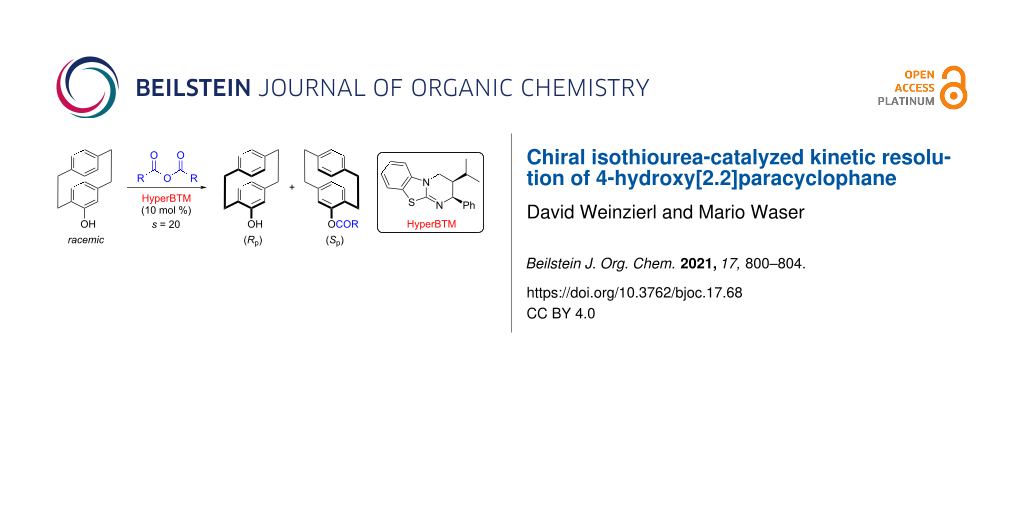
Abstract
We herein report a method for the kinetic resolution of racemic 4-hydroxy[2.2]paracyclophane by means of a chiral isothiourea-catalyzed acylation with isobutyric anhydride. This protocol allows for a reasonable synthetically useful s-factor of 20 and provides a novel entry to obtain this interesting planar chiral motive in an enantioenriched manner.

Graphical Abstract
Introduction
Substituted [2.2]paracyclophanes are fascinating planar chiral molecules [1-12] which have been systematically investigated since Brown and Farthing discovered the formation of the unsubstituted and achiral parent [2.2]paracyclophane (1) via gas phase pyrolysis of para-xylene in 1949 [5]. Over the years, these compounds established themselves as a unique class of “bent and battered” [6] strained molecules with remarkable chemical and physical properties [1-4,7-9]. Besides their potential applications in material and polymer chemistry [1,2,7-9], these planar chiral molecules have been very successfully used in asymmetric catalysis [3,4,10-12]. Accordingly, the development of methods for the asymmetric synthesis of enantiomerically pure, or at least enantiomerically enriched, derivatives that can be utilized as building blocks for more demanding ligands and catalysts became a task of high importance. Thus, several strategies to access enantioenriched [2.2]paracyclophanes have been reported, either relying on classical resolution approaches or, more recently, making use of asymmetric catalysis to carry out kinetic resolutions of easily accessed racemic precursors [3,4,13-15]. 4-Hydroxy[2.2]paracyclophane (2) is one of the commonly used building blocks, which is easily accessible in a racemic manner starting from 1 according to nowadays well-established procedures [16-18]. Over the last decades, it was shown that enantioenriched 2 may serve as a valuable building block to access more advanced chiral cyclophane ligands and catalysts [3,4,19-22] and therefore its asymmetric synthesis became an important task [3,4,18-27]. Several strategies to access 2 in an enantioenriched fashion have been developed. One commonly used method relies on the resolution of 4-formyl[2.2]paracyclophane via formation of a chiral Schiff base first, followed by a subsequent Dakin-type oxidation to alcohol 2 [18]. Alternatively, the direct resolution of rac-2 via transformation into diastereomers by esterification with chiral acid chlorides [19,20] as well as the kinetic resolution (KR) of racemic esters of 2 via an enzymatic hydrolysis [25-27] were very successfully used to access enantioenriched 2. Recently, Akiyama and co-workers reported the kinetic resolution of rac-PHANOL (4,12-dihydroxy[2.2]paracyclophane) by means of a chiral phosphoric acid-catalyzed esterification with achiral anhydrides [28]. This method allowed for high s-factors but was unfortunately not satisfyingly applicable to rac-4-hydroxy[2.2]paracyclophane (rac-2) [28].
Considering the interest in compound 2, we thus thought about developing an alternative organocatalytic kinetic resolution protocol to control the esterification of rac-2. Chiral isothioureas (ITUs) emerged as easily available and powerful catalysts for numerous applications [29-32] and have been very successfully used for the kinetic resolution of different racemic alcohols [33-37]. Inspired by this unique catalysis potential, we therefore became interested in testing those chiral catalysts for the, to the best of our knowledge, so far not investigated acylative kinetic resolution of 4-hydroxy[2.2]paracyclophane (2, Scheme 1).
![[1860-5397-17-68-i1]](/bjoc/content/inline/1860-5397-17-68-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Overview about established methods to access enantioenriched 2 and the herein investigated kinetic resolution (KR) with chiral isothiourea (ITU) catalysts.
Scheme 1: Overview about established methods to access enantioenriched 2 and the herein investigated kinetic ...
Results and Discussion
BTM (ITU 1 [33]) and HyperBTM (ITU 2 [38]) are amongst the most commonly used chiral ITUs and these nowadays commercially available catalysts were used to optimize the resolution of rac-2 with isobutyric anhydride (4a) (Table 1 gives an overview of the most significant results obtained in this screening). Anhydride 4a was chosen in a first instance as it proved successful in previous acylative resolutions reported by others [28,33,34,36,37] but we later on also tested other anhydrides and acid chlorides (vide infra, Scheme 2). First experiments with 10 mol % BTM (ITU 1) carried out in CHCl3 or toluene at room temperature (Table 1, entries 1 and 2) proved the general feasibility of this concept, resulting in s-factors around 6. When lowering the temperature, a slight improvement could be achieved at −15 °C (Table 1, entry 3) but unfortunately ITU 1 performed less selective at −78 °C (Table 1, entry 4). Instead, (2S,3R)-HyperBTM (ITU 2) resulted in an enhanced selectivity with s = 14.5 at −78 °C but conversion was relatively slow (Table 1, entry 5). Gratefully however, the obtained s-factor was almost the same at −40 °C and a reasonable conversion of around 30% could be observed after 4 h reaction time (Table 1, entry 6). Varying solvent and concentration at −40 °C next showed that toluene allows for higher selectivities than CHCl3 (compare Table 1, entries 6 and 7), while the use of other solvents like CH2Cl2 and THF resulted in almost no product formation and no reasonable selectivities (not mentioned in Table 1). In addition, higher concentrations lead to notably lower selectivities (Table 1, entry 9), while more diluted conditions did not allow for a significant improvement of the s-factor anymore (Table 1, entry 8). Lowering the catalyst loading from 10 to 5 mol % allowed for a similar conversion, but resulted in a slightly reduced selectivity (Table 1, entry 10).
Table 1: Identification of the optimum catalyst and best conditions for the resolution of rac-2 with anhydride 4aa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-68-i3.svg?max-width=637&scale=1.0)
|
||||||||
| Entry | ITU | Solvent | T [°C] | t [h] | Conv. (C) [%]b | ee (2) [%]c,d | ee (3a) [%]c | se |
| 1 | ITU 1 | CHCl3 | 25 | 1 | 41 | 42 | 60 | 6 |
| 2 | ITU 1 | toluene | 25 | 1 | 38 | 39 | 64 | 6.5 |
| 3 | ITU 1 | toluene | −15 | 1 | 34 | 38 | 74 | 10 |
| 4 | ITU 1 | toluene | −78 | 1 | 15 | 13 | 74 | 7.5 |
| 5 | ITU 2 | toluene | −78 | 1 | 16 | 16 | 85 | 14.5 |
| 6 | ITU 2 | toluene | −40 | 4 | 33 | 40 | 81 | 14 |
| 7 | ITU 2 | CHCl3 | −40 | 4 | 45 | 55 | 67 | 9 |
| 8 | ITU 2 | toluene (0.055 M) | −40 | 4 | 30 | 35 | 82 | 14.5 |
| 9 | ITU 2 | toluene (0.22 M) | −40 | 4 | 36 | 32 | 75 | 9.5 |
| 10f | ITU 2 | toluene | −40 | 4 | 30 | 34 | 79 | 12 |
| 11g | ITU 2 | toluene | −40 | 22 | 57 | 94 (39%)h | 71 (53%)h | 20 |
aAll reactions were carried out using 0.1 mmol rac-2 and 0.06 mmol 4a in the presence of 0.06 mmol Hünig’s base (diisopropylethylamine, DIPEA) and 10 mol % ITU in the indicated solvent (0.11 M with respect to 2) unless otherwise stated; bdetermined by 1H NMR of the crude product; isolated yields of 2 and 3 were almost quantitative in all cases; cdetermined by HPLC using a chiral stationary phase; dabsolute configuration of recovered 2 was assigned to be (Rp) by comparison of its (+)-optical rotation with previous reports [20,26,39]; ethe s-factor was calculated from the ee of recovered 2 and/or the ee of ester 3 [40-43]; fusing 5 mol % ITU 2; gusing 1.1 equiv of 4a; hisolated yield when carried out on 1 mmol rac-2 scale.
At this point, we decided to screen other anhydrides and acid chlorides 4, but, as outlined in Scheme 2, the initially used isobutyric anhydride 4a clearly outperformed its analogous acid chloride 4b, as well as the other derivatives 4c–f.
![[1860-5397-17-68-i2]](/bjoc/content/inline/1860-5397-17-68-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Use of alternative acylating agents 4 for the kinetic resolution of rac-2.
Scheme 2: Use of alternative acylating agents 4 for the kinetic resolution of rac-2.
Finally, the resolution of rac-2 was run for 22 h in the presence of 10 mol % HyperBTM (ITU 2) with 1.1 equivalents of anhydride 4a (instead of the previously used 0.6 equiv; Table 1, entry 11). Under these conditions it was possible to achieve a conversion of slightly above 50% combined with good enantioselectivities for both, the recovered alcohol 2 and the ester 3a (s = 20). With these optimum conditions the resolution was also successfully carried out on 1 mmol scale, resulting in an identical conversion and s-factor (s = 20; C = 57%) and allowing for the isolation of (Rp)-2 in 39% yield (94% ee) and (Sp)-3a in 53% yield (71% ee) (Table 1, entry 11). Mechanistically, this resolution process should proceed via the well-understood formation of a chiral acyl-transfer species between the isothiourea catalyst ITU 2 and the anhydride 4a [33-37], which then allows for the resolution of the enantiomers of alcohol 2. Unfortunately, however, the true nature of this enantiodiscriminating step has not yet been elucidated and will require detailed computational studies.
Conclusion
In conclusion, we identified conditions that allow for the kinetic resolution of racemic 4-hydroxy[2.2]paracyclophane (2) by means of an acylation with isobutyric anhydride (4a) in the presence of the chiral isothiourea catalyst HyperBTM (ITU 2). The reaction can be carried out with an s-factor around 20 and allows for the isolation of recovered (Rp)-2 and ester (Sp)-3a with reasonable enantiomeric excesses around 90%, depending on the conversion. These two compounds can easily be separated by silica gel column chromatography in almost quantitative yields, thus providing a novel entry to obtain these interesting planar chiral motives in an enantioenriched manner.
Experimental
General details
1H- and 13C NMR spectra were recorded on a Bruker Avance III 300 MHz spectrometer with a broad band observe probe and a sample changer for 16 samples. NMR spectra were referenced on the solvent peak and chemical shifts are given in ppm.
High-resolution mass spectra were obtained using a Thermo Fisher Scientific LTQ Orbitrap XL with an Ion Max API Source. Analyses were made in the positive ionization mode if not otherwise stated. HPLC was performed using a Thermo Scientific Dionex Ultimate 3000 system with diode array detector with a CHIRAL ART Cellulose-SB stationary phase. Optical rotations were recorded on a Schmidt + Haensch Polarimeter Model UniPol L1000 at 589 nm.
All chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. rac-2 was prepared from 1 according to a previously published procedure [16].
Optimized procedure for the KR of rac-2
Racemic 4-hydroxy[2.2]paracyclophane (rac-2; 250 mg; 1.115 mmol) and HyperBTM (ITU 2; 35 mg; 10 mol %) were dissolved in dry toluene (10 mL) in a Schlenk flask (Ar atmosphere), followed by the addition of Hünig’s base (DIPEA; 118 µL; 0.67 mmol; 0.6 equiv). The solution was then cooled to −40 °C and isobutyric anhydride (4a; 208 µL; 1.226 mmol; 1.1 equiv) was added and the mixture was stirred at −40 °C for 22 h. The reaction was quenched by addition of MeOH. The crude product was filtered over Na2SO4 and the solvent removed in vacuum. Recovered alcohol 2 and ester 3a were separated by silica gel column chromatography (heptanes/ethyl acetate 10:1), yielding (Sp)-3a in 53% (175 mg) and (Rp)-2 in 43% (98 mg) (39%).
(Rp)-2a: Analytical data match those reported in literature [18-20,26,28,39]. TLC (heptanes/ethyl acetate 10:1; Rf = 0.11). [α]D24 14.1 (c 1, CH2Cl2, 92% ee) and 12.1 (c 1, CHCl3, 92% ee); 1H NMR (300 MHz, CDCl3, 298.0 K) δ/ppm 7.00 (dd, J = 8, 1.8 Hz, 1H), 6.55 (dd, J = 8, 1.8 Hz, 1H), 6.45 (dd, J = 8, 1.8 Hz, 1H), 6.41–6.37 (m, 2H), 6.26 (dd, J = 8, 1.8 Hz, 1H), 5.54 (d, J = 1.6 Hz, 1H), 4.42 (s, 1H), 3.37–3.29 (m, 1H), 3.14–3.02 (m, 4H), 2.98–2.85 (m, 2H), 2.71–2.60 (m, 1H); 13C NMR (75 MHz, CDCl3, 298.0 K) δ/ppm 153.8 (1C, CAr), 142.1 (1C, CAr), 139.8 (1C, CAr), 139.0 (1C, CAr), 135.6 (1C, CAr), 133.8 (1C, CAr), 132.9 (1C, CAr), 132.0 (1C, CAr), 128.1 (1C, CAr), 125.6 (1C, CAr), 125.2 (1C, CAr), 122.7 (1C, CAr), 35.4 (1C, -CH2), 34.9 (1C, -CH2), 34.0 (1C, -CH2), 32.2 (1C, -CH2); HRMS (ESI) m/z: calcd for [C16H16O + H]+, 225.1274; found, 225.1280, HPLC: YMC Chiral ART Cellulose-SB, n-hexane/iPrOH 3:1, 1 mL/min, 10 °C; tR = 6.4 min [Sp; minor], 7.2 min [Rp; major].
(Sp)-3a: Analytical data match those reported in literature [28]. TLC (heptanes/ethyl acetate 10:1; Rf = 0.33). [α]D24 27.5 (c 1.0, CHCl3, 82% ee); mp 80–82 °C; 1H NMR (300 MHz, CDCl3, 298.0 K) δ/ppm 6.91 (dd, J = 7.8, 1.8 Hz, 1H), 6.56–6.43 (m, 5H), 6.00 (d, J = 1.7 Hz, 1H), 3.17–2.94 (m, 7H), 2.93–2.79 (m, 1H), 2.73–2.64 (m, 1H), 1.42 (d, J = 7 Hz, 3H), 1.38 (d, J = 7 Hz, 3H); 13C NMR (75 MHz, CDCl3, 298.0 K) δ/ppm 174.8 (1C, C=O), 149.1 (1C, CAr), 141.7 (1C, CAr), 139.6 (1C, CAr), 139.3 (1C, CAr), 135.4 (1C, CAr), 133.5 (1C, CAr), 133.1 (1C, CAr), 132.3 (1C, CAr), 131.1 (1C, CAr), 130.1 (1C, CAr), 129.6 (1C, CAr), 128.2 (1C, CAr), 35.4 (1C, -CH2), 35.0 (1C, -CH2), 34.4 (2C, -CH, -CH2), 31.8 (1C, -CH2), 19.4 (1C, -CH3), 19.1 (1C, -CH3); HRMS (ESI) m/z: calcd for [C20H22O2 + NH4]+, 312.1958; found, 312.1958, HPLC: YMC Chiral ART Cellulose-SB, n-hexane/iPrOH 3:1, 1 mL/min, 10 °C; tR = 7.3 min [Rp; minor], 8.4 min [Sp; major].
Supporting Information
| Supporting Information File 1: Copies of NMR spectra and HPLC chromatograms as well as analytical data of esters 3 obtained with the alternative acyl-transfer reagents 4. | ||
| Format: PDF | Size: 1.4 MB | Download |
Acknowledgements
We are grateful to Thomas Bögl (Institute of Analytical Chemistry, JKU Linz) for support with HRMS analysis.
Funding
This work was generously supported by the Austrian Science Funds (FWF): Project No. P31784. The used NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ program (project M00146, "RERI-uasb").
References
-
Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/3527603964
Return to citation in text: [1] [2] [3] -
Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969
Return to citation in text: [1] [2] [3] -
Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Brown, C. J.; Farthing, A. C. Nature 1949, 164, 915–916. doi:10.1038/164915b0
Return to citation in text: [1] [2] -
Cram, D. J.; Cram, J. M. Acc. Chem. Res. 1971, 4, 204–213. doi:10.1021/ar50042a003
Return to citation in text: [1] [2] -
Elacqua, E.; MacGillivray, L. R. Eur. J. Org. Chem. 2010, 6883–6894. doi:10.1002/ejoc.201000930
Return to citation in text: [1] [2] [3] -
Marrocchi, A.; Tomasi, I.; Vaccaro, L. Isr. J. Chem. 2012, 52, 41–52. doi:10.1002/ijch.201100091
Return to citation in text: [1] [2] [3] -
Mori, T.; Inoue, Y. Top. Curr. Chem. 2010, 298, 99–128.
Return to citation in text: [1] [2] [3] -
Rowlands, G. J. Isr. J. Chem. 2012, 52, 60–75. doi:10.1002/ijch.201100098
Return to citation in text: [1] [2] -
Paradies, J. Synthesis 2011, 3749–3766. doi:10.1055/s-0031-1289296
Return to citation in text: [1] [2] -
Gibson, S. E.; Knight, J. D. Org. Biomol. Chem. 2003, 1, 1256–1269. doi:10.1039/b300717k
Return to citation in text: [1] [2] -
Kotha, S.; Shirbhate, M. E.; Waghule, G. T. Beilstein J. Org. Chem. 2015, 11, 1274–1331. doi:10.3762/bjoc.11.142
Return to citation in text: [1] -
Rowlands, G. J. Org. Biomol. Chem. 2008, 6, 1527–1534. doi:10.1039/b800698a
Return to citation in text: [1] -
Tanaka, K. Bull. Chem. Soc. Jpn. 2018, 91, 187–194. doi:10.1246/bcsj.20170346
Return to citation in text: [1] -
Krohn, K.; Rieger, H.; Hopf, H.; Barrett, D. Chem. Ber. 1990, 123, 1729–1732. doi:10.1002/cber.19901230824
Return to citation in text: [1] [2] -
Kane, V. V.; Gerdes, A.; Grahn, W.; Ernst, L.; Dix, I.; Jones, P. G.; Hopf, H. Tetrahedron Lett. 2001, 42, 373–376. doi:10.1016/s0040-4039(00)01992-4
Return to citation in text: [1] -
Friedmann, C. J.; Ay, S.; Bräse, S. J. Org. Chem. 2010, 75, 4612–4614. doi:10.1021/jo100468s
Return to citation in text: [1] [2] [3] [4] -
Rozenberg, V.; Danilova, T.; Sergeeva, E.; Vorontsov, E.; Starikova, Z.; Korlyukov, A.; Hopf, H. Eur. J. Org. Chem. 2002, 468–477. doi:10.1002/1099-0690(20022)2002:3<468::aid-ejoc468>3.0.co;2-3
Return to citation in text: [1] [2] [3] [4] -
Zhang, T.-Z.; Dai, L.-X.; Hou, X.-L. Tetrahedron: Asymmetry 2007, 18, 251–259. doi:10.1016/j.tetasy.2007.01.020
Return to citation in text: [1] [2] [3] [4] [5] -
Vorontsova, N. V.; Zhuravsky, R. P.; Sergeeva, E. V.; Vorontsov, E. V.; Starikova, Z. A.; Rozenberg, V. I. Russ. Chem. Bull. 2007, 56, 2225–2231. doi:10.1007/s11172-007-0348-x
Return to citation in text: [1] [2] -
Wang, Y.; Yuan, H.; Lu, H.; Zheng, W.-H. Org. Lett. 2018, 20, 2555–2558. doi:10.1021/acs.orglett.8b00711
Return to citation in text: [1] [2] -
Hitchcock, P. B.; Rowlands, G. J.; Parmar, R. Chem. Commun. 2005, 4219–4221. doi:10.1039/b507394d
Return to citation in text: [1] -
Parmar, R.; Coles, M. P.; Hitchcock, P. B.; Rowlands, G. J. Synthesis 2010, 4177–4187. doi:10.1055/s-0030-1258286
Return to citation in text: [1] -
Cipiciani, A.; Fringuelli, F.; Mancini, V.; Piermatti, O.; Scappini, A. M.; Ruzziconi, R. Tetrahedron 1997, 53, 11853–11858. doi:10.1016/s0040-4020(97)00758-8
Return to citation in text: [1] [2] -
Pamperin, D.; Schulz, C.; Hopf, H.; Syldatk, C.; Pietzsch, M. Eur. J. Org. Chem. 1998, 1441–1445. doi:10.1002/(sici)1099-0690(199807)1998:7<1441::aid-ejoc1441>3.0.co;2-k
Return to citation in text: [1] [2] [3] [4] -
Pamperin, D.; Ohse, B.; Hopf, H.; Pietzsch, M. J. Mol. Catal. B: Enzym. 1998, 5, 317–319. doi:10.1016/s1381-1177(98)00063-0
Return to citation in text: [1] [2] -
Mori, K.; Kishi, H.; Akiyama, T. Synthesis 2017, 49, 365–370. doi:10.1055/s-0036-1588898
Return to citation in text: [1] [2] [3] [4] [5] -
Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/c2cs15288f
Return to citation in text: [1] -
Merad, J.; Pons, J.-M.; Chuzel, O.; Bressy, C. Eur. J. Org. Chem. 2016, 5589–5610. doi:10.1002/ejoc.201600399
Return to citation in text: [1] -
Birman, V. Aldrichimica Acta 2016, 49, 23–33.
Return to citation in text: [1] -
McLaughlin, C.; Smith, A. D. Chem. – Eur. J. 2021, 27, 1533–1555. doi:10.1002/chem.202002059
Return to citation in text: [1] -
Birman, V. B.; Li, X. Org. Lett. 2006, 8, 1351–1354. doi:10.1021/ol060065s
Return to citation in text: [1] [2] [3] [4] -
Birman, V. B.; Li, X. Org. Lett. 2008, 10, 1115–1118. doi:10.1021/ol703119n
Return to citation in text: [1] [2] [3] -
Li, X.; Jiang, H.; Uffman, E. W.; Guo, L.; Zhang, Y.; Yang, X.; Birman, V. B. J. Org. Chem. 2012, 77, 1722–1737. doi:10.1021/jo202220x
Return to citation in text: [1] [2] -
Qu, S.; Greenhalgh, M. D.; Smith, A. D. Chem. – Eur. J. 2019, 25, 2816–2823. doi:10.1002/chem.201805631
Return to citation in text: [1] [2] [3] -
Qu, S.; Smith, S. M.; Laina‐Martín, V.; Neyyappadath, R. M.; Greenhalgh, M. D.; Smith, A. D. Angew. Chem., Int. Ed. 2020, 59, 16572–16578. doi:10.1002/anie.202004354
Return to citation in text: [1] [2] [3] -
Joannesse, C.; Johnston, C. P.; Concellón, C.; Simal, C.; Philp, D.; Smith, A. D. Angew. Chem., Int. Ed. 2009, 48, 8914–8918. doi:10.1002/anie.200904333
Return to citation in text: [1] -
Cipiciani, A.; Fringuelli, F.; Mancini, V.; Piermatti, O.; Pizzo, F.; Ruzziconi, R. J. Org. Chem. 1997, 62, 3744–3747. doi:10.1021/jo962142a
Return to citation in text: [1] [2] -
Kagan, H. B.; Fiaud, J. C. Top. Stereochem. 1988, 18, 249–330. doi:10.1002/9780470147276.ch4
Return to citation in text: [1] -
Greenhalgh, M. D.; Taylor, J. E.; Smith, A. D. Tetrahedron 2018, 74, 5554–5560. doi:10.1016/j.tet.2018.05.069
Return to citation in text: [1] -
Calculation from recovered 2: s = ln[(1 − C)(1 − ee(2))] / ln[(1 − C)(1 + ee(2))].
Return to citation in text: [1] -
Calculation from isolated 3: s = ln[1 − C(1 + ee(3))] / ln[1 − C(1 − ee(3))].
Return to citation in text: [1]
| 1. | Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/3527603964 |
| 2. | Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969 |
| 3. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a |
| 4. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863 |
| 5. | Brown, C. J.; Farthing, A. C. Nature 1949, 164, 915–916. doi:10.1038/164915b0 |
| 6. | Cram, D. J.; Cram, J. M. Acc. Chem. Res. 1971, 4, 204–213. doi:10.1021/ar50042a003 |
| 7. | Elacqua, E.; MacGillivray, L. R. Eur. J. Org. Chem. 2010, 6883–6894. doi:10.1002/ejoc.201000930 |
| 8. | Marrocchi, A.; Tomasi, I.; Vaccaro, L. Isr. J. Chem. 2012, 52, 41–52. doi:10.1002/ijch.201100091 |
| 9. | Mori, T.; Inoue, Y. Top. Curr. Chem. 2010, 298, 99–128. |
| 10. | Rowlands, G. J. Isr. J. Chem. 2012, 52, 60–75. doi:10.1002/ijch.201100098 |
| 11. | Paradies, J. Synthesis 2011, 3749–3766. doi:10.1055/s-0031-1289296 |
| 12. | Gibson, S. E.; Knight, J. D. Org. Biomol. Chem. 2003, 1, 1256–1269. doi:10.1039/b300717k |
| 1. | Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/3527603964 |
| 2. | Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969 |
| 7. | Elacqua, E.; MacGillivray, L. R. Eur. J. Org. Chem. 2010, 6883–6894. doi:10.1002/ejoc.201000930 |
| 8. | Marrocchi, A.; Tomasi, I.; Vaccaro, L. Isr. J. Chem. 2012, 52, 41–52. doi:10.1002/ijch.201100091 |
| 9. | Mori, T.; Inoue, Y. Top. Curr. Chem. 2010, 298, 99–128. |
| 28. | Mori, K.; Kishi, H.; Akiyama, T. Synthesis 2017, 49, 365–370. doi:10.1055/s-0036-1588898 |
| 1. | Gleiter, R.; Hopf, H., Eds. Modern Cyclophane Chemistry; Wiley-VCH: Weinheim, Germany, 2004. doi:10.1002/3527603964 |
| 2. | Hopf, H. Angew. Chem., Int. Ed. 2008, 47, 9808–9812. doi:10.1002/anie.200800969 |
| 3. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a |
| 4. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863 |
| 7. | Elacqua, E.; MacGillivray, L. R. Eur. J. Org. Chem. 2010, 6883–6894. doi:10.1002/ejoc.201000930 |
| 8. | Marrocchi, A.; Tomasi, I.; Vaccaro, L. Isr. J. Chem. 2012, 52, 41–52. doi:10.1002/ijch.201100091 |
| 9. | Mori, T.; Inoue, Y. Top. Curr. Chem. 2010, 298, 99–128. |
| 29. | Taylor, J. E.; Bull, S. D.; Williams, J. M. J. Chem. Soc. Rev. 2012, 41, 2109–2121. doi:10.1039/c2cs15288f |
| 30. | Merad, J.; Pons, J.-M.; Chuzel, O.; Bressy, C. Eur. J. Org. Chem. 2016, 5589–5610. doi:10.1002/ejoc.201600399 |
| 31. | Birman, V. Aldrichimica Acta 2016, 49, 23–33. |
| 32. | McLaughlin, C.; Smith, A. D. Chem. – Eur. J. 2021, 27, 1533–1555. doi:10.1002/chem.202002059 |
| 6. | Cram, D. J.; Cram, J. M. Acc. Chem. Res. 1971, 4, 204–213. doi:10.1021/ar50042a003 |
| 25. | Cipiciani, A.; Fringuelli, F.; Mancini, V.; Piermatti, O.; Scappini, A. M.; Ruzziconi, R. Tetrahedron 1997, 53, 11853–11858. doi:10.1016/s0040-4020(97)00758-8 |
| 26. | Pamperin, D.; Schulz, C.; Hopf, H.; Syldatk, C.; Pietzsch, M. Eur. J. Org. Chem. 1998, 1441–1445. doi:10.1002/(sici)1099-0690(199807)1998:7<1441::aid-ejoc1441>3.0.co;2-k |
| 27. | Pamperin, D.; Ohse, B.; Hopf, H.; Pietzsch, M. J. Mol. Catal. B: Enzym. 1998, 5, 317–319. doi:10.1016/s1381-1177(98)00063-0 |
| 28. | Mori, K.; Kishi, H.; Akiyama, T. Synthesis 2017, 49, 365–370. doi:10.1055/s-0036-1588898 |
| 3. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a |
| 4. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863 |
| 19. | Rozenberg, V.; Danilova, T.; Sergeeva, E.; Vorontsov, E.; Starikova, Z.; Korlyukov, A.; Hopf, H. Eur. J. Org. Chem. 2002, 468–477. doi:10.1002/1099-0690(20022)2002:3<468::aid-ejoc468>3.0.co;2-3 |
| 20. | Zhang, T.-Z.; Dai, L.-X.; Hou, X.-L. Tetrahedron: Asymmetry 2007, 18, 251–259. doi:10.1016/j.tetasy.2007.01.020 |
| 21. | Vorontsova, N. V.; Zhuravsky, R. P.; Sergeeva, E. V.; Vorontsov, E. V.; Starikova, Z. A.; Rozenberg, V. I. Russ. Chem. Bull. 2007, 56, 2225–2231. doi:10.1007/s11172-007-0348-x |
| 22. | Wang, Y.; Yuan, H.; Lu, H.; Zheng, W.-H. Org. Lett. 2018, 20, 2555–2558. doi:10.1021/acs.orglett.8b00711 |
| 18. | Friedmann, C. J.; Ay, S.; Bräse, S. J. Org. Chem. 2010, 75, 4612–4614. doi:10.1021/jo100468s |
| 16. | Krohn, K.; Rieger, H.; Hopf, H.; Barrett, D. Chem. Ber. 1990, 123, 1729–1732. doi:10.1002/cber.19901230824 |
| 17. | Kane, V. V.; Gerdes, A.; Grahn, W.; Ernst, L.; Dix, I.; Jones, P. G.; Hopf, H. Tetrahedron Lett. 2001, 42, 373–376. doi:10.1016/s0040-4039(00)01992-4 |
| 18. | Friedmann, C. J.; Ay, S.; Bräse, S. J. Org. Chem. 2010, 75, 4612–4614. doi:10.1021/jo100468s |
| 19. | Rozenberg, V.; Danilova, T.; Sergeeva, E.; Vorontsov, E.; Starikova, Z.; Korlyukov, A.; Hopf, H. Eur. J. Org. Chem. 2002, 468–477. doi:10.1002/1099-0690(20022)2002:3<468::aid-ejoc468>3.0.co;2-3 |
| 20. | Zhang, T.-Z.; Dai, L.-X.; Hou, X.-L. Tetrahedron: Asymmetry 2007, 18, 251–259. doi:10.1016/j.tetasy.2007.01.020 |
| 3. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a |
| 4. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863 |
| 13. | Kotha, S.; Shirbhate, M. E.; Waghule, G. T. Beilstein J. Org. Chem. 2015, 11, 1274–1331. doi:10.3762/bjoc.11.142 |
| 14. | Rowlands, G. J. Org. Biomol. Chem. 2008, 6, 1527–1534. doi:10.1039/b800698a |
| 15. | Tanaka, K. Bull. Chem. Soc. Jpn. 2018, 91, 187–194. doi:10.1246/bcsj.20170346 |
| 3. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a |
| 4. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863 |
| 10. | Rowlands, G. J. Isr. J. Chem. 2012, 52, 60–75. doi:10.1002/ijch.201100098 |
| 11. | Paradies, J. Synthesis 2011, 3749–3766. doi:10.1055/s-0031-1289296 |
| 12. | Gibson, S. E.; Knight, J. D. Org. Biomol. Chem. 2003, 1, 1256–1269. doi:10.1039/b300717k |
| 3. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Lahann, J.; Bräse, S. Chem. Soc. Rev. 2018, 47, 6947–6963. doi:10.1039/c7cs00803a |
| 4. | Hassan, Z.; Spuling, E.; Knoll, D. M.; Bräse, S. Angew. Chem., Int. Ed. 2020, 59, 2156–2170. doi:10.1002/anie.201904863 |
| 18. | Friedmann, C. J.; Ay, S.; Bräse, S. J. Org. Chem. 2010, 75, 4612–4614. doi:10.1021/jo100468s |
| 19. | Rozenberg, V.; Danilova, T.; Sergeeva, E.; Vorontsov, E.; Starikova, Z.; Korlyukov, A.; Hopf, H. Eur. J. Org. Chem. 2002, 468–477. doi:10.1002/1099-0690(20022)2002:3<468::aid-ejoc468>3.0.co;2-3 |
| 20. | Zhang, T.-Z.; Dai, L.-X.; Hou, X.-L. Tetrahedron: Asymmetry 2007, 18, 251–259. doi:10.1016/j.tetasy.2007.01.020 |
| 21. | Vorontsova, N. V.; Zhuravsky, R. P.; Sergeeva, E. V.; Vorontsov, E. V.; Starikova, Z. A.; Rozenberg, V. I. Russ. Chem. Bull. 2007, 56, 2225–2231. doi:10.1007/s11172-007-0348-x |
| 22. | Wang, Y.; Yuan, H.; Lu, H.; Zheng, W.-H. Org. Lett. 2018, 20, 2555–2558. doi:10.1021/acs.orglett.8b00711 |
| 23. | Hitchcock, P. B.; Rowlands, G. J.; Parmar, R. Chem. Commun. 2005, 4219–4221. doi:10.1039/b507394d |
| 24. | Parmar, R.; Coles, M. P.; Hitchcock, P. B.; Rowlands, G. J. Synthesis 2010, 4177–4187. doi:10.1055/s-0030-1258286 |
| 25. | Cipiciani, A.; Fringuelli, F.; Mancini, V.; Piermatti, O.; Scappini, A. M.; Ruzziconi, R. Tetrahedron 1997, 53, 11853–11858. doi:10.1016/s0040-4020(97)00758-8 |
| 26. | Pamperin, D.; Schulz, C.; Hopf, H.; Syldatk, C.; Pietzsch, M. Eur. J. Org. Chem. 1998, 1441–1445. doi:10.1002/(sici)1099-0690(199807)1998:7<1441::aid-ejoc1441>3.0.co;2-k |
| 27. | Pamperin, D.; Ohse, B.; Hopf, H.; Pietzsch, M. J. Mol. Catal. B: Enzym. 1998, 5, 317–319. doi:10.1016/s1381-1177(98)00063-0 |
| 38. | Joannesse, C.; Johnston, C. P.; Concellón, C.; Simal, C.; Philp, D.; Smith, A. D. Angew. Chem., Int. Ed. 2009, 48, 8914–8918. doi:10.1002/anie.200904333 |
| 33. | Birman, V. B.; Li, X. Org. Lett. 2006, 8, 1351–1354. doi:10.1021/ol060065s |
| 34. | Birman, V. B.; Li, X. Org. Lett. 2008, 10, 1115–1118. doi:10.1021/ol703119n |
| 35. | Li, X.; Jiang, H.; Uffman, E. W.; Guo, L.; Zhang, Y.; Yang, X.; Birman, V. B. J. Org. Chem. 2012, 77, 1722–1737. doi:10.1021/jo202220x |
| 36. | Qu, S.; Greenhalgh, M. D.; Smith, A. D. Chem. – Eur. J. 2019, 25, 2816–2823. doi:10.1002/chem.201805631 |
| 37. | Qu, S.; Smith, S. M.; Laina‐Martín, V.; Neyyappadath, R. M.; Greenhalgh, M. D.; Smith, A. D. Angew. Chem., Int. Ed. 2020, 59, 16572–16578. doi:10.1002/anie.202004354 |
| 28. | Mori, K.; Kishi, H.; Akiyama, T. Synthesis 2017, 49, 365–370. doi:10.1055/s-0036-1588898 |
| 16. | Krohn, K.; Rieger, H.; Hopf, H.; Barrett, D. Chem. Ber. 1990, 123, 1729–1732. doi:10.1002/cber.19901230824 |
| 18. | Friedmann, C. J.; Ay, S.; Bräse, S. J. Org. Chem. 2010, 75, 4612–4614. doi:10.1021/jo100468s |
| 19. | Rozenberg, V.; Danilova, T.; Sergeeva, E.; Vorontsov, E.; Starikova, Z.; Korlyukov, A.; Hopf, H. Eur. J. Org. Chem. 2002, 468–477. doi:10.1002/1099-0690(20022)2002:3<468::aid-ejoc468>3.0.co;2-3 |
| 20. | Zhang, T.-Z.; Dai, L.-X.; Hou, X.-L. Tetrahedron: Asymmetry 2007, 18, 251–259. doi:10.1016/j.tetasy.2007.01.020 |
| 26. | Pamperin, D.; Schulz, C.; Hopf, H.; Syldatk, C.; Pietzsch, M. Eur. J. Org. Chem. 1998, 1441–1445. doi:10.1002/(sici)1099-0690(199807)1998:7<1441::aid-ejoc1441>3.0.co;2-k |
| 28. | Mori, K.; Kishi, H.; Akiyama, T. Synthesis 2017, 49, 365–370. doi:10.1055/s-0036-1588898 |
| 39. | Cipiciani, A.; Fringuelli, F.; Mancini, V.; Piermatti, O.; Pizzo, F.; Ruzziconi, R. J. Org. Chem. 1997, 62, 3744–3747. doi:10.1021/jo962142a |
| 40. | Kagan, H. B.; Fiaud, J. C. Top. Stereochem. 1988, 18, 249–330. doi:10.1002/9780470147276.ch4 |
| 41. | Greenhalgh, M. D.; Taylor, J. E.; Smith, A. D. Tetrahedron 2018, 74, 5554–5560. doi:10.1016/j.tet.2018.05.069 |
| 42. | Calculation from recovered 2: s = ln[(1 − C)(1 − ee(2))] / ln[(1 − C)(1 + ee(2))]. |
| 43. | Calculation from isolated 3: s = ln[1 − C(1 + ee(3))] / ln[1 − C(1 − ee(3))]. |
| 33. | Birman, V. B.; Li, X. Org. Lett. 2006, 8, 1351–1354. doi:10.1021/ol060065s |
| 34. | Birman, V. B.; Li, X. Org. Lett. 2008, 10, 1115–1118. doi:10.1021/ol703119n |
| 35. | Li, X.; Jiang, H.; Uffman, E. W.; Guo, L.; Zhang, Y.; Yang, X.; Birman, V. B. J. Org. Chem. 2012, 77, 1722–1737. doi:10.1021/jo202220x |
| 36. | Qu, S.; Greenhalgh, M. D.; Smith, A. D. Chem. – Eur. J. 2019, 25, 2816–2823. doi:10.1002/chem.201805631 |
| 37. | Qu, S.; Smith, S. M.; Laina‐Martín, V.; Neyyappadath, R. M.; Greenhalgh, M. D.; Smith, A. D. Angew. Chem., Int. Ed. 2020, 59, 16572–16578. doi:10.1002/anie.202004354 |
| 28. | Mori, K.; Kishi, H.; Akiyama, T. Synthesis 2017, 49, 365–370. doi:10.1055/s-0036-1588898 |
| 33. | Birman, V. B.; Li, X. Org. Lett. 2006, 8, 1351–1354. doi:10.1021/ol060065s |
| 34. | Birman, V. B.; Li, X. Org. Lett. 2008, 10, 1115–1118. doi:10.1021/ol703119n |
| 36. | Qu, S.; Greenhalgh, M. D.; Smith, A. D. Chem. – Eur. J. 2019, 25, 2816–2823. doi:10.1002/chem.201805631 |
| 37. | Qu, S.; Smith, S. M.; Laina‐Martín, V.; Neyyappadath, R. M.; Greenhalgh, M. D.; Smith, A. D. Angew. Chem., Int. Ed. 2020, 59, 16572–16578. doi:10.1002/anie.202004354 |
| 20. | Zhang, T.-Z.; Dai, L.-X.; Hou, X.-L. Tetrahedron: Asymmetry 2007, 18, 251–259. doi:10.1016/j.tetasy.2007.01.020 |
| 26. | Pamperin, D.; Schulz, C.; Hopf, H.; Syldatk, C.; Pietzsch, M. Eur. J. Org. Chem. 1998, 1441–1445. doi:10.1002/(sici)1099-0690(199807)1998:7<1441::aid-ejoc1441>3.0.co;2-k |
| 39. | Cipiciani, A.; Fringuelli, F.; Mancini, V.; Piermatti, O.; Pizzo, F.; Ruzziconi, R. J. Org. Chem. 1997, 62, 3744–3747. doi:10.1021/jo962142a |
© 2021 Weinzierl and Waser; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)