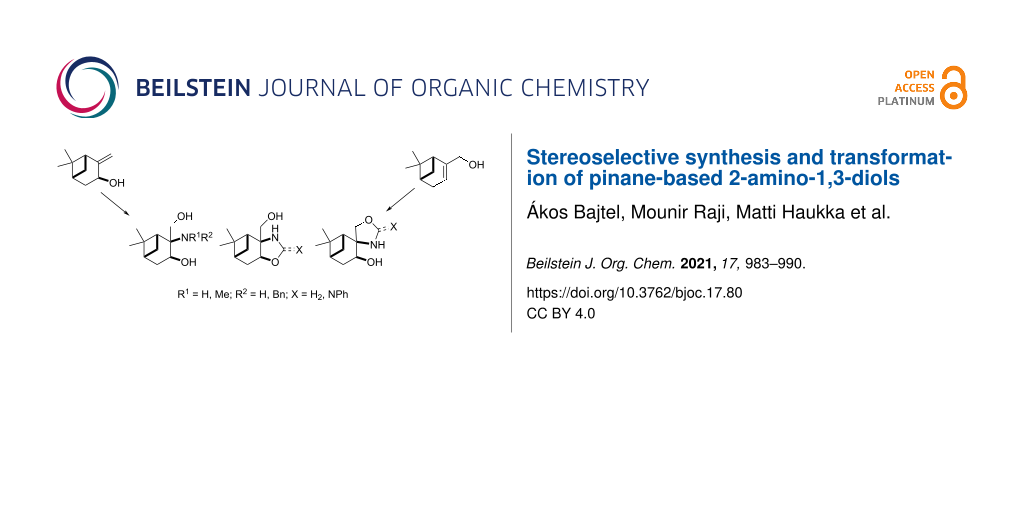
Abstract
A library of pinane-based 2-amino-1,3-diols was synthesised in a stereoselective manner. Isopinocarveol prepared from (−)-α-pinene was converted into condensed oxazolidin-2-one in two steps by carbamate formation followed by a stereoselective aminohydroxylation process. The relative stereochemistry of the pinane-fused oxazolidin-2-one was determined by 2D NMR and X-ray spectroscopic techniques. The regioisomeric spiro-oxazolidin-2-one was prepared in a similar way starting from the commercially available (1R)-(−)-myrtenol (10). The reduction or alkaline hydrolysis of the oxazolidines, followed by reductive alkylation resulted in primary and secondary 2-amino-1,3-diols, which underwent a regioselective ring closure with formaldehyde or benzaldehyde delivering pinane-condensed oxazolidines. During the preparation of 2-phenyliminooxazolidine, an interesting ring–ring tautomerism was observed in CDCl3.

Graphical Abstract
Introduction
The best known 2-amino-1,3-diol derivative sphingosine (1) plays a crucial role in intracellular signaling as second messenger, and its derivatives called sphingolipids are also critical for cell growth, cell differentiation, cell recognition, and apoptosis [1-7]. Due to its involvement in a wide range of cellular processes, significant efforts have been made in the last two decades targeting sphingosine analogues signalling as a therapeutic strategy. For instance, FTY720-P (3), the phosphate of FTY720 (2, fingolimod), proved to be a very good agonist for the S1P1 receptor (Figure 1). Sphingosine 1-phosphate (S1P, 4), in turn, performed critical regulator functions in many physiological and pathological treatments, such as Alzheimer’s disease [8,9], cancer [10-13], multiple sclerosis [14], and inflammation [15].
![[1860-5397-17-80-1]](/bjoc/content/figures/1860-5397-17-80-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Biologically active 2-amino-1,3-diols.
Figure 1: Biologically active 2-amino-1,3-diols.
Due to the lack of a readily available natural sources and the high biological importance of sphingolipid analogues, their synthesis has been the subject of numerous studies [16]. The key step for the synthesis is the stereoselective construction of the 2-amino-1,3-diol moiety of the molecules. Generally, two main synthetic strategies are used to prepare these analogues. One requires the insertion of the alcohol and amino groups in the α,β position with the correct stereochemistry [17-20]. The second strategy involves a bond formation between two chiral centers to produce the targeted 2-amino-1,3-diol [21,22]. For instance, deoxoprosophylline (5) as a cyclic 2-amino-1,3-diol target molecule was prepared by Kokatla et al. in an 8 step synthesis starting from Perlin aldehydes, via Pd(OH)2-catalyzed reductive azidoketon cyclisation [23]. Another synthetic pathway involves a stereoselective aminohydroxylation process starting from allylic carbamates usually carried out in the presence of potassium osmate [24-28].
In recent years, we have extensively studied the stereoselective synthesis, as well as catalytic and pharmacological applications of monoterpene-based 3-amino-1,2-diols, which are the regioisomers of potential monoterpenic 2-amino-1,3-diols [29-33]. These trifunctionalized terpenoids may also possess diverse biological activities and could successfully applied as chiral catalysts in enantioselective transformations [34]. In the present study, our aim was to synthesize novel, cyclic potentially analogues of sphingosine, incorporating a lipophilic natural pinane skeleton, starting from commercially available monoterpene-based allylic alcohols via a stereoselective hydroxyamination in the presence of a potassium osmate(VI) catalyst. We also planned to explore the regioselectivity of the ring closure of the resulting 2-amino-1,3-diols to obtain promising 1,3-heterocycles. To reach our goal, (1S)-(−)-α-pinene (6) and (1R)-(−)-myrtenol (10), two naturally occurring monoterpenoids were selected as precursors, as both are commercially available, cheap starting materials.
Results and Discussion
Synthesis of regioisomeric oxazolidinones from (1S)-(−)-α-pinene (6) and (1R)-myrtenol (10)
The synthesis of isopinocarveol (7), the key intermediate allylic alcohol, was performed according to a literature procedure in good yield [35]. The first step was the stereoselective epoxidation of (−)-α-pinene (6), carried out with meta-chloroperoxybenzoic acid (MCPBA), followed by a base-catalyzed allylic rearrangement mediated by aluminium isopropoxide (Al(OiPr)3). The resulting allylic alcohol 7 was reacted with trichloroacetyl isocyanate, followed by alkaline treatment, delivering carbamate 8 in good yield [27,28,36]. In the next step, the aminohydroxylation was accomplished by potassium osmate(VI) as the catalyst and t-BuOCl in the presence of DIPEA affording oxazolidine-2-one 9 [27]. The reaction was found to be highly stereoselective, giving exclusively the diexo-fused tricyclic 9 ring system (Scheme 1).
![[1860-5397-17-80-i1]](/bjoc/content/inline/1860-5397-17-80-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Stereoselective synthesis of the pinane-fused oxazolidin-2-one 9.
Scheme 1: Stereoselective synthesis of the pinane-fused oxazolidin-2-one 9.
The absolute configuration of compound 9 was determined by 2D NMR spectroscopic techniques. Clear NOE signals were observed between the H-7a and Me-10 as well as the Ha-9 and Me-10 protons. Beside NOESY experiments, the structure was also elucidated by X-ray crystallography (Figure 2).
![[1860-5397-17-80-2]](/bjoc/content/figures/1860-5397-17-80-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: NOESY experiments and X-ray structure elucidation of oxazolidin-2-one 9.
Figure 2: NOESY experiments and X-ray structure elucidation of oxazolidin-2-one 9.
To synthesize the regioisomeric spiro-oxazolidinone derivative 12, (1R)-(−)-myrtenol (10) was chosen as starting material (Scheme 2). The synthetic method was similar to that mentioned above for (−)-isopinocarveol. In the first step, carbamate 11 was prepared [37], then the aminohydroxylation was carried out catalyzed by potassium osmate(VI), which led to the formation of the spiro-oxazolidine-2-one 12 in a highly regio- and stereoselective manner. Based on the NMR spectroscopic measurements of the crude product, the spiro derivative 12 was obtained exclusively with the relative configuration depicted in Scheme 2. Beside 2D NMR spectroscopic studies, the absolute configuration of compound 12 was determined by its transformation into the corresponding aminodiols 13 and 14, comparing the products with those obtained from the regioisomer 9 (discussed in Scheme 3).
![[1860-5397-17-80-i2]](/bjoc/content/inline/1860-5397-17-80-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Stereoselective synthesis of the pinane spiro-fused oxazolidin-2-one 12.
Scheme 2: Stereoselective synthesis of the pinane spiro-fused oxazolidin-2-one 12.
Synthesis and transformations of pinane-based 2-amino-1,3-diols
To obtain a library of pinane-based 2-amino-1,3-diols, the oxazolidine-2-ones 9 and 12 were applied as starting materials. The alkaline hydrolysis of both 9 and 12 resulted in the same primary aminodiol 13 [38]. According to the NMR spectra and other physical and chemical properties, there was no difference between the products of the two reactions. Since the relative configuration of compound 9 was clarified by NMR spectroscopy and X-ray crystallographic results, we were able to assign the stereochemistry of spiro-derivate 12, too. In a similar manner, the LiAlH4 (LAH) reduction of both 9 and 12 gave the same N-methylaminodiol 14 with modest yield (Scheme 3).
![[1860-5397-17-80-i3]](/bjoc/content/inline/1860-5397-17-80-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Parallel synthesis of 2-amino-1,3-diols.
Scheme 3: Parallel synthesis of 2-amino-1,3-diols.
Subsequently, compound 13 was reacted with benzaldehyde. In this process, the Schiff' base 15A was generated in situ. Our efforts to reduce it with sodium borohydride failed, since we did not observe the formation of the expected N-benzylaminodiol either at room temperature or under reflux conditions, probably due to the strong steric hindrance of the bicyclic system and the hydroxymethyl group. The 1H NMR spectroscopic measurements in CDCl3 clearly showed that the crude product was a five-component tautomeric mixture containing condensed oxazolidine 15E as the main component. Additional minor components included the other condensed oxazolidine (15D), spiro compounds 15B and 15C as well as the Schiff’ base 15A existing in a ratio of 15A/15B/15C/15D/15E = 4:<1:4:12:79 (Scheme 4) [39,40]. The structures of the five components 15A–E were determined by 2D NMR spectroscopic techniques (NOESY and HMBC). Since this finding is quite unusual in the case of Schiff’ bases, we decided to study the ring/chain tautomeric mixture (15A–E) in the reaction of 13 with benzaldehyde by 1H NMR spectroscopy. When a time-dependent 1H NMR spectroscopic measurement was accomplished, we observed that the equilibrium composition was established rapidly, without any significant change in the ratio of the tautomers. The equilibrium shifting strongly to product 15E can account of the difficulty of the reduction process and the necessity to use a stronger reducing agent and more severe conditions. The reduction step, therefore, was performed by applying LAH, a stronger reducing agent, and longer reflux, resulting in N-benzyl-2-amino-1,3-diol 16 (Scheme 4).
![[1860-5397-17-80-i4]](/bjoc/content/inline/1860-5397-17-80-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of N-benzyl-2-amino-1,3-diol 16.
Scheme 4: Synthesis of N-benzyl-2-amino-1,3-diol 16.
When compound 16 was treated with formaldehyde at room temperature, pinane-fused oxazolidine 17 was obtained regioselectively (Scheme 5), as it was indicated by clear HMBC correlations between the CH2 of the oxazolidine ring and the anellation carbons, in contrast to the results observed in the case of the regioisomeric 3-amino-1,2-diols, where spiro-oxazolidines formed exclusively [41]. The configuration of oxazolidine 17 was determined by 2D NMR spectroscopic techniques. Clear NOE signals were observed between the H-7a and Me-10 as well as the Ha-9 and Me-10 protons. In addition to NOESY experiments, the structure was also elucidated by X-ray crystallography (Figure 3).
![[1860-5397-17-80-i5]](/bjoc/content/inline/1860-5397-17-80-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of 2-amino-1,3-diols.
Scheme 5: Synthesis of 2-amino-1,3-diols.
![[1860-5397-17-80-3]](/bjoc/content/figures/1860-5397-17-80-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: NOESY experiments and X-ray structure proofment of the structure of oxazolidine 17.
Figure 3: NOESY experiments and X-ray structure proofment of the structure of oxazolidine 17.
The LAH reduction of oxazolidine 17 gave N-benzyl-N-methyl analogue 18 which, alternatively, was prepared directly from 2-oxazolidinone 9 via N-benzylation followed by LAH reduction in 2 steps.
When compound 13 was reacted with phenylisothiocyanate, thiourea 20 was obtained, which underwent a regioselective ring closure resulting in 21A. The structure of 21A was determined by 1H (whereas the CH-OH gave a dublet in DMSO-d6 while the CH2-OH of 21B could be detected as triplet) and 2D NMR spectroscopic techniques (HMBC). It is important to mention that this regioselectivity is the opposite to that observed in the reaction of aminodiols 13 and 16 with aldehydes (see Scheme 4 and Scheme 5), but it is similar to that observed in our earlier study with pinane-based 3-amino-1,2-diols [41]. During the NMR spectroscopic study of 21A in CDCl3 for 30 days, an unknown slow ring–ring tautomerization was observed, forming a 1:1 mixture of the two regioisomers 21A and 21B. Compound 21B could be isolated from the mixture by column chromatography in pure form.
The synthesis of the heteroanalogue 2-phenyliminothiazolidines 22A and 22B failed, even when the reaction was attempted under acidic or even milder conditions (Scheme 6).
![[1860-5397-17-80-i6]](/bjoc/content/inline/1860-5397-17-80-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of 2-phenyliminooxazolidines.
Scheme 6: Synthesis of 2-phenyliminooxazolidines.
The proposed reaction pathway for the ring–ring tautomerism of 21A and 21B is presented in Figure 4 and it explains why the acidic environment (present generally in CDCl3 solution) is necessary. In a similar manner, an oxazolidine–1,3-oxazine tautomerism of pulegone-based 3-amino-1,2-diols was recently reported [31]. When compound 21A or 21B were treated in less protic solvents such as DMSO-d6 or CD3OD, tautomerization was not observed.
![[1860-5397-17-80-4]](/bjoc/content/figures/1860-5397-17-80-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Proposed pathway for the ring–ring tautomerism.
Figure 4: Proposed pathway for the ring–ring tautomerism.
Conclusion
A small library of pinane-based 2-amino-1,3-diols was synthesized in a stereoselective manner starting from (1R)-(−)-myrtenol and isopinocarveol prepared from α-pinene. Pinane-condensed or spiro-oxazolidin-2-ones were formed in three steps by a stereoselective hydroxyamination process. The relative stereochemistry of new compounds was determined by 2D NMR spectroscopic and X-ray techniques. The resulting primary and secondary 2-amino-1,3-diols underwent a regioselective ring closure with formaldehyde and benzaldehyde producing pinane-condensed oxazolidines. In the case of 2-phenyliminooxazolidine, an interesting ring–ring tautomerism was observed in CDCl3. The prepared trifunctional compounds may serve as chiral catalysts in enantioselective transformations, while the 2-phenyliminooxazolidines could be interesting in the field of antiproliferative or antioxidants studies based on our former studies on 2-imino-1,3-heterocycles [42,43].
Supporting Information
| Supporting Information File 1: Experimental part, analytical data, NMR spectra and X-ray data of the prepared compounds. | ||
| Format: PDF | Size: 2.2 MB | Download |
References
-
Posse de Chaves, E.; Sipione, S. FEBS Lett. 2010, 584, 1748–1759. doi:10.1016/j.febslet.2009.12.010
Return to citation in text: [1] -
Hannun, Y. A.; Obeid, L. M. J. Biol. Chem. 2002, 277, 25847–25850. doi:10.1074/jbc.r200008200
Return to citation in text: [1] -
Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U. T. Biochem. J. 2001, 359, 335. doi:10.1042/0264-6021:3590335
Return to citation in text: [1] -
Kolesnick, R. N.; Goñi, F. M.; Alonso, A. J. Cell. Physiol. 2000, 184, 285–300. doi:10.1002/1097-4652(200009)184:3<285::aid-jcp2>3.0.co;2-3
Return to citation in text: [1] -
Perry, D. K.; Hannun, Y. A. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1998, 1436, 233–243. doi:10.1016/s0005-2760(98)00145-3
Return to citation in text: [1] -
Igarashi, Y. J. Biochem. 1997, 122, 1080–1087. doi:10.1093/oxfordjournals.jbchem.a021865
Return to citation in text: [1] -
Spiegel, S.; Foster, D.; Kolesnick, R. Curr. Opin. Cell Biol. 1996, 8, 159–167. doi:10.1016/s0955-0674(96)80061-5
Return to citation in text: [1] -
Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; Lee, V. M.- Y.; Trojanowski, J. Q.; Tomita, T.; Iwatsubo, T. J. Neurosci. 2011, 31, 6850–6857. doi:10.1523/jneurosci.6467-10.2011
Return to citation in text: [1] -
Prager, B.; Spampinato, S. F.; Ransohoff, R. M. Trends Mol. Med. 2015, 21, 354–363. doi:10.1016/j.molmed.2015.03.006
Return to citation in text: [1] -
Heffernan-Stroud, L. A.; Obeid, L. M. Sphingosine Kinase 1 in Cancer. Advances in Cancer Research; Elsevier, 2013; Vol. 117, pp 201–235. doi:10.1016/b978-0-12-394274-6.00007-8
Return to citation in text: [1] -
Pyne, N. J.; Tonelli, F.; Lim, K. G.; Long, J. S.; Edwards, J.; Pyne, S. Biochem. Soc. Trans. 2012, 40, 94–100. doi:10.1042/bst20110602
Return to citation in text: [1] -
Plano, D.; Amin, S.; Sharma, A. K. J. Med. Chem. 2014, 57, 5509–5524. doi:10.1021/jm4011687
Return to citation in text: [1] -
Pyne, N. J.; Pyne, S. Nat. Rev. Cancer 2010, 10, 489–503. doi:10.1038/nrc2875
Return to citation in text: [1] -
Chi, H. Trends Pharmacol. Sci. 2011, 32, 16–24. doi:10.1016/j.tips.2010.11.002
Return to citation in text: [1] -
Maceyka, M.; Spiegel, S. Nature 2014, 510, 58–67. doi:10.1038/nature13475
Return to citation in text: [1] -
Howell, A. R.; So, R. C.; Richardson, S. K. Tetrahedron 2004, 60, 11327–11347. doi:10.1016/j.tet.2004.09.056
Return to citation in text: [1] -
Cardillo, G.; Orena, M.; Porzi, G.; Sandri, S. J. Chem. Soc., Chem. Commun. 1981, 465. doi:10.1039/c39810000465
Return to citation in text: [1] -
Azuma, H.; Takao, R.; Niiro, H.; Shikata, K.; Tamagaki, S.; Tachibana, T.; Ogino, K. J. Org. Chem. 2003, 68, 2790–2797. doi:10.1021/jo0206824
Return to citation in text: [1] -
He, L.; Byun, H.-S.; Bittman, R. J. Org. Chem. 2000, 65, 7627–7633. doi:10.1021/jo001226n
Return to citation in text: [1] -
Sasai, H.; Tokunaga, T.; Watanabe, S.; Suzuki, T.; Itoh, N.; Shibasaki, M. J. Org. Chem. 1995, 60, 7388–7389. doi:10.1021/jo00128a005
Return to citation in text: [1] -
Masui, M.; Shioiri, T. Tetrahedron Lett. 1998, 39, 5199–5200. doi:10.1016/s0040-4039(98)01020-x
Return to citation in text: [1] -
Ma, N.; Ma, D. Tetrahedron: Asymmetry 2003, 14, 1403–1406. doi:10.1016/s0957-4166(03)00275-1
Return to citation in text: [1] -
Kokatla, H. P.; Lahiri, R.; Kancharla, P. K.; Doddi, V. R.; Vankar, Y. D. J. Org. Chem. 2010, 75, 4608–4611. doi:10.1021/jo100489k
Return to citation in text: [1] -
Han, H.; Cho, C.-W.; Janda, K. D. Chem. – Eur. J. 1999, 5, 1565–1569. doi:10.1002/(sici)1521-3765(19990503)5:5<1565::aid-chem1565>3.0.co;2-j
Return to citation in text: [1] -
Rudolph, J.; Sennhenn, P. C.; Vlaar, C. P.; Sharpless, K. B. Angew. Chem., Int. Ed. Engl. 1996, 35, 2810–2813. doi:10.1002/anie.199628101
Return to citation in text: [1] -
Donohoe, T. J.; Johnson, P. D.; Helliwell, M.; Keenan, M. Chem. Commun. 2001, 2078–2079. doi:10.1039/b107253f
Return to citation in text: [1] -
Donohoe, T. J.; Johnson, P. D.; Cowley, A.; Keenan, M. J. Am. Chem. Soc. 2002, 124, 12934–12935. doi:10.1021/ja0276117
Return to citation in text: [1] [2] [3] -
Hovey, M. T.; Eklund, E. J.; Pike, R. D.; Mainkar, A. A.; Scheerer, J. R. Org. Lett. 2011, 13, 1246–1249. doi:10.1021/ol200155p
Return to citation in text: [1] [2] -
Szakonyi, Z.; Hetényi, A.; Fülöp, F. Tetrahedron 2008, 64, 1034–1039. doi:10.1016/j.tet.2007.07.065
Return to citation in text: [1] -
Csillag, K.; Németh, L.; Martinek, T. A.; Szakonyi, Z.; Fülöp, F. Tetrahedron: Asymmetry 2012, 23, 144–150. doi:10.1016/j.tetasy.2012.01.020
Return to citation in text: [1] -
Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Tetrahedron: Asymmetry 2016, 27, 480–486. doi:10.1016/j.tetasy.2016.04.009
Return to citation in text: [1] [2] -
Szakonyi, Z.; Csőr, Á.; Csámpai, A.; Fülöp, F. Chem. – Eur. J. 2016, 22, 7163–7173. doi:10.1002/chem.201600749
Return to citation in text: [1] -
Le, T. M.; Csámpai, A.; Fülöp, F.; Szakonyi, Z. Chem. – Eur. J. 2018, 24, 13607–13615. doi:10.1002/chem.201802484
Return to citation in text: [1] -
El Alami, M. S. I.; El Amrani, M. A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chem. – Eur. J. 2015, 21, 1398–1413. doi:10.1002/chem.201404303
Return to citation in text: [1] -
Lavallee, P.; Bouthillier, G. J. Org. Chem. 1986, 51, 1362–1365. doi:10.1021/jo00358a041
Return to citation in text: [1] -
Miller, K. E.; Wright, A. J.; Olesen, M. K.; Hovey, M. T.; Scheerer, J. R. J. Org. Chem. 2015, 80, 1569–1576. doi:10.1021/jo502493e
Return to citation in text: [1] -
Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363–2365. doi:10.1039/c2ob07190h
Return to citation in text: [1] -
Byun, H.-S.; Bittman, R. Chem. Phys. Lipids 2012, 165, 794–801. doi:10.1016/j.chemphyslip.2012.10.002
Return to citation in text: [1] -
Lázár, L.; Fülöp, F. Eur. J. Org. Chem. 2003, 3025–3042. doi:10.1002/ejoc.200300142
Return to citation in text: [1] -
Hetényi, A.; Szakonyi, Z.; Klika, K. D.; Pihlaja, K.; Fülöp, F. J. Org. Chem. 2003, 68, 2175–2182. doi:10.1021/jo026428t
Return to citation in text: [1] -
Szakonyi, Z.; Hetényi, A.; Fülöp, F. ARKIVOC 2007, No. iii, 33–42. doi:10.3998/ark.5550190.0009.305
Return to citation in text: [1] [2] -
Szakonyi, Z.; Zupkó, I.; Sillanpää, R.; Fülöp, F. Molecules 2014, 19, 15918–15937. doi:10.3390/molecules191015918
Return to citation in text: [1] -
Firpo, G.; Ramírez, M. L.; Faillace, M. S.; Mendes de Brito, M. d. R.; Correia Lima e Silva, A. P. S.; Pereira Costa, J.; Rodríguez, M. C.; Argüello, G. A.; Szakonyi, Z.; Fülöp, F.; Peláez, W. J. Antioxidants 2019, 8, 197. doi:10.3390/antiox8060197
Return to citation in text: [1]
| 41. | Szakonyi, Z.; Hetényi, A.; Fülöp, F. ARKIVOC 2007, No. iii, 33–42. doi:10.3998/ark.5550190.0009.305 |
| 38. | Byun, H.-S.; Bittman, R. Chem. Phys. Lipids 2012, 165, 794–801. doi:10.1016/j.chemphyslip.2012.10.002 |
| 39. | Lázár, L.; Fülöp, F. Eur. J. Org. Chem. 2003, 3025–3042. doi:10.1002/ejoc.200300142 |
| 40. | Hetényi, A.; Szakonyi, Z.; Klika, K. D.; Pihlaja, K.; Fülöp, F. J. Org. Chem. 2003, 68, 2175–2182. doi:10.1021/jo026428t |
| 1. | Posse de Chaves, E.; Sipione, S. FEBS Lett. 2010, 584, 1748–1759. doi:10.1016/j.febslet.2009.12.010 |
| 2. | Hannun, Y. A.; Obeid, L. M. J. Biol. Chem. 2002, 277, 25847–25850. doi:10.1074/jbc.r200008200 |
| 3. | Kågedal, K.; Zhao, M.; Svensson, I.; Brunk, U. T. Biochem. J. 2001, 359, 335. doi:10.1042/0264-6021:3590335 |
| 4. | Kolesnick, R. N.; Goñi, F. M.; Alonso, A. J. Cell. Physiol. 2000, 184, 285–300. doi:10.1002/1097-4652(200009)184:3<285::aid-jcp2>3.0.co;2-3 |
| 5. | Perry, D. K.; Hannun, Y. A. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 1998, 1436, 233–243. doi:10.1016/s0005-2760(98)00145-3 |
| 6. | Igarashi, Y. J. Biochem. 1997, 122, 1080–1087. doi:10.1093/oxfordjournals.jbchem.a021865 |
| 7. | Spiegel, S.; Foster, D.; Kolesnick, R. Curr. Opin. Cell Biol. 1996, 8, 159–167. doi:10.1016/s0955-0674(96)80061-5 |
| 27. | Donohoe, T. J.; Johnson, P. D.; Cowley, A.; Keenan, M. J. Am. Chem. Soc. 2002, 124, 12934–12935. doi:10.1021/ja0276117 |
| 14. | Chi, H. Trends Pharmacol. Sci. 2011, 32, 16–24. doi:10.1016/j.tips.2010.11.002 |
| 37. | Kamon, T.; Shigeoka, D.; Tanaka, T.; Yoshimitsu, T. Org. Biomol. Chem. 2012, 10, 2363–2365. doi:10.1039/c2ob07190h |
| 10. | Heffernan-Stroud, L. A.; Obeid, L. M. Sphingosine Kinase 1 in Cancer. Advances in Cancer Research; Elsevier, 2013; Vol. 117, pp 201–235. doi:10.1016/b978-0-12-394274-6.00007-8 |
| 11. | Pyne, N. J.; Tonelli, F.; Lim, K. G.; Long, J. S.; Edwards, J.; Pyne, S. Biochem. Soc. Trans. 2012, 40, 94–100. doi:10.1042/bst20110602 |
| 12. | Plano, D.; Amin, S.; Sharma, A. K. J. Med. Chem. 2014, 57, 5509–5524. doi:10.1021/jm4011687 |
| 13. | Pyne, N. J.; Pyne, S. Nat. Rev. Cancer 2010, 10, 489–503. doi:10.1038/nrc2875 |
| 35. | Lavallee, P.; Bouthillier, G. J. Org. Chem. 1986, 51, 1362–1365. doi:10.1021/jo00358a041 |
| 8. | Takasugi, N.; Sasaki, T.; Suzuki, K.; Osawa, S.; Isshiki, H.; Hori, Y.; Shimada, N.; Higo, T.; Yokoshima, S.; Fukuyama, T.; Lee, V. M.- Y.; Trojanowski, J. Q.; Tomita, T.; Iwatsubo, T. J. Neurosci. 2011, 31, 6850–6857. doi:10.1523/jneurosci.6467-10.2011 |
| 9. | Prager, B.; Spampinato, S. F.; Ransohoff, R. M. Trends Mol. Med. 2015, 21, 354–363. doi:10.1016/j.molmed.2015.03.006 |
| 27. | Donohoe, T. J.; Johnson, P. D.; Cowley, A.; Keenan, M. J. Am. Chem. Soc. 2002, 124, 12934–12935. doi:10.1021/ja0276117 |
| 28. | Hovey, M. T.; Eklund, E. J.; Pike, R. D.; Mainkar, A. A.; Scheerer, J. R. Org. Lett. 2011, 13, 1246–1249. doi:10.1021/ol200155p |
| 36. | Miller, K. E.; Wright, A. J.; Olesen, M. K.; Hovey, M. T.; Scheerer, J. R. J. Org. Chem. 2015, 80, 1569–1576. doi:10.1021/jo502493e |
| 23. | Kokatla, H. P.; Lahiri, R.; Kancharla, P. K.; Doddi, V. R.; Vankar, Y. D. J. Org. Chem. 2010, 75, 4608–4611. doi:10.1021/jo100489k |
| 29. | Szakonyi, Z.; Hetényi, A.; Fülöp, F. Tetrahedron 2008, 64, 1034–1039. doi:10.1016/j.tet.2007.07.065 |
| 30. | Csillag, K.; Németh, L.; Martinek, T. A.; Szakonyi, Z.; Fülöp, F. Tetrahedron: Asymmetry 2012, 23, 144–150. doi:10.1016/j.tetasy.2012.01.020 |
| 31. | Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Tetrahedron: Asymmetry 2016, 27, 480–486. doi:10.1016/j.tetasy.2016.04.009 |
| 32. | Szakonyi, Z.; Csőr, Á.; Csámpai, A.; Fülöp, F. Chem. – Eur. J. 2016, 22, 7163–7173. doi:10.1002/chem.201600749 |
| 33. | Le, T. M.; Csámpai, A.; Fülöp, F.; Szakonyi, Z. Chem. – Eur. J. 2018, 24, 13607–13615. doi:10.1002/chem.201802484 |
| 42. | Szakonyi, Z.; Zupkó, I.; Sillanpää, R.; Fülöp, F. Molecules 2014, 19, 15918–15937. doi:10.3390/molecules191015918 |
| 43. | Firpo, G.; Ramírez, M. L.; Faillace, M. S.; Mendes de Brito, M. d. R.; Correia Lima e Silva, A. P. S.; Pereira Costa, J.; Rodríguez, M. C.; Argüello, G. A.; Szakonyi, Z.; Fülöp, F.; Peláez, W. J. Antioxidants 2019, 8, 197. doi:10.3390/antiox8060197 |
| 21. | Masui, M.; Shioiri, T. Tetrahedron Lett. 1998, 39, 5199–5200. doi:10.1016/s0040-4039(98)01020-x |
| 22. | Ma, N.; Ma, D. Tetrahedron: Asymmetry 2003, 14, 1403–1406. doi:10.1016/s0957-4166(03)00275-1 |
| 34. | El Alami, M. S. I.; El Amrani, M. A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chem. – Eur. J. 2015, 21, 1398–1413. doi:10.1002/chem.201404303 |
| 17. | Cardillo, G.; Orena, M.; Porzi, G.; Sandri, S. J. Chem. Soc., Chem. Commun. 1981, 465. doi:10.1039/c39810000465 |
| 18. | Azuma, H.; Takao, R.; Niiro, H.; Shikata, K.; Tamagaki, S.; Tachibana, T.; Ogino, K. J. Org. Chem. 2003, 68, 2790–2797. doi:10.1021/jo0206824 |
| 19. | He, L.; Byun, H.-S.; Bittman, R. J. Org. Chem. 2000, 65, 7627–7633. doi:10.1021/jo001226n |
| 20. | Sasai, H.; Tokunaga, T.; Watanabe, S.; Suzuki, T.; Itoh, N.; Shibasaki, M. J. Org. Chem. 1995, 60, 7388–7389. doi:10.1021/jo00128a005 |
| 41. | Szakonyi, Z.; Hetényi, A.; Fülöp, F. ARKIVOC 2007, No. iii, 33–42. doi:10.3998/ark.5550190.0009.305 |
| 16. | Howell, A. R.; So, R. C.; Richardson, S. K. Tetrahedron 2004, 60, 11327–11347. doi:10.1016/j.tet.2004.09.056 |
| 24. | Han, H.; Cho, C.-W.; Janda, K. D. Chem. – Eur. J. 1999, 5, 1565–1569. doi:10.1002/(sici)1521-3765(19990503)5:5<1565::aid-chem1565>3.0.co;2-j |
| 25. | Rudolph, J.; Sennhenn, P. C.; Vlaar, C. P.; Sharpless, K. B. Angew. Chem., Int. Ed. Engl. 1996, 35, 2810–2813. doi:10.1002/anie.199628101 |
| 26. | Donohoe, T. J.; Johnson, P. D.; Helliwell, M.; Keenan, M. Chem. Commun. 2001, 2078–2079. doi:10.1039/b107253f |
| 27. | Donohoe, T. J.; Johnson, P. D.; Cowley, A.; Keenan, M. J. Am. Chem. Soc. 2002, 124, 12934–12935. doi:10.1021/ja0276117 |
| 28. | Hovey, M. T.; Eklund, E. J.; Pike, R. D.; Mainkar, A. A.; Scheerer, J. R. Org. Lett. 2011, 13, 1246–1249. doi:10.1021/ol200155p |
| 31. | Gonda, T.; Szakonyi, Z.; Csámpai, A.; Haukka, M.; Fülöp, F. Tetrahedron: Asymmetry 2016, 27, 480–486. doi:10.1016/j.tetasy.2016.04.009 |
© 2021 Bajtel et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)