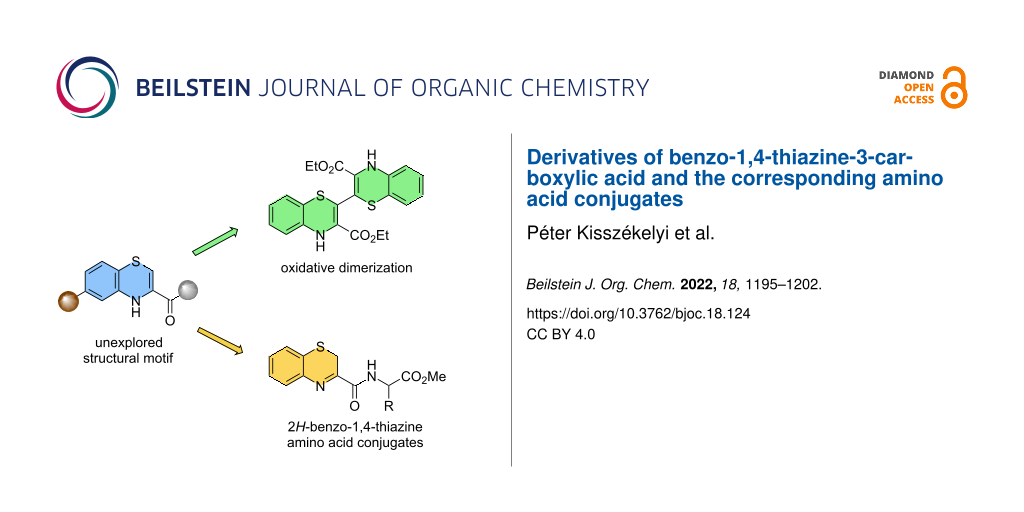
Abstract
Herein, we present the synthesis and utilization of derivatives of 4H-benzo[b][1,4]thiazine-3-carboxylic acid. These benzothiazine compounds were assembled via the coupling of aminothiols and bromopyruvates. Oxidative dimerization of these starting materials was also observed and the corresponding benzothiazine dimers were isolated. Moreover, the coupling of benzothiazines with amino acids was realized. In doing so, an enantioselective synthesis of the nonproteinogenic amino acid 2-amino-3-propylhexanoic acid was accomplished.

Graphical Abstract
Introduction
Heterocyclic compounds with a benzothiazine moiety are attractive building blocks in medicinal chemistry. Benzo-1,4-thiazine derivatives possess a wide range of biological and pharmacological properties, such as anticancer and antitumor, antioxidant, antimicrobial, antibacterial, antifungal, antiviral, antimalarial, antidiabetic, antihypertensive, anti-inflammatory, analgesic, anti-rheumatic, or anti-allergic properties [1-5].
Several methods for the preparation of 4H-benzo-1,4-thiazines have been described in the literature. Methods for the synthesis of 2,3-disubstituted 4H-benzo-1,4-thiazines 1 (Figure 1) are the most studied and described. Such benzothiazine derivatives are typically prepared by reactions of various carbonyl or carboxyl compounds with 2-aminothiophenols [6,7] or the respective dimers (2,2'-disulfanediyldianilines) [8]. These disulfides are often formed in situ from the corresponding aminothiols [9-13].
![[1860-5397-18-124-1]](/bjoc/content/figures/1860-5397-18-124-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Typical benzothiazine structures.
Figure 1: Typical benzothiazine structures.
Green chemistry methods for benzo-1,4-thiazine synthesis have also been described in the literature. 2,3-Disubstituted-1,4-benzothiazines were prepared in high yield (83–96%) by oxidative cyclocondensation of 2-aminobenzenethiols and 1,3-dicarbonyl compounds using a catalytic amount of hydrazine hydrate without solvent in a short reaction time (10 min) [14]. Reactions of 2-aminothiophenols with β-keto esters and β-diketones under microwave irradiation (MWI) using basic alumina as heterogeneous catalyst without solvent afforded 4H-benzo-1,4-thiazines in a yield of 69–85% within 6–11 min [15]. Furthermore, baker’s yeast as whole-cell biocatalyst catalyzed the reaction of 2-aminothiophenols with 1,3-dicarbonyl compounds in methanol and the corresponding 4H-benzo-1,4-thiazines were prepared in 51–82% yield. These reactions were significantly accelerated by ultrasonic irradiation [16]. Cyclocondensation of 1,3-dicarbonyl compounds with substituted diaryl disulfides in water in the presence of β-cyclodextrin gave 2,3-disubstituted benzo-1,4-thiazines in 70–91% yield in 50 min [17]. A highly efficient visible-light-mediated one-pot, three-component procedure was also explored for the preparation of 3-aryl-4H-benzo-1,4-thiazin-2-amines [18].
3-Aryl- and 3-alkyl-4H-benzo[b][1,4]thiazine-4-carbonitriles 2 (Figure 1) were synthesized in high yield from the corresponding 2-aminobenzothiazoles using the copper–organic framework Cu–MOF-74 as a catalyst [19]. The reactions of 2-aminobenzenethioles with ethyl 2-bromoalkanoates [20], 2-chloroacetic acid [21,22], or diethyl 2-bromo-2-methylmalonate [23] gave 2H-benzo[b][1,4]thiazin-3(4H)-one derivatives 3 (Figure 1), which also have interesting biological properties. 2H-Benzo-1,4-thiazin-3(4H)-ones were also prepared by cyclization of 1,2-diaryldisulfanes with dialkyl but-2-ynedioates [24,25]. N-Substituted benzo-1,4-thiazine-2-carboxylates 4 (Figure 1) were prepared by m-CPBA-mediated oxidative ring expansion of substituted benzothiazoles [26], or via copper-catalyzed intramolecular amination of aryl bromides [27].
Recently, Nguyen and Retailleau introduced a TFA-catalyzed umpolung strategy with 2-aminothiophenols, preparing several 2H-benzo-1,4-thiazine derivatives 5 in high yield [28]. 3-Phenyl-2H-benzo-1,4-thiazine, an earlier representative of this family, was found to transform into a green-blue chromophore in the presence of peroxides or redox-active metal ions under acidic conditions, creating a potential detection method for such entities [29]. Additionally, the same structure was used for the preparation of a benzo-1,4-thiazine-based cyanine chromophore, which showed a reversible acidochromic behavior [30]. Zhao et al. demonstrated a three-component transition-metal-free aerobic method using a KI/DMSO/O2 system for the facile generation of iminobenzo-1,4-thiazines in moderate to good yield [31]. 3,4-Dihydro-2H-benzo-1,4-thiazine derivatives 6 were also successfully prepared [32]. The protocol, using NaI as a catalyst and K2S2O8 as an oxidant, tolerated a broad range of substrates with good stereoselectivity.
Interestingly, structurally related benzothiazine derivatives with a carboxylic function in the C-3 position are only seldomly described in the literature. Syntheses and utilization of the corresponding 4H-benzo[b][1,4]thiazine-3-carboxylic acids 7 are very rare.
Part of our research program is the construction of chiral heterocyclic compounds of medicinal interest [33,34]. Recently, we have been involved in the synthesis of potential SARS-CoV-2 protease inhibitors. Given the potential usefulness of the benzothiazine scaffold as a biologically active unit and the peptidomimetic nature of many SARS-CoV-2 protease inhibitors [35], we decided to investigate the viability of attaching a 4H-benzo[b][1,4]thiazine-3-carboxylic core to amino acids.
Results and Discussion
We began our work with the condensation reactions of 2-aminothiophenols 8 and bromopyruvic acid and esters 9 to form 4H-benzo-1,4-thiazines 10, having a carboxylic acid or an ester function at the C-3 position (Scheme 1).
![[1860-5397-18-124-i1]](/bjoc/content/inline/1860-5397-18-124-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Condensation reactions of 2-aminothiophenols 8 and bromopyruvic acid and esters 9.
Scheme 1: Condensation reactions of 2-aminothiophenols 8 and bromopyruvic acid and esters 9.
The reaction of thiol 8a with bromo-substituted acid 9a in diethyl ether at 0 °C for 1 h gave acid 10aa in 75% yield, while complete decomposition was observed at reflux temperature after only 10 min (Table 1, entries 1 and 2). Acid 10aa did not form when the reaction was performed in ethanol, CH2Cl2, THF, DMF, or ethyl acetate at different temperatures under classical conditions as well as under MWI. The acid 10aa could not be formed even when 2,2'-disulfanediyldianiline was used as the starting material in DMF or ethanol at room temperature or under reflux. Thiol 8a reacted with the keto ester 9c in ethanol to form the ester 10ac with a yield of 51% and 29%, respectively, under different conditions (Table 1, entries 3 and 4). Reactions in ethanol under MWI and in CH2Cl2 with classical stirring at room temperature only resulted in oxidative dimerization, forming derivative 11b in 15–28% yield (Table 1, entries 5–7). Neither did the reaction proceed in ethyl acetate nor in CH2Cl2. Like acid 9a, 2,2'-disulfanediyldianiline did also not react with the ester 9c. The reaction of 2-amino-4-chlorobenzenethiol (8b) with keto acid 9a in ethanol provided acid 10ba in 43% and 66% yield, depending on the reaction time (Table 1, entries 8 and 9). Similarly, ester 10bb was formed in 50% yield in ethanol and 30% in diethyl ether (Table 1, entries 10 and 11). Reactions with thiol 8c, having an electron-donating methoxy group, and acid 9a or esters 9b,c only gave unidentifiable decomposition products in various solvents (ethanol, diethyl ether, methanol, and CH2Cl2) at different temperatures under classical conditions and even under MWI (Table 1, entries 12 and 13). Also, reactions with 6,6'-disulfanediylbis(3-methoxyaniline) were unsuccessful.
Table 1: Conditions applied for the condensation reactions.a
| entry | X | R | conditions | yield of 10 (%) | yield of 11b (%) |
| 1 | H | H | Et2O, 0 °C, 1 h | 75 (10aa) | — |
| 2 | H | H | Et2O, reflux, 10 min | decomposition | |
| 3 | H | Et | EtOH, rt, 40 min | 51 (10ac) | — |
| 4 | H | Et | EtOH, 0 °C → rt, 2 h | 29 (10ac) | — |
| 5 | H | Et | EtOH, MWI, 100 °C, 20 min | — | 28 |
| 6 | H | Et | EtOH, MWI, 60 °C, 1 h | — | 20 |
| 7 | H | Et | CH2Cl2, rt, 2 h | — | 15 |
| 8 | Cl | H | EtOH, rt, 0.5 h | 43 (10ba) | — |
| 9 | Cl | H | EtOH, rt, 1 h | 66 (10ba) | — |
| 10 | Cl | Me | EtOH, rt, 0.5 h | 50 (10bb) | — |
| 11 | Cl | Me | Et2O, rt, 1 h | 30 (10bb) | — |
| 12 | MeO | H | Et2O, 0 °C, 1 h | decomposition | |
| 13 | MeO | Et | EtOH, 0 °C → rt, 1 h | decomposition | |
aRefer to Supporting Information File 1 for all explored reaction conditions.
Having observed dimer formation during the syntheses of benzothiazines, we have attempted to synthesize the corresponding dimer directly. To that end, we attempted reactions from aminothiol 8a as well as from the corresponding disulfide, both at room temperature and under MWI. In these experiments, the yield of dimer 11a was in the range of 10–34%. We also tried to enhance the oxidative dimerization using a mild oxidizing agent. The use of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in 1,4-dioxane afforded the dimer 11a in a slightly better yield of 46% (Scheme 2).
![[1860-5397-18-124-i2]](/bjoc/content/inline/1860-5397-18-124-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Direct synthesis of dimer 11a under oxidative reaction conditions.
Scheme 2: Direct synthesis of dimer 11a under oxidative reaction conditions.
For all the prepared benzothiazine derivatives 10 we observed some degree of instability. The derivatives were reasonably stable in the solid state but usually decomposed in solution. During the preparation of 10ac, dimers 11a and 11b were also detected in most experiments (TLC analysis), and they could be isolated by crystallization from MeOH or hexane. Performing the reaction in the dark with freshly degassed ethanol at lower temperature did bring about somewhat better results, but the oxidative dimerization was still a competitive reaction. NMR analyses of the isolated product 10ac also revealed the presence of the corresponding 2H-isomer in a low amount, but it was not isolated separately.
Quantum chemical calculations (ωB97xD/6-31G(d)//MN15/6-311+G(2d,p)) revealed that the stability of the two isomers was very similar, with a difference in ΔG of only 2.6 kJ⋅mol−1 and 2H-benzo-1,4-thiazine 11b being more stable (Figure 2). It seems likely that initially enamine dimer 11a formed, which then tautomerized to the more stable imine form 11b.
![[1860-5397-18-124-2]](/bjoc/content/figures/1860-5397-18-124-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: DFT (ωB97xD/6-31G*)-calculated structures of enamine and imine tautomers 11a and 11b.
Figure 2: DFT (ωB97xD/6-31G*)-calculated structures of enamine and imine tautomers 11a and 11b.
As a continuation of our work, we aimed to utilize the carboxylic acid function of the prepared 4H-benzo-1,4-thiazines 10 by attaching them to nonproteinogenic amino acid 16a and ʟ-phenylalanine. Preparation of 3-propylnorleucin methyl ester (16a) started with the condensation reaction of heptan-4-one (12) and methyl isocyanoacetate (13). The palladium-catalyzed hydrogenation of intermediate 14 gave the racemic N-formyl-protected amino acid methyl ester 15 in good yield. Using either concentrated HCl (aq) or in situ-formed HCl from the reaction of MeOH and acetyl chloride, compound 15 could easily be deprotected to gain either the salt 16a·HCl or the free amine 16a in good to excellent yield (Scheme 3).
![[1860-5397-18-124-i3]](/bjoc/content/inline/1860-5397-18-124-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Preparation of racemic 3-propylnorleucin 16a.
Scheme 3: Preparation of racemic 3-propylnorleucin 16a.
We also explored the asymmetric catalytic hydrogenation of adduct 14. Our first attempt at the reduction using organocatalyzed transfer hydrogenation was unsuccessful (see Supporting Information File 1). The (R)-Ru(OAc)2(BINAP)-assisted hydrogenation with H2 pressure up to 50 bar was also found to be ineffective. By changing the metal complex to Rh(COD)2BF4, we successfully realized the saturation of the double bond. Chiral ligands (R)-BINAP (L1) and (R,R)-phenyl-BPE (L4) gave unsatisfactory selectivity (Table 2, entries 1 and 4). Application of ligand (S,S)-methyl-DUPHOS (L3) gave increased ee in the hydrogenation reaction, but the best result (90% ee) was achieved using 6 mol % Josiphos ligand L2 at 35 °C.
Table 2: Stereoselective catalytic hydrogenation reactions of dehydroamino acid ester 14.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-124-i6.svg?max-width=637&scale=1.0)
|
||||
| entry | ligand (mol %) | T (°C) | yielda | ee (%) |
| 1 | L1 (5)b | 40 | not determined | 21 |
| 2 | L2 (5) | 40 | 90 | 83 |
| 3 | L3 (5) | 40 | 85 | 73 |
| 4 | L4 (5) | 40 | 87 | 19 |
| 5 | L2 (6) | 35 | 90 | 90 |
| 6 | L3 (6) | 35 | 83 | 69 |
aIsolated yield after purification by flash chromatography. bNo complete conversion.
Following the synthesis of 3-propylnorleucin methyl ester (16a), we carried on with the amine couplings. Several attempts were made to combine the benzothiazine motif with amino acid methyl esters (Scheme 4) using coupling agents (EDC, COMU, T3P®, etc.) or through acid chloride (SOCl2 and (COCl)2) in a flask with stirring or under ball-milling conditions. However, we were unable to isolate the desired amides (see Supporting Information File 1 for detailed reaction conditions) since decomposition of the benzothiazine core was observed in all coupling reactions.
![[1860-5397-18-124-i4]](/bjoc/content/inline/1860-5397-18-124-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Unsuccessful direct coupling of amino acid methyl esters with the benzothiazine motif and retrosynthetic analysis of alternative linear reaction routes.
Scheme 4: Unsuccessful direct coupling of amino acid methyl esters with the benzothiazine motif and retrosynt...
To avoid the issues related to the instable benzothiazine ring, we explored other possible linear reaction routes (Scheme 4) as an alternative to the original convergent synthesis plan. Instead of the classical amide coupling, bromo derivative Int1 should allow the formation of the benzothiazine ring as the final step. As an α-halocarbonyl compound, we tried to prepare Int1 through the coupling of bromopyruvic acid and ester, respectively, with the amino acids. However, these reactions proved to be unsuccessful due to the high reactivity of the bromo derivative. On the contrary, coupling of the less reactive amides formed from pyruvic acid and the amino acids 16a and 16b was accomplished (Scheme 5a), and several different conditions were tested (see Supporting Information File 1). Next, compounds 17a and 17b were brominated in the α-position using Br2 under acidic conditions. Finally, cyclization reaction with 2-aminothiophenol (8a) in dry diethyl ether at 0 °C gave benzo-1,4-thiazines 19a and 19b in good yield. Interestingly, the isolated products were not the expected 4H-, rather the 2H-benzo-1,4-thiazines. 3D renderings of derivatives 19a and 19b obtained at the ωB97xD/6-31G* level are depicted in Scheme 5b.
![[1860-5397-18-124-i5]](/bjoc/content/inline/1860-5397-18-124-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: a) Synthesis of 2H-benzo-1,4-thiazine amino acid conjugates 19a and 19b and b) 3D renderings of 19a and 19b obtained by DFT calculations (ωB97xD/6-31G*).
Scheme 5: a) Synthesis of 2H-benzo-1,4-thiazine amino acid conjugates 19a and 19b and b) 3D renderings of 19a...
Conclusion
In conclusion, we have described the synthesis of rarely explored 4H-benzo[b][1,4]thiazine-3-carboxylic esters and amides with amino acids. Benzothiazine derivatives with a carboxylic function in the C-3 position exhibit low stability under acidic as well as basic conditions, which complicates the synthetic utilization. As such, direct coupling of 4H-benzo[b][1,4]thiazine-3-carboxylic acid with amino acids failed. However, we have managed the synthesis of benzothiazine–amino acid conjugates via a linear synthesis in which the benzothiazine moiety was assembled from pyruvic acid attached to an amino acid. Oxidative dimerization of benzothiazine derivatives was also observed, and potentially useful benzothiazine dimers were isolated.
Supporting Information
| Supporting Information File 1: Experimental procedures and characterization data, additional experimental results, pictures of NMR and HRMS spectra. | ||
| Format: PDF | Size: 4.4 MB | Download |
Funding
This work was supported by the Slovak Research and Development Agency under the Contract no. PP-COVID-20-0010. This study was supported by the Operation Program of Integrated Infrastructure for the project Advancing University Capacity and Competence in Research, Development and Innovation, ITMS2014+: 313021X329, co-financed by the European Regional Development Fund.
References
-
Fringuelli, R.; Milanese, L.; Schiaffella, F. Mini-Rev. Med. Chem. 2005, 5, 1061–1073. doi:10.2174/138955705774933365
Return to citation in text: [1] -
Ajani, O. O. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 841–851. doi:10.1002/ardp.201200140
Return to citation in text: [1] -
Badshah, S. L.; Naeem, A. Molecules 2016, 21, 1054. doi:10.3390/molecules21081054
Return to citation in text: [1] -
Rai, A.; Singh, A. K.; Raj, V.; Saha, S. Mini-Rev. Med. Chem. 2018, 18, 42–57. doi:10.2174/1389557517666170529075556
Return to citation in text: [1] -
Sharma, P. K.; Amin, A.; Kumar, M. Open Med. Chem. J. 2020, 14, 71–82. doi:10.2174/1874104502014010071
Return to citation in text: [1] -
Rai, A.; Raj, V.; Singh, A. K.; Keshari, A. K.; Kumar, U.; Kumar, D.; Saha, S. Cogent Chem. 2017, 3, 1303909. doi:10.1080/23312009.2017.1303909
Return to citation in text: [1] -
Ingle, R. D.; Bhingolikar, V. E.; Bondge, S. P.; Mane, R. A. Indian J. Chem. 2003, 42B, 695–698.
Return to citation in text: [1] -
Sheibani, H.; Islami, M. R.; Hassanpour, A.; Hosseininasab, F. A. ARKIVOC 2006, No. xv, 68–75. doi:10.3998/ark.5550190.0007.f09
Return to citation in text: [1] -
Rathore, B. S.; Kumar, M. Bioorg. Med. Chem. 2006, 14, 5678–5682. doi:10.1016/j.bmc.2006.04.009
Return to citation in text: [1] -
Kumar, G.; Gupta, V.; Gautam, D. C.; Gupta, R. R. Heterocycl. Commun. 2002, 8, 381–384. doi:10.1515/hc.2002.8.4.381
Return to citation in text: [1] -
Kachhee, T. L.; Gupta, V.; Gupta, R.; Gautam, D. C. Heterocycl. Commun. 2002, 8, 579–582. doi:10.1515/hc.2002.8.6.579
Return to citation in text: [1] -
Thomas, L.; Gupta, A.; Gupta, V. J. Fluorine Chem. 2003, 122, 207–213. doi:10.1016/s0022-1139(03)00092-7
Return to citation in text: [1] -
Khandelwal, N.; Abhilasha, G.; Gautam, N.; Gautam, D. C. J. Chem. Sci. 2013, 125, 85–93. doi:10.1007/s12039-013-0363-4
Return to citation in text: [1] -
Munde, S. B.; Bondge, S. P.; Bhingolikar, V. E.; Mane, R. A. Green Chem. 2003, 5, 278–279. doi:10.1039/b212042a
Return to citation in text: [1] -
Paul, S.; Gupta, R.; Loupy, A.; Rani, B.; Dandia, A. Synth. Commun. 2001, 31, 711–717. doi:10.1081/scc-100103260
Return to citation in text: [1] -
Pratap, U. R.; Jawale, D. V.; Londhe, B. S.; Mane, R. A. J. Mol. Catal. B: Enzym. 2011, 68, 94–97. doi:10.1016/j.molcatb.2010.09.018
Return to citation in text: [1] -
Londhe, B. S.; Padwal, S. L.; Bhosale, M. R.; Mane, R. A. J. Iran. Chem. Soc. 2016, 13, 443–447. doi:10.1007/s13738-015-0752-3
Return to citation in text: [1] -
Yadav, N.; Sagir, H.; Ansari, M. D.; Siddiqui, I. R. Catal. Lett. 2018, 148, 1676–1685. doi:10.1007/s10562-018-2388-2
Return to citation in text: [1] -
Dang, H. V.; Le, Y. T. N.; Tran, D. T. M.; Phan, A. N. Q.; Phan, N. T. S. Catal. Lett. 2018, 148, 1383–1395. doi:10.1007/s10562-018-2358-8
Return to citation in text: [1] -
Koóš, M. Monatsh. Chem. 1994, 125, 1011–1016. doi:10.1007/bf00812716
Return to citation in text: [1] -
Gowda, J.; Khader, A. M. A.; Kalluraya, B.; Shree, P.; Shabaraya, A. R. Eur. J. Med. Chem. 2011, 46, 4100–4106. doi:10.1016/j.ejmech.2011.06.010
Return to citation in text: [1] -
Guarda, V. L. d. M.; Perrissin, M.; Thomasson, F.; Ximenes, E. A.; Galdino, S. L.; Pitta, I. R.; Luu-Duc, C.; Barbe, J. Eur. J. Med. Chem. 2003, 38, 769–773. doi:10.1016/s0223-5234(03)00127-2
Return to citation in text: [1] -
Leskovšek Cizej, V.; Urleb, U. J. Heterocycl. Chem. 1996, 33, 97–101. doi:10.1002/jhet.5570330117
Return to citation in text: [1] -
Islami, M. R.; Mollazehi, F.; Badiei, A.; Sheibani, H. ARKIVOC 2005, No. xv, 25–29. doi:10.3998/ark.5550190.0006.f04
Return to citation in text: [1] -
Heravi, M. M.; Nami, N.; Oskooie, H. A.; Hekmatshoar, R. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 1873–1878. doi:10.1080/104265090889585
Return to citation in text: [1] -
Pi, H.-J.; Liu, H.; Du, W.; Deng, W.-P. Tetrahedron Lett. 2009, 50, 4529–4531. doi:10.1016/j.tetlet.2009.05.083
Return to citation in text: [1] -
Melkonyan, F.; Topolyan, A.; Karchava, A.; Yurovskaya, M. Tetrahedron 2011, 67, 6826–6832. doi:10.1016/j.tet.2011.06.081
Return to citation in text: [1] -
Nguyen, T. B.; Retailleau, P. Adv. Synth. Catal. 2020, 362, 5380–5384. doi:10.1002/adsc.202000964
Return to citation in text: [1] -
Leone, L.; Crescenzi, O.; Amorati, R.; Valgimigli, L.; Napolitano, A.; Barone, V.; d’Ischia, M. Org. Lett. 2013, 15, 4944–4947. doi:10.1021/ol402161j
Return to citation in text: [1] -
Alfieri, M. L.; Panzella, L.; d’Ischia, M.; Napolitano, A. Molecules 2020, 25, 3817. doi:10.3390/molecules25173817
Return to citation in text: [1] -
Zhao, J.; Luo, Z.; Xu, J. Adv. Synth. Catal. 2020, 362, 1988–1992. doi:10.1002/adsc.202000201
Return to citation in text: [1] -
Alom, N.-E.; Kaur, N.; Wu, F.; Saluga, S. J.; Li, W. Chem. – Eur. J. 2019, 25, 6902–6906. doi:10.1002/chem.201900549
Return to citation in text: [1] -
Rodriguez, L.; Fišera, R.; Gaálová, B.; Koči, K.; Bujdáková, H.; Mečiarová, M.; Górová, R.; Jurdáková, H.; Šebesta, R. Eur. J. Org. Chem. 2020, 2565–2575. doi:10.1002/ejoc.202000235
Return to citation in text: [1] -
Modrocká, V.; Veverková, E.; Mečiarová, M.; Šebesta, R. J. Org. Chem. 2018, 83, 13111–13120. doi:10.1021/acs.joc.8b01847
Return to citation in text: [1] -
de Almeida, S. M. V.; Santos Soares, J. C.; dos Santos, K. L.; Alves, J. E. F.; Ribeiro, A. G.; Jacob, Í. T. T.; da Silva Ferreira, C. J.; dos Santos, J. C.; de Oliveira, J. F.; de Carvalho Junior, L. B.; de Lima, M. d. C. A. Bioorg. Med. Chem. 2020, 28, 115757. doi:10.1016/j.bmc.2020.115757
Return to citation in text: [1]
| 30. | Alfieri, M. L.; Panzella, L.; d’Ischia, M.; Napolitano, A. Molecules 2020, 25, 3817. doi:10.3390/molecules25173817 |
| 28. | Nguyen, T. B.; Retailleau, P. Adv. Synth. Catal. 2020, 362, 5380–5384. doi:10.1002/adsc.202000964 |
| 29. | Leone, L.; Crescenzi, O.; Amorati, R.; Valgimigli, L.; Napolitano, A.; Barone, V.; d’Ischia, M. Org. Lett. 2013, 15, 4944–4947. doi:10.1021/ol402161j |
| 1. | Fringuelli, R.; Milanese, L.; Schiaffella, F. Mini-Rev. Med. Chem. 2005, 5, 1061–1073. doi:10.2174/138955705774933365 |
| 2. | Ajani, O. O. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 841–851. doi:10.1002/ardp.201200140 |
| 3. | Badshah, S. L.; Naeem, A. Molecules 2016, 21, 1054. doi:10.3390/molecules21081054 |
| 4. | Rai, A.; Singh, A. K.; Raj, V.; Saha, S. Mini-Rev. Med. Chem. 2018, 18, 42–57. doi:10.2174/1389557517666170529075556 |
| 5. | Sharma, P. K.; Amin, A.; Kumar, M. Open Med. Chem. J. 2020, 14, 71–82. doi:10.2174/1874104502014010071 |
| 14. | Munde, S. B.; Bondge, S. P.; Bhingolikar, V. E.; Mane, R. A. Green Chem. 2003, 5, 278–279. doi:10.1039/b212042a |
| 26. | Pi, H.-J.; Liu, H.; Du, W.; Deng, W.-P. Tetrahedron Lett. 2009, 50, 4529–4531. doi:10.1016/j.tetlet.2009.05.083 |
| 9. | Rathore, B. S.; Kumar, M. Bioorg. Med. Chem. 2006, 14, 5678–5682. doi:10.1016/j.bmc.2006.04.009 |
| 10. | Kumar, G.; Gupta, V.; Gautam, D. C.; Gupta, R. R. Heterocycl. Commun. 2002, 8, 381–384. doi:10.1515/hc.2002.8.4.381 |
| 11. | Kachhee, T. L.; Gupta, V.; Gupta, R.; Gautam, D. C. Heterocycl. Commun. 2002, 8, 579–582. doi:10.1515/hc.2002.8.6.579 |
| 12. | Thomas, L.; Gupta, A.; Gupta, V. J. Fluorine Chem. 2003, 122, 207–213. doi:10.1016/s0022-1139(03)00092-7 |
| 13. | Khandelwal, N.; Abhilasha, G.; Gautam, N.; Gautam, D. C. J. Chem. Sci. 2013, 125, 85–93. doi:10.1007/s12039-013-0363-4 |
| 27. | Melkonyan, F.; Topolyan, A.; Karchava, A.; Yurovskaya, M. Tetrahedron 2011, 67, 6826–6832. doi:10.1016/j.tet.2011.06.081 |
| 8. | Sheibani, H.; Islami, M. R.; Hassanpour, A.; Hosseininasab, F. A. ARKIVOC 2006, No. xv, 68–75. doi:10.3998/ark.5550190.0007.f09 |
| 23. | Leskovšek Cizej, V.; Urleb, U. J. Heterocycl. Chem. 1996, 33, 97–101. doi:10.1002/jhet.5570330117 |
| 6. | Rai, A.; Raj, V.; Singh, A. K.; Keshari, A. K.; Kumar, U.; Kumar, D.; Saha, S. Cogent Chem. 2017, 3, 1303909. doi:10.1080/23312009.2017.1303909 |
| 7. | Ingle, R. D.; Bhingolikar, V. E.; Bondge, S. P.; Mane, R. A. Indian J. Chem. 2003, 42B, 695–698. |
| 24. | Islami, M. R.; Mollazehi, F.; Badiei, A.; Sheibani, H. ARKIVOC 2005, No. xv, 25–29. doi:10.3998/ark.5550190.0006.f04 |
| 25. | Heravi, M. M.; Nami, N.; Oskooie, H. A.; Hekmatshoar, R. Phosphorus, Sulfur Silicon Relat. Elem. 2005, 180, 1873–1878. doi:10.1080/104265090889585 |
| 18. | Yadav, N.; Sagir, H.; Ansari, M. D.; Siddiqui, I. R. Catal. Lett. 2018, 148, 1676–1685. doi:10.1007/s10562-018-2388-2 |
| 33. | Rodriguez, L.; Fišera, R.; Gaálová, B.; Koči, K.; Bujdáková, H.; Mečiarová, M.; Górová, R.; Jurdáková, H.; Šebesta, R. Eur. J. Org. Chem. 2020, 2565–2575. doi:10.1002/ejoc.202000235 |
| 34. | Modrocká, V.; Veverková, E.; Mečiarová, M.; Šebesta, R. J. Org. Chem. 2018, 83, 13111–13120. doi:10.1021/acs.joc.8b01847 |
| 17. | Londhe, B. S.; Padwal, S. L.; Bhosale, M. R.; Mane, R. A. J. Iran. Chem. Soc. 2016, 13, 443–447. doi:10.1007/s13738-015-0752-3 |
| 21. | Gowda, J.; Khader, A. M. A.; Kalluraya, B.; Shree, P.; Shabaraya, A. R. Eur. J. Med. Chem. 2011, 46, 4100–4106. doi:10.1016/j.ejmech.2011.06.010 |
| 22. | Guarda, V. L. d. M.; Perrissin, M.; Thomasson, F.; Ximenes, E. A.; Galdino, S. L.; Pitta, I. R.; Luu-Duc, C.; Barbe, J. Eur. J. Med. Chem. 2003, 38, 769–773. doi:10.1016/s0223-5234(03)00127-2 |
| 35. | de Almeida, S. M. V.; Santos Soares, J. C.; dos Santos, K. L.; Alves, J. E. F.; Ribeiro, A. G.; Jacob, Í. T. T.; da Silva Ferreira, C. J.; dos Santos, J. C.; de Oliveira, J. F.; de Carvalho Junior, L. B.; de Lima, M. d. C. A. Bioorg. Med. Chem. 2020, 28, 115757. doi:10.1016/j.bmc.2020.115757 |
| 16. | Pratap, U. R.; Jawale, D. V.; Londhe, B. S.; Mane, R. A. J. Mol. Catal. B: Enzym. 2011, 68, 94–97. doi:10.1016/j.molcatb.2010.09.018 |
| 31. | Zhao, J.; Luo, Z.; Xu, J. Adv. Synth. Catal. 2020, 362, 1988–1992. doi:10.1002/adsc.202000201 |
| 15. | Paul, S.; Gupta, R.; Loupy, A.; Rani, B.; Dandia, A. Synth. Commun. 2001, 31, 711–717. doi:10.1081/scc-100103260 |
| 19. | Dang, H. V.; Le, Y. T. N.; Tran, D. T. M.; Phan, A. N. Q.; Phan, N. T. S. Catal. Lett. 2018, 148, 1383–1395. doi:10.1007/s10562-018-2358-8 |
| 32. | Alom, N.-E.; Kaur, N.; Wu, F.; Saluga, S. J.; Li, W. Chem. – Eur. J. 2019, 25, 6902–6906. doi:10.1002/chem.201900549 |
© 2022 Kisszékelyi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.