Recent advances in palladium-catalysed asymmetric 1,4–additions of arylboronic acids to conjugated enones and chromones
1,
1,
1,
1,
2,
1 and
1
Jan Bartáček
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
Department of Biological and Biochemical Sciences, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
1Institute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic,
2Department of Biological and Biochemical Sciences, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 532 10 Pardubice, Czech Republic
Corresponding author email
Associate Editor: B. Stoltz Beilstein J. Org. Chem.2021,17, 1048–1085.https://doi.org/10.3762/bjoc.17.84 Received 14 Feb 2021,
Accepted 17 Apr 2021,
Published 10 May 2021
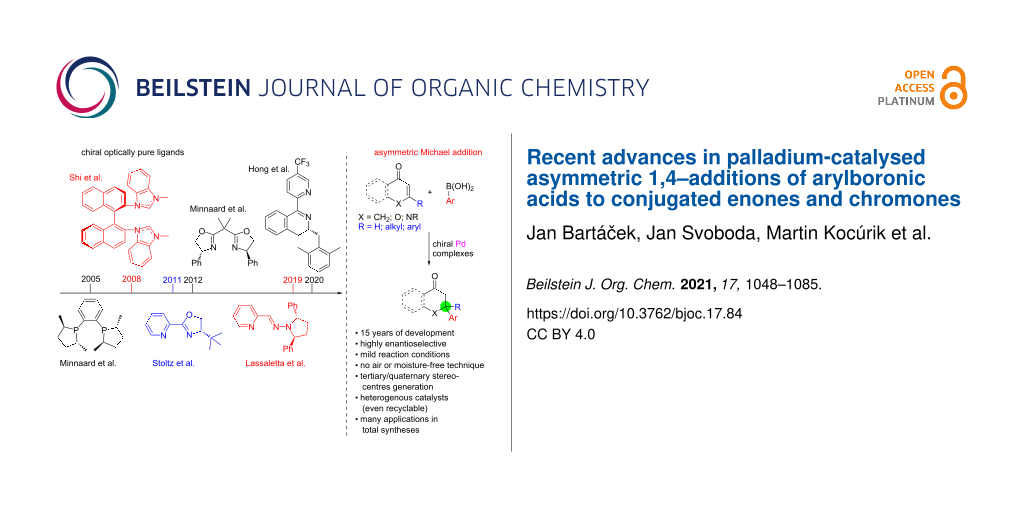
The transition metal (palladium)-catalysed asymmetric 1,4-addition of arylboronic acids to conjugated enones belong to the most important and emerging strategies for the construction of C–C bonds in an asymmetric fashion. This review covers known catalytic systems used for this transformation. For clarity, we are using the type of ligand as a sorting criterion. Finally, we attempted to create a flowchart facilitating the selection of a suitable ligand for a given combination of enone and arylboronic acid.
The asymmetric 1,4-addition of arylboronic acids to conjugated cyclic enones and chromones is a very important reaction nowadays. For illustration, the addition products are very promising in medicinal chemistry research [1-7] and in natural products total syntheses [8-16]. Chiral complexes of Rh [17-24] and Pd usually catalyse the reaction, however, palladium holds a special place in this area. There are several review articles partially covering this topic [25-31]. However, a comprehensive review is missing. In the following sections, we attempt to fill this gap. As a sorting criterion, the type of ligand (phosphines, NHC-carbenes, bisoxazolines, pyridine-oxazolines, and miscellaneous) is used.
Review
Catalytic systems based on phosphine ligands
A pioneering work on the enantioselective addition of boron-derived carbon nucleophiles to cyclic enones was published by the group of Miyaura et al. in 2005 [32]. Specifically, they have dealt with the addition of potassium aryltrifluoroborates to conjugated cyclic enones differing in ring size [32]. The catalysts PdL1a,b exhibited great conversion and enantioselectivities (up to 99% and up to 96% ee) for various combinations of nucleophiles and enones (Table 1). The authors also studied the possibility of the addition of boronic acids. The reaction of phenylboronic acid with 2-cyclohexenone catalysed by 5% of achiral [Pd(dppe)(PhCN)2](BF4)2 at −5 °C gave the product in 21% yield. When 1 equiv of BF3·OEt2 was added, the yield was increased to 74%. This result led to the conclusion that in this catalytic system, much better results were obtained when aryltrifluoroborates are used. The system also worked well for linear enone electrophiles (entries 12–20, Table 1). The main disadvantage of this approach is the necessity of sub-zero temperatures [32,33].
Table 1:
First example of asymmetric addition of organoboron reagents to cyclic enones [32,33].
entry
cyclic substrate
Ar
cat.
temp. (°C)
yield (%)
ee (%)
n
1
0
Ph
PdL1a
−5
60
95 (S)
2
1
Ph
PdL1b
−15
95
93 (R)
3
1
4-MeO-C6H4
PdL1b
−5
89
85 (R)
4
1
3-MeO-C6H4
PdL1b
−15
97
95 (R)
5
1
4-Me-C6H4
PdL1b
−5
70
90 (R)
6
1
3-Me-C6H4
PdL1b
−5
96
93 (R)
7
1
4-F-C6H4
PdL1b
−5
99
92 (R)
8
1
3-F-C6H4
PdL1b
−15
81
96 (R)
9
1
4-CF3-C6H4
PdL1b
−5
33
87 (R)
10
1
4-CF3-C6H4
PdL1b
−5
66a
92a (R)
11
2
Ph
PdL1b
−15
91
89 (R)
acyclic substrate
R1
R2
12
n-C5H11
iPr
Ph
PdL1a
−15
93
87
13
n-C5H11
Cy
Ph
PdL1a
−15
98
88
14
n-C5H11
Ph
Ph
PdL1a
−15
99
89
15
iPr
Me
3-MeO-C6H4
PdL1a
−5
65
83
16
Cy
Me
Ph
PdL1a
−5
22
78
17
Ph
Me
3-MeO-C6H4
PdL1a
0
90
95
18
Ph
n-Bu
3-MeO-C6H4
PdL1a
5
91
99
19
Ph
Ph
3-MeO-C6H4
PdL1a
−5
94
97
20
2-naphthyl
Me
3-MeO-C6H4
PdL1a
0
73
96
aNo water added.
A follow-up report of the Miyaura group in 2007 provided an experimental protocol that allowed the addition of arylboronic acids instead of aryltrifluoroborates [34]. The previously used catalysts PdL1a,b were combined with additional silver salts (AgBF4 or AgSbF6) that greatly accelerated the transmetalation of the boronic acid to Pd. This enhanced catalytic system showed a great turnover number (TON) up to 9,900. The authors described additions to cyclic substrates with high yields (90–99%) and enantioselectivities (89–94% ee; entries 1–5, Table 2). Also, a library of linear enones was tested giving excellent yields and enantioselectivities in most of the cases (with up to 99% yield and 99% ee; entries 6–24, Table 2). Several substrates did not even require the addition of Ag(I) salts to achieve high yields (entries 7, 10, 12, 17, 22, and 23, Table 2) [34,35].
Table 2:
Addition of arylboronic acids to enones accelerated by silver salts [34,35].
entry
cyclic substrates
Ar
additive (equiv)
temp. (°C)
yield (%)
ee (%)
n (catalyst)
1
0 (PdL1b)
Ph
AgBF4
0
94
94 (S)
2
1 (PdL1a)
Ph
AgBF4
0
90
92 (R)
3
1 (PdL1a)
Ph
AgBF4 (0.05)
20
99a
89 (R)
4
1 (PdL1a)
3-MeO-C6H4
AgBF4 (0.05)
20
98a
91 (R)
5
2 (PdL1a)
Ph
AgBF4
0
92
89 (R)
acyclic substrates
R1
R2
6
Ph
Ph
4-Me-C6H4
AgBF4 (0.1)
20
73
95
7
Ph
Me
3-Cl-C6H4
–
25
90
93
8
Ph
Me
3-MeO-C6H4
AgBF4 (0.1)
0
96
95
9
Ph
Me
4-MeO-C6H4
AgBF4 (0.1)
0
75
94
10
Ph
Me
3,4-(CH2O2)-C6H3
–
0
77
95
11
Ph
Me
4-MeS-C6H4
AgBF4 (0.1)
25
<10
–
12
Ph
Me
4-Ac-C6H4
–
0
95
93
13
Ph
n-Bu
3-MeO-C6H4
AgBF4 (0.1)
0
66
99
14
Ph
iPr
3-MeO-C6H4
AgBF4 (0.1)
0
80
95
15
Ph
Cy
3-MeO-C6H4
AgSbF6 (0.05)
0
93
95
16
Ph
Ph
3-MeO-C6H4
AgBF4 (0.1)
0
86
97
17
Ph
Ph
4-Me-C6H4
–
0
91
95
18
Ph
4-MeO-C6H4
3-MeO-C6H4
AgSbF6 (0.1)
0
73
95
19
Ph
3-NO2-C6H4
3-MeO-C6H4
AgSbF6 (0.2)
0
44
92
20
4-MeO-C6H4
Ph
3-MeO-C6H4
AgBF4 (0.1)
0
75
99
21
2-naphthyl
Me
3-MeO-C6H4
AgBF4 (0.1)
0
99
96
22
2-BnO-5-Me-C6H3
Me
Ph
–
0
97
96
23
Ph
Ph
–
0
86
98
24
n-C5H11
Me
Ph
AgBF4
0
99
80
aReaction time: 48 h.
An interesting finding was that β-(2-hydroxyaryl)enones underwent cyclization to ketals (chromanols) after the addition of boronic acid. The prepared chromanols afforded the chromenes through elimination upon treatment with p-TsOH. A series of different β-(2-hydroxyaryl)enones and boronic acids was tested and provided the substituted chromenes in excellent yields (89–94%) and enantioselectivities (95–99% ee; Table 3). It is worth mentioning that a free phenolic hydroxy group did not interfere with the Pd complex and did not affect the enantioselectivity of the reaction.
Table 3:
Synthesis of chromenes by the 1,4-addition of boronic acids to β-(2-hydroxyaryl)enones [34].
entry
R1
R2
R3
Ar
additive (equiv)
yield A + B (%)
ratio A/B
yield C (%)
ee C (%)
1
H
Me
Me
Ph
–
99 (1:13)
90
96
2
H
Me
Me
4-MeO-C6H4
AgBF4 (0.1)
96 (1:13)
90
97
3
H
Me
Me
3-MeO-C6H4
AgBF4 (0.1)
96 (1:13)
94
97
4
H
Me
Me
3,4-(CH2O2)-C6H3
AgBF4 (0.1)
99 (1:16)
89
98
5
H
Me
Me
4-Me-C6H4
–
99 (1:13)
94
97
6
H
Me
Me
4-Ac-C6H4
AgBF4 (0.1)
99 (1:16)
90
96
7
H
H
Ph
Ph
–
99 (2:1)
92
99
8
H
OMe
Me
Ph
–
99 (1:16)
94
95
9
t-Bu
t-Bu
Me
Ph
–
94 (1:99)
90
–
The authors also demonstrated that the product mixture obtained after the addition of the boronic acid to the β-(2-hydroxyaryl)enone could be oxidized to afford optically pure 4-phenylchroman-2-one (Scheme 1).
Scheme 1:
Synthesis of optically pure 4-phenylchroman-2-one [34].
Scheme 1:
Synthesis of optically pure 4-phenylchroman-2-one [34].
Also in 2007, Miyaura and co-workers presented the synthesis of enantioenriched 1-aryl-1H-indenes by a tandem 1,4-addition of arylboronic acids to enones and aldol condensation [36]. The catalytic system for this transformation was adapted from earlier works [34,36] and included the addition of a 42% aqueous solution of HBF4 that facilitated consequent cyclization. A series of various β-(2-acylphenyl)enones and arylboronic acids was tested. Almost every combination provided the product in an excellent yield (60–99%) and enantioselectivity (up to 97% ee; Table 4), the only exception being the addition of an ortho-substituted boronic acid (entry 5, Table 4) [36].
Table 4:
Synthesis of enantiomerically enriched 1-aryl-1H-indenes [36].
entry
R1
R2
Ar
yield (%)
ee (%)
1
Me
Me
Ph
95
90
2
Me
Me
4-Cl-C6H4
91
90
3
Me
Me
3-Cl-C6H4
88
91
4
Me
Me
4-Me-C6H4
94
93
5
Me
Me
2-MeO-C6H4
60
24
6
Me
Me
3-MeO-C6H4
91
93
7
Me
Me
4-MeO-C6H4
90
96
8
Me
Me
3,4-(CH2O2)-C6H3
76
93
9
Me
Me
4-(4-MeO-C6H4)-C6H4
91
97
10
Me
Me
3-BnO-C6H4
90
94
11
Ph
Me
4-MeO-C6H4
99
92
12
Ph
4-MeO-C6H4
4-MeO-C6H4
79
90
13
Ph
4-MeO-C6H4
3,4-(CH2O2)-C6H3
81
90
14
Me
Et
4-MeO-C6H4
99
93
15
H
Me
4-MeO-C6H4
60
90
In 2008, the same group further expanded the substrate scope of the addition reaction to electron-rich chalcones. The products obtained after the addition reaction with arylboronic acids were further subjected to a regioselective Bayer–Villiger oxidation (Table 5) [3].
Table 5:
Stepwise addition of arylboronic acids to electron-rich chalcones and Bayer–Villiger oxidation [3].
entry
Ar1
Ar2
yield A (%)
ee A (%)
yield B (%)
ee B (%)
1
Ph
3-MeO-C6H4
99
95
73
95
2
4-iPr-C6H4
3-MeO-C6H4
90
95
0
–
3
4-MeO-C6H4
3,4-diMeO-C6H4
86
95
72
97
4
3,4-(CH2O2)-C6H3
3,4-diMeO-C6H4
74a
97
67
95
5
2-BnO-5-Me-C6H3
Ph
91 (83)b
95 (99)b
–
aReaction performed in MeOH/water 10:1 instead of acetone/water 10:1; bafter recrystallization.
An enhanced protocol for the synthesis of 4-aryldihydrocoumarins (Table 6) was also presented [3], which was already mentioned above (Scheme 1) [34].
Table 6:
Synthesis of 4-aryldihydrocoumarins by stepwise 1,4-addition and Bayer–Villiger oxidation [3].
entry
Ar
yield (%)
ee (%)
1
Ph
83
96
2
4-MeO-C6H4
75
98
3
3,4-(CH2O2)-C6H3
70
97
4
4-MeO-3,5-diMe-C6H2
74
97
Both presented methods were used in the synthesis of an antimuscarinic drug (R)-tolterodine (Scheme 2) [3].
A plausible catalytic cycle has been proposed (Scheme 3). The usual cross-coupling of an organoboron to Pd(0) requires a base. In the case of Pd(II) this reaction smoothly progresses under neutral conditions. The authors postulated that the vacancy on the square-planar Pd(II) species allows a faster alkene insertion in comparison to Pd(0). The cationic Pd(II) enolate exists as a dynamic mixture of C- and O-bound enolate and is highly susceptible to hydrolysis. This means that in the presence of water, it is selectively converted to the 1,4-addition product instead of undergoing a β-hydride elimination leading to an oxidative Heck product [3,26,35].
Scheme 3:
Catalytic cycle of the Pd(II)-catalysed 1,4-addition of organoboron reagents to enones [3,26,35].
Scheme 3:
Catalytic cycle of the Pd(II)-catalysed 1,4-addition of organoboron reagents to enones [3,26,35].
In 2005, one month after the very first report of the addition of aryltrifluoroborates to enones by Miyaura [32], the Minnaard group reported a protocol for the addition of boronic acids to enones [37]. At first, they tested the combination of Pd(OAc)2 with triflic acid (TfOH) to obtain a Pd(II) complex with a weakly coordinating anion that is necessary for a fast Pd–C bond cleavage and thus avoiding the undesired β-hydride elimination. However, the obtained yields were inconsistent. The usage of Pd(TFA)2 led to a better reproducibility of the results. From the various diphosphine ligands tested, (R,R)-MeDuPhos (L2) was identified as the one leading to the best level of enantioselectivity (up to 99% yield and up to 99% ee; Table 7) [37].
Table 7:
First report of the Pd-catalysed enantioselective addition of boronic acids to cyclic enones [37].
entry
Ar
time (h)
yield (%)
ee (%)
1
Ph
6
80
98
2
2-MeO-C6H4
18
80
99
3
2-Me-C6H4
18
>99
99
4
3-Me-C6H4
18
>99
97
5
3-MeO-C6H4
18
98
97
6
4-Me-C6H4
18
90
98
7
3-NO2-C6H4
24
0
–
8
3-Cl-C6H4
24
40
98
Furthermore, water was discovered to be a crucial additive in the reaction, increasing the yield without impact on the enantioselectivity [37]. The presented catalytic system worked well in the case of electron-rich arylboronic acids (entries 1–6, Table 7). Electron-poor arylboronic acids reacted much slower or did not react at all due to the slow transmetalation to Pd (entries 7 and 8, Table 7) [37]. The addition of phenylboronic acid (or aprotic triphenylboroxine with slow addition of water to the reaction mixture) was also tested in combination with enones differing in ring size, unsaturated lactone, N-protected dihydropyridone and one example of a linear substrate. In all cases a decreased reactivity was observed, however, good to excellent enantioselectivity levels were maintained (81–99% ee; Table 8) [37].
Table 8:
Addition of boron-derived C-nucleophiles to cyclic enones, catalysed by L2/Pd(TFA)2[37].
entry
substrate
C-nucleophile
time (h)
yield (%)
ee (%)
1
A
PhB(OH)2
6
75
82
2
B
PhB(OH)2
18
55
86
3
C
PhB(OH)2
22
60
>99
4
D
(PhBO)3
(slow addition of water)
5
75
94
5
E
(PhBO)3
(slow addition of water)
18
45(60%a)
81
aConversion.
To our best knowledge, at this time only one method for the enantioselective β-arylation of cyclic ketones is known [38]. In 2017, Hu et al. presented the possibility of an enantioselective β-arylation of cyclohexanone using the above mentioned ligand L2. Cyclohexanone was in situ oxidized by 2-iodoxybenzoic acid (IBX) to 2-cyclohexenone, that subsequently underwent addition of phenylboronic acid (Scheme 4). The complex L2/Pd(OAc)2 was used to obtain the product with excellent enantioselectivity (95% ee) but only poor yield (12%) (Scheme 4) [38].
Scheme 4:
Enantioselective β-arylation of cyclohexanone [38].
Scheme 4:
Enantioselective β-arylation of cyclohexanone [38].
A catalytic system based on L2/Pd(OAc)2 was recently used by Khatua et al. for the synthesis of ar-macrocarpenes with excellent yields and enantioselectivities (89–92%; 91–99% ee; Scheme 5) [8].
Scheme 5:
Application of L2/Pd(OAc)2 in the total synthesis of terpenes [8].
Scheme 5:
Application of L2/Pd(OAc)2 in the total synthesis of terpenes [8].
In 2007, the group of Ito described the application of ferrocenylphosphines for the palladium-catalysed addition of arylboronic acids to 2-cyclohexenone at various temperatures giving the products with high conversions but only very low enantioselectivities (25–71% ee; Table 9) [39].
Table 9:
Asymmetric addition of arylboronic acids to 2-cyclohexenone catalysed by L3/Pd(dba)2[39].
entry
Ar
temp. (°C)
yield (%)
ee (%)
1
Ph
80
82
42
2
Ph
60
83
46
3
Ph
25
79
66
4
4-Me-C6H4
80
88
61
5
4-Me-C6H4
25
90
71
6
2-Me-C6H4
80
93
25
7
3-Me-C6H4
80
63
58
The same group continued their work on this catalytic system under different reaction conditions with the cheaper base K2CO3 and without the addition of water. The observed yields were excellent (45–94%) although the enantioselectivities were only average to poor (4–79% ee; entries 1–9, Table 10). Also several linear enones were tested giving the products with varying yields (53–99%) and only moderate enantioselectivities (42–52% ee; entries 10–13, Table 10) [40]. Additionally, the authors proposed a plausible catalytic cycle for the reaction (Scheme 6) [40].
Table 10:
Additions to different enones catalysed by L3/Pd(dba)2[40].
entry
cyclic substrates
Ar
yield (%)
ee (%)
n
1
0
Ph
94
54
2
1
Ph
92
76
3
1
4-Me-C6H4
89
78
4
1
4-MeO-C6H4
83
76
5
1
4-t-Bu-C6H4
92
79
6
1
4-CF3-C6H4
81
4
7
1
4-F-C6H4
45
68
8
1
1-naphthyl
80
42
9
2
Ph
90
38
acyclic substrates
R1
R2
10
Me
Me
Ph
53
44
11
Me
Et
Ph
62
47
12
iPr
Me
Ph
70
52
13
n-C5H11
Me
Ph
99
42
Scheme 6:
Plausible catalytic cycle for the addition of phenylboronic acid to 2-cyclohexenone catalysed by L3/Pd(dba)2[40].
Scheme 6:
Plausible catalytic cycle for the addition of phenylboronic acid to 2-cyclohexenone catalysed by L3...
A different approach using microwave irradiation was explored by the group of Toma et al. [41]. After an initial tuning of the reaction conditions of a catalytic system based on Pd(OAc)2/2,2’-bipy several optically pure phosphoramidite and diphosphine ligands in combination with Pd2(dba)3·CHCl3 were tested [41]. The obtained yields were within the range of 12–37% with enantioselectivities 12–85% ee. The best level of enantioselectivity was achieved using diphosphine ligand L4 (Scheme 7). The results in terms of both yield and enantioselectivity were very poor (37%; 85% ee), but the reaction times were very short (Scheme 7) [41].
Scheme 7:
Microwave-assisted addition of phenylboronic acid to 2-cyclohexenone catalysed by L4/Pd2(dba)3·CHCl3[41].
Scheme 7:
Microwave-assisted addition of phenylboronic acid to 2-cyclohexenone catalysed by L4/Pd2(dba)3·CHCl3...
In 2011, the groups of Hayashi and Chujo studied Pd complexes of diphosphacrown ethers [42]. The macrocyclic Pd complex PdL5 in combination with AgSbF6 or AgOTf was tested for the addition reaction of various arylboronic acids to 2-cyclopentenone. In the case of the addition of phenylboronic acid, high yields and enantioselectivities were achieved (83–92% ee; entries 1–4, Table 11). However, in the case of substituted boronic acids decreased enantioselectivities were observed (72–82% ee; entries 5–8, Table 11) [42].
Table 11:
Addition of arylboronic acid on 2-cyclopentenone catalysed by PdL5[42].
entry
Ar
temp. (°C)
yield (%)
ee (%)
1
Ph
30
90
85
2
Ph
0
89
87
3
Ph
30
>99a
83
4
Ph
0
36a
92
5
4-MeO-C6H4
30
94
82
6
4-CF3-C6H4
30
91
72
7
4-Br-C6H4
30
95
78
8
2-Me-C6H4
30
94
72
aAgOTf 6 mol % instead of AgSbF6.
The most recent systematic study of phosphine-based Pd complexes was done by Wong et al. in 2014. The palladacycle PdL6 was used in combination with triphenylphosphine and K3PO4 acting as a base. The highest enantioselectivity of 99% ee of a model addition of phenylboronic acid to 2-cyclohexenone was achieved in dioxane as the solvent, but the yield was only 22%. Therefore, the authors used toluene as the best compromise between yield and enantioselectivity for the next study (Table 12). The addition reaction using the five-membered enone provided the product in moderate yield and enantioselectivity (64%; 50% ee; entry 1, Table 12). On the other hand, the addition of phenylboronic acid to six and seven-membered cycles as well as linear substrates provided the products with high yields (72–97%) and enantioselectivities (78–92% ee; entries 2, 3, 4–12, Table 12). In reactions with substituted arylboronic acids and selected acyclic enones comparable enantioselectivities were observed, while the yields were slightly lower in most cases (56–93% ee, 47–97%; entries 13–21, Table 12) [43].
Table 12:
Application of dimeric palladacycle PdL6 in the addition reactions of arylboronic acids to various enones [43].
entry
cyclic substrates
Ar
yield (%)
ee (%)
n
1
0
Ph
64
50 (S)
2
1
Ph
89
92 (R)
3
2
Ph
72
87 (R)
acyclic substrates
R1
R2
4
4-F-C6H4
Ph
Ph
88
81
5
4-Cl-C6H4
Ph
Ph
92
78
6
4-Br-C6H4
Ph
Ph
88
78
7
4-MeO-C6H4
Ph
Ph
95
81
8
4-Me-C6H4
Ph
Ph
97
81
9
4-CF3-C6H4
Ph
Ph
92
69
10
2-naphthyl
Ph
Ph
88
85
11
4-Ph-C6H4
Ph
Ph
85
79
12
3,4-(CH2O2)-C6H3
Ph
Ph
95
81
13
Ph
Me
4-Me-C6H4
63
87
14
Me
Me
Ph
56
93
15
Ph
Ph
2-naphthyl
97
77
16
Ph
Ph
4-F-C6H4
92
79
17
Ph
Ph
4-Cl-C6H4
56
82
18
Ph
Ph
4-Br-C6H4
88
56
19
Ph
Ph
4-Me-C6H4
89
69
20
Ph
Ph
4-MeO-C6H4
83
85
21
Ph
Ph
4-CF3-C6H4
47
80
Furthermore, the authors proposed a catalytic cycle (Scheme 8) [43] and stated that the rate-determining step (RDS) was the protonolysis of the O-bound enolate in the presence of PPh3 that leads to the regeneration of the catalytically active hydroxopalladium species and the addition product (Scheme 8) [43]. The presence of PPh3 ensures the preference of hydrolysis instead of a β-hydride elimination, which would lead to an oxidative Heck-type product. The authors stated that as a result of the coordination with PPh3, there is a steric hindrance disfavouring the β-hydride elimination [43].
Scheme 8:
Plausible catalytic cycle of the addition of phenylboronic acid to 2-cyclohexenone catalysed by palladacycle PdL6[43].
Scheme 8:
Plausible catalytic cycle of the addition of phenylboronic acid to 2-cyclohexenone catalysed by pal...
Historically, the second type of ligands used were N-heterocyclic carbenes (NHC). The first use was reported in a work Shi and co-workers in 2008 who studied the addition of arylboronic acids to 2-cyclohexenone catalysed by Pd complexes of axially chiral NHC carbenes with two other weakly coordinating ligands [44,45]. The complexes with acetates (PdL7a), trifluoroacetates (PdL7b), and diaquo complex (PdL7c) provided similar results in the reactions with simple enones (Table 13). The authors discussed the need for the presence of KOH as a base [44,45]. Without the base the reaction did not give any product.
Table 13:
Addition reaction of boronic acids to 2-cyclohexenone, catalysed by Pd-NHC complexes PdL7a–c[44,45].
entry
Ar
catalyst
yield (%)
ee (%)
1
Ph
PdL7a
95
93
2
Ph
PdL7b
97
96
3
Ph
PdL7c
98
95
4
3-Me-C6H4
PdL7b
97
97
5
3-Me-C6H4
PdL7c
95
92
6
4-Me-C6H4
PdL7b
89
92
7
4-Me-C6H4
PdL7c
83
90
8
3-MeO-C6H4
PdL7a
92
94
9
3-MeO-C6H4
PdL7b
90
97
10
3-MeO-C6H4
PdL7c
90
97
11
4-MeO-C6H4
PdL7b
82
84
12
2-naphtyl
PdL7a
98
96
13
2-naphtyl
PdL7b
99
97
14
2-naphtyl
PdL7c
99
96
15
4-Ph-C6H4
PdL7b
97
93
16
3-Cl-C6H4
PdL7b
78
88
17
3-Cl-C6H4
PdL7c
78
86
18
3,5-diMe-C6H3
PdL7b
90
92
19
3,5-diMe-C6H3
PdL7c
95
88
The broadening of the reaction scope showed that the catalysts were also suitable for reactions with seven-membered cyclic enones. However, the effectiveness was decreased in the case of five-membered rings or heterocyclic six-membered rings as the substrates (Table 14) [44].
Table 14:
Addition reaction of arylboronic acids to different enones catalysed by Pd-NHC complexes PdL7a–c[44,45].
entry
substrate
Ar
catalyst
yield (%)
ee (%)
1
A
Ph
PdL7a
85
94
2
A
Ph
PdL7b
88
91
3
A
Ph
PdL7c
85
94
4
A
4-Me-C6H4
PdL7b
90
91
5
A
3-MeO-C6H4
PdL7b
86
96
6
A
3-MeO-C6H4
PdL7c
84
96
7
A
2-naphthyl
PdL7a
84
96
8
A
2-naphthyl
PdL7b
99
97
9
A
2-naphthyl
PdL7c
93
94
10
B
Ph
PdL7b
53a
81
11
C
Ph
PdL7b
62a
38
12
D
Ph
PdL7b
58
32
areaction temperature 50 °C.
The unsatisfactory result obtained for substrate B (entry 10, Table 14) was overcome in the next work that focused on the optimisation of the reaction conditions for the addition of arylboronic acids to substituted dihydropyridones. Under the optimised conditions, 1,4-dioxane was used instead of THF as a solvent. The obtained results for the additions of various boronic acids to a series of alkyl 4-oxo-3,4-dihydropyridine-1(2H)-carboxylates were excellent in terms of both conversion (72–96%) and enantioselectivities (87–99% ee; Table 15) [45]. In addition, the authors proposed a catalytic cycle for this reaction (Scheme 9).
Table 15:
Addition reaction of arylboronic acids to various 4-oxo-3,4-dihydropyridine-1(2H)-carboxylates catalysed by Pd-NHC complexes PdL7a–c[45].
entry
R
Ar
catalyst
yield (%)
ee (%)
1
Bn
Ph
PdL7a
86
99
2
Bn
Ph
PdL7b
88
>99
3
Bn
Ph
PdL7c
88
>99
4
Bn
4-Me-C6H4
PdL7b
85
96
5
Bn
4-Me-C6H4
PdL7c
82
95
6
Bn
3-Me-C6H4
PdL7b
80
95
7
Bn
3-Me-C6H4
PdL7c
80
98
8
Bn
4-MeO-C6H4
PdL7b
78
>99
9
Bn
4-MeO-C6H4
PdL7c
82
>99
10
Bn
3-MeO-C6H4
PdL7b
76
99
11
Bn
3-MeO-C6H4
PdL7c
72
90
12
Bn
2-naphthyl
PdL7b
85
98
13
Bn
2-naphthyl
PdL7c
86
97
14
Bn
4-Ph-C6H4
PdL7b
94
97
15
Bn
4-Ph-C6H4
PdL7c
96
98
16
Et
Ph
PdL7b
92
87
17
Et
Ph
PdL7c
90
98
18
Et
2-naphthyl
PdL7b
85
97
19
Et
4-Ph-C6H4
PdL7b
95
97
20
t-Bu
Ph
PdL7b
82
99
21
t-Bu
Ph
PdL7c
80
98
22
t-Bu
2-naphthyl
PdL7b
80
97
23
t-Bu
4-Ph-C6H4
PdL7b
95
>99
Scheme 9:
Proposed catalytic cycle for the addition of phenylboronic acids to 2-cyclohexenone catalysed by Pd-NHC complex PdL7b[44].
Scheme 9:
Proposed catalytic cycle for the addition of phenylboronic acids to 2-cyclohexenone catalysed by Pd...
In 2013, the most recent NHC-Pd based system has been developed by Mullick et al. who used ligands derived from trans-9,10-dihydro-9,10-ethanoanthracene-11,12-diyl (DEA) and trans-9,10-dihydro-9,10-ethanoanthracene-11,12-diylmethanediyl (DEAM) in form of Pd-bisNHC complexes [46]. The catalysts were prepared in situ and tested for the addition reaction of various boronic acids to five and six-membered enones (Table 16). The results were unsatisfactory in terms of yield and enantioselectivity (24–98%; 30–51% ee) and most of the studied combinations gave no product or the authors were not able to determine the enantioselectivity. A selection of some interesting results is summarised in Table 16[46].
Table 16:
Addition reactions of boronic acids to five and six-membered enones catalysed by in situ-prepared Pd-bisNHC complex PdL8[46].
entry
n
Ar
yield (%)
ee (%)
1
0
2-Me-C6H4
36
50
2
0
2-MeO-C6H4
35
51
3
0
4-MeO-C6H4
30
35
4
0
1-naphthyl
24
30
5
1
Ph
98
51
6
1
2-Me-C6H4
62
33
7
1
1-naphthyl
48
30
Catalytic systems based on pyridine-oxazolines ligands
Currently, the most studied ligand class is focused on pyridine-oxazolines (PyOx). The first report for the use of this type of ligand for the asymmetric addition of arylboronic acids to cyclic enones was published by the Stoltz group in 2011 [47]. The most efficient catalytic system was identified as a combination of (S)-t-Bu-PyOx (L9) with Pd(TFA)2 (Table 17). This system exhibited a remarkable tolerance for water and air. It was demonstrated by the addition of 10 equiv of water into the reaction mixture that caused only a very small decrease of the enantioselectivity from 93% ee to 91% ee (entries 1 and 2, Table 17). Additional deuteration experiments demonstrated that water acted as a proton source in the catalytic cycle [48]. Furthermore, only a very low conversion was achieved without water, especially in large-scale experiments. Proton sources other than water were tested too. The use of MeOH or t-BuOH resulted in a 10 to 15% decrease of enantioselectivity and 2,2,2-trifluoroethanol (TFE) had only a minimal impact on the enantioselectivity. The benefit of using TFE instead of water was its miscibility with the reaction medium (DCE) [48].
A series of different arylboronic acids was tested for the addition reaction to 3-methyl-2-cyclohexenone (Table 17). Electron-poor arylboronic acids gave generally better enantioselectivities than electron-rich arylboronic acids [47,49].
Table 17:
Addition reaction of arylboronic acids to 3-methyl-2-cyclohexenone catalysed by L9/Pd(TFA)2[47,49].
entry
Ar
temp. (°C)
time (h)
yield (%)
ee (%)
1
Ph
60
12
99
93
2
Ph
60
12
99
91a
3
4-Me-C6H4
60
12
99
87
4
4-Et-C6H4
60
12
90
85
5
4-MeO-C6H4
40
24
58
69
6
4-BnO-C6H4
60
18
96
74
7
4-TBSO-C6H4
40
24
52
82
8
4-Ac-C6H4
60
18
99
96
9
4-Cl-C6H4
60
12
94
95
10
4-F-C6H4
80
12
84
92
11
2-F-C6H4
60
12
32
77
12
4-CF3-C6H4
60
12
99
96
13
3-Me-C6H4
60
24
99
91
14
3-Cl-C6H4
60
18
55
96
15
3-Br-C6H4
60
24
44
85
16
3-MeOOC-C6H4
60
24
91
95
17
3-NO2-C6H4
60
18
40
92
aAddition of 10 equiv of water.
Different enone substrates varying in ring size and substitution in the 3-position were also tested. The products were usually obtained with a high degree of enantioselectivity in good yields (up to 96%; up to 93% ee; Table 18) [47,49].
Table 18:
Addition reactions of phenylboronic acid to various 3-substituted enones catalysed by L9/Pd(TFA)2[47,49].
entry
n
R
yield (%)
ee (%)
1
0
Me
84
91
2
2
Me
85
93
3
1
Et
96
92
4
1
n-Bu
95
91
5
1
Bn
74
91
6
1
Cy
86
85
8
1
iPr
86
79
7
1
cyclopropyl
68
88
9
1
(CH2)3OBn
65
91
An interesting finding was the effect of non-coordinating hexafluorophosphate anions. The addition of 30 mol % NH4PF6 increased the catalytic activity and allowed to run the reaction at a lower temperature [48]. This can be very useful for substrates that can react with traces of Pd(0) that are formed by minor side reactions. The authors suspected that hexafluorophosphate anions stabilize the cationic Pd species and result in its increased solubility. The impact of the addition of 30 mol % NH4PF6 caused that the product yield was almost doubled even when the temperature was 20 °C lower (Table 19) [48], while there was only a minimal to no effect on the enantioselectivity (Table 19). Scale-up to a gram-scale was possible, without a major loss of either yield or enantioselectivity (entry 7, Table 19) [50].
Table 19:
Effect of ammonium hexafluorophosphate as additive on the addition reactions of arylboronic acids to 3-methyl-2-cyclohexanone catalysed by L9/Pd(TFA)2[48,50].
The substrate scope was further expanded with addition reactions of arylboronic acids to 3-acetyl-2-cyclohexenone. The products were isolated in moderate to good yields and excellent enantioselectivities (57–92%; 90–95% ee). Furthermore, no 2-arylated products have been detected (Table 20) [49].
Table 20:
Addition reactions of arylboronic acids to 3-acetyl-2-cyclohexanone catalysed by L9/Pd(TFA)2[49].
entry
n
Ar
yield (%)
ee (%)
1
1
4-Cl-C6H4
85
96
2
1
4-F-C6H4
92
90
3
1
3-Me-C6H4
66
92
4
1
3-(CF3CONH)-4-Me-C6H3
73
91
5
0
Ph
72
93
6
0
3-Me-C6H4
72
90
7
0
4-F-C6H4
57
92
Next, the substrate scope was further expanded with the addition reactions of N-protected aminophenylboronic acids. The best results in terms of enantioselectivity were achieved when trifluoroacetyl was used as the N-protecting group (Table 21) [49].
Table 21:
Addition reactions of N-protected aminophenylboronic acids to 3-methyl-2-cyclohexanone catalysed by L9/Pd(TFA)2[49].
entry
Ar
yield (%)
ee (%)
1
4-(Cbz-NH)-C6H4
45
76
2
4-(Boc-NH)-C6H4
72
78
3
4-(CF3CONH)-C6H4
98
89
4
4-(CF3CONH)-3-Me-OC6H3
75
88
5
4-(CF3CONH)-3,5-diMeO-C6H2
93
90
6
3-(CF3CONH)-C6H4
60
92
7
3-(CF3CONH)-4-MeO-C6H3
77
88
In other experiments, Stoltz and co-workers showed the ineffectiveness of the L9/Pd(TFA)2 catalytic system for the addition of phenylboronic acid to nonsubstituted 2-cyclohexenone, yielding the product with very low enantioselectivity (18%; entry 1, Table 22). Furthermore, the addition reaction to a 6,6,3-trimethylated substrate gave the product in only very low yield (9%), but with high enantioselectivity (90% ee; entry 3, Table 22) [48]. The application of the catalytic system in the addition reaction to an unsaturated lactone yielded the product with both low yield and enantioselectivity (49%; 57% ee; entry 4, Table 22) [48]. Finally, the catalytic system failed in the addition reaction with 2-methylchromone and did not yield the expected product, however, it proved to be highly effective for the addition reaction to unsubstituted chromone (91%; 94% ee; entry 5, Table 22) [51].
Table 22:
Addition reaction of phenylboronic acid to various enones, lactones, and chromones catalysed by L9/Pd(TFA)2[48,51].
entry
substrate
yield (%)
ee (%)
1
A
87 (no NH4PF6)
18
2
B
99
93
3
C
9a
90
4
D
49a
57
5
E
91
94
6
F
0
–
aReaction temperature 40 °C.
According to these findings, Stoltz and co-workers tested the catalytic system with a library of different chromones for the addition of various boronic acids. The substituted flavanones were obtained with moderate to good yields (36–96%) and usually very high levels of enantioselectivity (up to 98% ee; entries 1–29, Table 23) [51]. Also, the addition reaction to the structurally similar N-Cbz-4-quinolone was tested, resulting in the corresponding products with only low to moderate yields (31–65%) and moderate to good enantioselectivities (40–89% ee; entries 30–38, Table 23) [51].
Table 23:
Addition reactions of arylboronic acids to substituted chromones and N-Cbz-4-quinolones catalysed by L9/Pd(TFA)2[51].
entry
X
R
Ar
yield (%)
ee (%)
1
O
H
Ph
91
94
2
O
H
2-F-C6H4
50
76
3
O
H
3-Me-C6H4
66
90
4
O
H
3-MeOOC-MeC6H4
72
93
5
O
H
3-Br-C6H4
40
89
6
O
H
3-(CF3CONH)-C6H4
77
98
7
O
H
3-Cl-C6H4
52
94
8
O
H
4-Me-C6H4
64
94
9
O
H
4-Et-C6H4
36
85
10
O
H
4-F-C6H4
51
90
11
O
H
3,5-diMeO-C6H3
69
95
12
O
H
dibenzofuran-4-yl
64
77
13
O
6-Ac-5,7-diMe
Ph
98
90
14
O
6-Ac-5,7-diMe
3-Me-C6H4
76
88
15
O
6-Ac-5,7-diMe
4-Et-C6H4
45
86
16
O
6-Ac-5,7-diMe
Ph
79
95
17
O
6-Ac-5,7-diMe
3-Me-C6H4
84
86
18
O
6-Ac-5,7-diMe
3-Br-C6H4
65
95
19
O
6-Ac-5,7-diMe
4-F-C6H4
68
91
20
O
6-Ac-5,7-diMe
3-MeOOC-C6H4
90
86
21
O
6-Ac-5,7-diMe
dibenzofuran-4-yl
70
83
22
O
5,7-diMe
Ph
84
93
23
O
5,7-diMe
4-(CF3CONH)-3-MeO-C6H3
80
95
24
O
7-OAc
Ph
77
92
25
O
7-OH
Ph
77
93
26
O
7-OH
3-Me-C6H4
66
90
27
O
7-OH
4-F-C6H4
50
93
28
O
7-MeO
Ph
96
94
29
O
7-MeO
3-MeOOC-C6H4
81
96
30
NCbz
H
Ph
50
80
31
NCbz
H
3-(CF3CONH)-4-Me-C6H3
45
85
32
NCbz
H
3-Me-C6H4
51
85
33
NCbz
H
3,5-diMeO-C6H3
50
85
34
NCbz
H
3-MeOOC-C6H4
34
60
35
NCbz
H
4-F-C6H4
65
89
36
NCbz
H
4-Me-C6H4
45
67
37
NCbz
H
4-MeO-C6H4
36
54
38
NCbz
H
dibenzofuran-4-yl
31
40
In 2018, Wang et al. applied the optimised reaction conditions for the synthesis of various compounds that could be potentially usable for the treatment of cystic fibrosis (Scheme 10) [5].
Scheme 10:
Usage of addition reactions of boronic acids to various chromones in the syntheses of potentially active substances in medicinal chemistry [5].
Scheme 10:
Usage of addition reactions of boronic acids to various chromones in the syntheses of potentially a...
In 2019, another expansion of the substrate scope for the synthesis of substituted flavanones was done by Liu et al. (Table 24). The prepared flavanones were further tested for their cancerostatic activity [7].
Table 24:
Addition reactions of arylboronic acids to substituted chromones catalysed by L9/Pd(TFA)2[7].
entry
R
Ar
yield (%)
ee (%)
1
H
Ph
88
94
2
H
3,4-diMeO-C6H3
58
89
3
H
4-MeO-C6H4
68
95
4
H
3-MeO-C6H4
62
86
5
H
3,4,5-triOMe-C6H2
70
92
6
H
piperonyl
59
89
7
H
4-NO2-C6H4
52
77
8
H
4-Me-C6H4
63
91
9
H
3-Me-C6H4
70
83
10
H
4-Cl-C6H4
50
96
11
H
3-Cl-C6H4
58
92
12
H
4-Br-C6H4
49
86
13
H
4-F-C6H4
46
75
14
H
1-naphthyl
59
78
15
H
2-furyl
55
74
16
H
thiophene-2-yl
45
87
17
H
4-Me2N-C6H4
43
83
18
H
4-Et-C6H4
58
77
19
H
4-MeS-C6H4
72
90
20
H
4-t-Bu-C6H4
66
91
21
7-MeO
4-MeO-C6H4
76
90
22
7-OBn
4-MeO-C6H4
83
74
23
7-Br
4-MeO-C6H4
70
93
24
7-F
4-MeO-C6H4
52
66
25
7-Me
4-MeO-C6H4
80
82
26
6-Cl-7-Me
4-MeO-C6H4
68
79
27
7-Cl-6-Me
4-MeO-C6H4
57
70
28
6-Cl
4-MeO-C6H4
70
95
29
6-Br
4-MeO-C6H4
59
76
30
6-F
4-MeO-C6H4
60
80
31
6-MeO
4-MeO-C6H4
87
94
32
6-Me
4-MeO-C6H4
44
79
33
6-NO2
4-MeO-C6H4
67
95
34
6,7-diMeO
4-MeO-C6H4
48
85
35
5-MeO
4-MeO-C6H4
75
94
36
5,7-diOMe
4-MeO-C6H4
65
89
37
6,8-diCl
4-MeO-C6H4
83
93
38
benzo[f]
4-MeO-C6H4
88
77
39
5,7-bis(MEM)
4-MeO-C6H4
74
88
40
7-OCH2OMe
4-MeO-C6H4
47
81
41
5,7-diOH
4-MeO-C6H4
86
–
42
5-OH
4-MeO-C6H4
89
–
In 2019, Timmerman et al. applied the asymmetric addition of phenylboronic acid to a chromone derivative for the total syntheses of (−)-caesalpinnone A and (−)-caesalpinflavan B (Scheme 12) [9].
Scheme 12:
Application of the asymmetric addition of phenylboronic acid to a chromone derivative for the total syntheses of the natural products (−)-caesalpinnone A and (−)-caesalpinflavan B [9].
Scheme 12:
Application of the asymmetric addition of phenylboronic acid to a chromone derivative for the total...
Mechanistic studies of this catalytic system were also made by Stoltz’s group. A linear relationship between the ee of the catalyst and the product has been found [48]. That means that the catalytically relevant species is monomeric Pd–PyOx. This was further supported by a mass spectrometric study [52]. The catalytic cycle was also suggested in accordance with DFT calculations and mechanistic studies (Scheme 13) [48,49]. The key step for both, the enantioselectivity and turnover, is the migratory insertion via TS1 (Scheme 13). The stereochemistry is controlled mainly by the hydrogen repulsion of the methylene group neighbouring the keto group of the enone with the t-Bu group of the ligand L9.
Scheme 13:
Plausible catalytic cycle for the addition of phenylboronic acid to 3-methyl-2-cyclohexenone catalysed by L9/Pd(TFA)2[48,49].
Scheme 13:
Plausible catalytic cycle for the addition of phenylboronic acid to 3-methyl-2-cyclohexenone cataly...
Another interesting example for the application of this reaction in the preparation of precursors of natural molecules was reported by Li et al. in 2014. They presented the synthesis of terpenoid precursors ((+)-taiwaniaquinone H and (+)-dichroanone) [10] starting from 3-methyl-2-cyclohexenone using the L9/Pd(TFA)2 catalytic system. The precursors were prepared in good yields (42–98%) with high enantioselectivities (85–99% ee; entry 1; Table 25) and used in the total synthesis of terpenoids (Scheme 14) [10].
Table 25:
Addition of various highly functionalized arylboronic acids to 3-methylcyclohexanone for the synthesis of terpenoids [10,11].
entry
R1
R2
R3
yield (%)
ee (%)
1
MeO
H
iPr
89a
85
2
MeO
MeO
iPr
trace
–
3
PivO
PivO
Ac
93
94
4
PivO
PivO
I
42
92
5
PivO
PivO
Br
98
>99
6
PivO
PivO
Cl
94
>99
aReaction performed at 60 °C for 48 h.
In the same year, these terpenoids were also prepared by the Stoltz group [11]. Arylboronic acids bearing the appropriate functional groups were identified and the addition reactions to 3-methyl-2-cyclohexenone were studied (entries 2–6, Table 25) [11]. The product, which was obtained in an almost quantitative yield and practically maximal possible enantioselectivity (entry 5 in Table 25), was subsequently converted to suitable intermediates for the synthesis of naturally occurring terpenoids (Scheme 14) [11].
Scheme 14:
Total syntheses of naturally occurring terpenoids [10,11].
Scheme 14:
Total syntheses of naturally occurring terpenoids [10,11].
Another possible use of this catalytic system was demonstrated by the groups of Lautens and Hashmi [4]. The starting enone, prepared by the Au(I)-catalysed Rautenstrauch rearrangement, was subjected to the addition reaction with phenylboronic acid (Scheme 15). Without isolation of the intermediate, the protecting group was removed and the product was obtained in 88% yield and 80% ee. The enantiomeric excess of the obtained (S)-3-(hydroxymethyl)-3-phenyl-2-cyclopentanone could be increased by double recrystallization to up to 97% ee (Scheme 15) [4].
Scheme 15:
Use of the L9/Pd(TFA)2 catalytic system for the synthesis of intermediates of biologically active compounds [4].
Scheme 15:
Use of the L9/Pd(TFA)2 catalytic system for the synthesis of intermediates of biologically active c...
The catalytic system L9/Pd(TFA)2 was further used in the work published in 2020 by Bisai et al. for the addition of 4-tolylboronic acid to 3-methyl-2-cyclohexenone in the total synthesis of the aromatic sesquiterpene (−)-ar-tenuifolene (Scheme 16) [12].
Scheme 16:
Usage of a Michael addition catalysed by L9/Pd(TFA)2 in the total synthesis of (–)-ar-tenuifolene [12].
Scheme 16:
Usage of a Michael addition catalysed by L9/Pd(TFA)2 in the total synthesis of (–)-ar-tenuifolene [12].
Later in 2020, Bisai et al. published the application of the L9/Pd(TFA)2 catalytic system for the preparation of the enantiomers of other sesquiterpenoids by the addition reactions of tolylboronic acids to 3-methyl-2-cyclopentenone (Scheme 17) [13].
Scheme 17:
Synthesis of terpenoids by Michael addition to 3-methyl-2-cyclopentenone [13].
Scheme 17:
Synthesis of terpenoids by Michael addition to 3-methyl-2-cyclopentenone [13].
Also in 2020, Ochi et al. expanded the synthetic usability of 3-alkyl-3-arylcyclopentanones by developing a method for their Rh-catalysed isomerisation to 1-tetralones with >99% stereoretention (Scheme 18) [53].
Scheme 18:
Rh-catalysed isomerisation of 3-alkyl-3-arylcyclopentanones to 1-tetralones [53].
Scheme 18:
Rh-catalysed isomerisation of 3-alkyl-3-arylcyclopentanones to 1-tetralones [53].
To obtain the starting material for the transformation (Scheme 18), the authors have described the addition of arylboronic acids to 3-substituted-2-cyclopentenones (Table 26) either by using Stoltz’s catalytic system L9/Pd(TFA)2 or by its simple modification (temperature, catalyst loading) combined with the iterative addition of boronic acids (1 equiv immediately and 1 equiv after 3 hours) [49].
Table 26:
Addition reactions of arylboronic acids to 3-alkyl-2-cyclopentenones catalysed by L9/Pd(TFA)2[53].
entry
R
Ar
temp. (°C)
conditions
yield (%)
ee (%)
1
Et
Ph
25
A
95
94
2
Et
4-Me-C6H4
25
B
67
91
3
Et
4-MeO-C6H4
25
A
49
84
4
Et
4-MeO-C6H4
60
B
63
74
5
Et
4-Bu-C6H4
60
B
91
84
6
Et
4-Cl-C6H4
60
B
78
93
7
Et
4-F-C6H4
60
B
84
92
8
Et
4-CF3-C6H4
25
A
6
95
9
Et
4-CF3-C6H4
60
B
99
94
10
Et
4-MeOOC-C6H4
60
B
99
94
11
Et
3-Me-C6H4
60
B
97
91
12
Bu
Ph
25
A
82
96
13
Cy
Ph
60
A
91
96
14
(CH2)2COOMe
Ph
60
A
86
97
Following Stoltz's works [11,27,47-49,51,52], Stanley et al. published the first example for the formation of all-carbon quaternary stereocentres, in an aqueous medium (Scheme 19) [54] by the addition of phenylboronic acid to 3-methyl-2-cyclohexenone using the L9/Pd(TFA)2 catalytic system. Compared to the reaction in DCE (93% yield, 92% ee,) [47], a slightly lower yield and significantly lower enantioselectivity were obtained in water as the solvent (86% yield, 71% ee, Scheme 19) [54].
Scheme 19:
Addition reaction of phenylboronic acid to 3-methyl-2-cyclohexenone catalysed by L9/Pd(TFA)2 in water [54].
Scheme 19:
Addition reaction of phenylboronic acid to 3-methyl-2-cyclohexenone catalysed by L9/Pd(TFA)2 in wat...
Significant successes of the Stanley group were achieved in the subsequent study of the as yet unexplored asymmetric addition of arylboronic acids to 3-aryl-2-cyclohexenones, where double benzyl quaternary stereogenic centres were formed [55]. The initial studies showed the formation of significant amounts of protodeborylation products, small amounts of boronic acid homocoupling products, and the corresponding phenols as boronic acid oxidation products. To optimise the yields, the amount of the boronic acid was increased to 3 equiv, which was added gradually (1 equiv every 3 hours) [55]. The authors presented interesting results and expanded the range of compounds that could be prepared by this methodology. The obtained results were excellent both in terms of enantioselectivity (up to 91% ee) and conversion (92%; Table 27) [55].
Table 27:
Addition reactions of arylboronic acids to 3-aryl-2-cyclohexenones catalysed by L9/Pd(TFA)2[55].
entry
n
Ar
R
yield (%)
ee (%)
1a
1
4-MeO-C6H4
4-Me
83 (81)b,c
89 (87)b,c
2a
1
4-MeO-C6H4
H
70c
87c
3
1
4-MeO-C6H4
4-Ph
92
90
4
1
4-MeO-C6H4
4-Cl
55
83
5
1
4-MeO-C6H4
4-F
49
91
6
1
4-MeO-C6H4
4-COOMe
39
87
7
1
4-MeO-C6H4
4-CF3
38
82
8
1
4-MeO-C6H4
3-Me
88
90
9
1
4-MeO-C6H4
3-MeO
60
90
10
1
4-MeO-C6H4
3-Cl
35
85
11
1
4-MeO-C6H4
3-F
18
84
12
1
4-MeO-C6H4
2-F
23
81
13
1
4-MeO-C6H4
3-F-4-MeO
66
88
14
1
4-MeO-C6H4
3,4-(CH2O2)
44
90
15
1
4-MeO-C6H4
3,4-diMe
36c
85
16
1
4-MeO-C6H4
3,5-diMe
38c
90
17
1
4-MeO-C6H4
3,4,5-triMeO
67
78
18
1
Ph
4-Me
70
87
19
1
4-NMe2-C6H4
4-Me
36
91
20
1
4-F-C6H4
4-Me
74
89
21
1
4-CF3-C6H4
4-Me
54
90
22
1
3-MeO-C6H4
4-Me
72
93
23
1
2-MeO-C6H4
4-Me
28
80
24
1
1H-indol-3-yl
4-Me
41
77
25
1
Ph
3-Me
76
88
26
1
Ph
4-MeO
44
80
27
1
4-Me-C6H4
H
70c
88
28
0
4-MeO-C6H4
4-Me
60
87
a5 mol % Pd catalyst were used; bon a 1 mmol scale; cin the presence of 5 equiv H2O.
In 2018, the very first heterogeneous catalytic system for the addition of arylboronic acids to cyclic enones was introduced by O’Reilly and co-workers [56]. The micellar nanoreactor was tested for the preparation of flavanones. The main advantages of such catalytic system were short reaction times in an aqueous medium and with a very small amount of the catalyst needed (Table 28). The heterogeneous catalyst PdL10b system worked significantly better than the conventional homogeneous synthesis, even when using a significantly higher amount of the PdL10a catalytic species in the homogeneous system. The results were excellent both in terms of enantioselectivities and conversions (up to 98%; up to 83% ee; Table 28). The reuse of the heterogeneous catalyst has not been studied in this case.
Table 28:
Micellar nanoreactor for the synthesis of substituted flavanones [56].
homogeneous system with PdL10a
heterogeneous system with PdL10b
R
Ar
entry
time (h)
yield (%)
ee (%)
entry
time (h)
yield (%)
ee (%)
H
Ph
1
24
98
84
5
24
90
80
H
Ph
2
24
95a
79
6
92
94
82
H
4-Cl-C6H4
3
24
94
81
7
24
68
76
6-Cl
4-Cl-C6H4
4
24
80
83
8
24
32
71
a30 mol % NH4PF6.
In 2020, our group reported the first heterogeneous polystyrene-supported recyclable catalyst for the asymmetric conjugate additions of arylboronic acids to five and six-membered enones (Table 29) [57]. For most of the substrates, the enantioselectivity was similar to the values reported for the homogeneous L9/Pd(TFA)2 system. The conversions obtained were a bit worse, especially for the more sterically demanding boronic acids (Table 29).
Table 29:
Polystyrene-supported Pd complex PdL11 as catalyst for addition reactions of arylboronic acids to cyclic enones [57].
Under the optimised conditions, we were able to use the catalyst in 6 runs with no significant drop in the enantioselectivity and only a small decrease in the conversion (Table 30). The main issues with transferring into heterogeneous conditions were the impossibility of using water as a proton source and the observed reduction of Pd(II) to Pd(0). HFIP was used as a proton source instead and Pd(0) was reoxidised to Pd(II) by p-chloranil between the individual cycles. The ratio PS-PyOx:Pd(TFA)2 showed a crucial role in the enantioselectivity. Using a higher excess of PS-PyOx allowed achieving a higher ee, however, it also caused a faster loss of catalytic activity.
Table 30:
Recyclisation of the polystyrene-supported Pd complex PdL11[57].
conversion % (ee %)
cycle
1st
2nd
3rd
4th
5th
6th
PyOx:Pd
ratio
1:2
95 (70)
95 (80)
84 (82)
89 (82)
66 (83)
96 (83)a
PyOx:Pd
ratio
2:1
93 (89)
54 (90)
PyOx:Pd
ratio
1.3:1
99 (73)
90 (87)a
99 (88)a
89 (89)a
54 (89)a
69 (87)a
aReoxidation with p-chloranil before cycle.
Later in 2020, Zhou et al. used an analogous heterogeneous system as O’Reilly (cf. Table 28) [56,58]. A RAFT polymerisation reaction, in this case, led to a polymeric backbone with terminal catalytic centres [58] (Scheme 20). The results obtained were consistent with those reported by O’Reilly using a polymeric backbone with catalytic centres inside the chain [56].
Scheme 20:
Micellar nanoreactor PdL10c for the synthesis of flavanones [58].
Scheme 20:
Micellar nanoreactor PdL10c for the synthesis of flavanones [58].
The authors outlined the possibility of recycling the catalyst based on the lower critical solution temperature (LCST) of the catalytic polymer system. The catalyst precipitated and was recovered by centrifugation and discarding the supernatant liquid. This process was complicated by a low catalyst loading and high phase-transition temperature leading to the loss of mass during this procedure. The authors, however, did not try the preparation of a polymer with a lower phase-transition temperature. The loss of mass was compensated by the addition of 10% of fresh catalyst. By this method, they were able to reuse the catalyst in 6 cycles with only a very small decrease in the yield (98, >97, >97, >96, >95, >91%). Unfortunately, the enantioselectivity was not estimated after each cycle [58].
In 2019, Lee et al. focused on the enantioselective desymmetrisation of polycyclic cyclohexenediones [59]. The variously substituted pyridine-oxazolines L9 and L12a,b were tested as ligands in combination with Pd(OAc)2 or Pd(TFA)2 (Table 31). As a suitable solvent was chosen DMF, although the use of polar aprotic solvents usually leads to products of the oxidative Heck reaction. The authors noticed a significant reduction of Pd(II) to Pd(0) (by secondary processes such as oxidative homocoupling or oxidation of boronic acid to the corresponding phenol). The Pd(0) reduced in this way was reoxidized to Pd(II) by adding oxygen to the reaction mixture. Excellent enantiomeric excesses were observed (80–96% ee), but the conversions were low (13–83%), especially for boronic acids with electron-acceptor substituents (Table 31). The authors also proposed a plausible catalytic cycle as outlined in Scheme 21[59].
Table 31:
Addition reactions of various boronic acids to polycyclic cyclohexenediones [59].
entry
conditions
substrate
Ar
yield (%)
ee (%)
1
I
A
4-MeO-C6H4
80a
84
2
I
A
4-HO-C6H4
65
80
3
I
B
4-MeO-C6H4
70
94
4
I
B
3-MeO-C6H4
58
94
5
I
B
2-MeO-C6H4
46b
84
6
I
B
4-HO-C6H4
65
96
7
I
B
Ph
83b
94
8
I
B
4-Me-C6H4
81b
94
9
I
B
3-Cl-4-MeO-C6H4
51b
94
10
I
B
4-F-C6H4
57b (80)c
88
11
I
B
4-(AcNH)-C6H4
42b,d (60)c
96
12
I
B
4-EtOOC-C6H4
13e
90
13
I
C
4-MeO-C6H4
73b
86
14
IIa
D
4-MeO-C6H4
64
90
15
I
E
4-MeO-C6H4
43b
94
16
IIa
E
4-MeO-C6H4
68
90
17
I
F
4-MeO-C6H4
68
88
18
I
G
4-MeO-C6H4
72 (60)f
84 (86)f
19
IIb
H
4-HO-C6H4
65
70
aTemperature 30 °C; bL9 11 mol % and Pd(OAc2) 10 mol %; cNMR yield; dtime 92 h; etemperature 50 °C and double amount of catalyst (50% added at the beginning, 50% added after 24 h); f10× larger amount (1 mmol).
Scheme 21:
Plausible catalytic cycle for the desymmetrisation of polycyclic cyclohexenediones by the addition of arylboronic acids [59].
Scheme 21:
Plausible catalytic cycle for the desymmetrisation of polycyclic cyclohexenediones by the addition ...
The latest ligand derived from pyridine-oxazolines is β-carbolino-oxazoline, whose Pd(II) complex was studied mainly as a catalyst for the addition of arylboronic acids to nitrostyrenes. It also showed to be a highly active catalyst for the addition to enones, under conditions similar to those developed by Stoltz et al. for pyridine-oxazolines (Table 32) [60].
Table 32:
Addition reactions of arylboronic acids to 3-methyl-2-cyclohexenone catalysed by L13/Pd(TFA)2[60].
entry
Ar
yield (%)
ee (%)
1
Ph
88
95
2
4-MeO-C6H4
75
70
3
4-Me-C6H4
72
91
4
1-naphthyl
88
89
5
4-CF3-C6H4
86
96
6
4-F-C6H4
81
95
7
3-Me-C6H4
73
88
8
3-Cl-C6H4
88
99
Catalytic systems based on bisoxazoline ligands
In 2012, the Minnaard group followed up their pioneering work with the phosphine ligand L2 to expand the substrate scope to 3-substituted enones [14]. At first, they have tried their original catalytic system L2/Pd(TFA)2 for the addition of phenylboronic acid to 3-methyl-2-cyclohexenone (Scheme 22) that provided the product with an excellent enantioselectivity of 96% but in a very poor yield <5%.
Scheme 22:
Attempt to use the catalytic system L2/Pd(TFA)2 for the addition of phenylboronic acid to 3-methyl-2-cyclohexenone [14].
Scheme 22:
Attempt to use the catalytic system L2/Pd(TFA)2 for the addition of phenylboronic acid to 3-methyl-...
The previously used ligand was changed to bisoxazoline L14. At first, they tested in situ-generated complexes of L14 and Pd(TFA)2 in methanol or acetone, but the reduction to catalytically inactive Pd(0) occurred faster. The reoxidation by Cu(BF4)2·6H2O led to the loss of enantioselectivity presumably because of the complexation of the bisoxazoline by Cu(II). This problem could be solved by using a higher amount of the ligand (27 mol %) [14].
The second more favourable solution was the preparation of the bisoxazoline complex with PdCl2 followed by dehalogenation. The use of AgSbF6 as the dehalogenating agent allowed the complete conversion in the model reaction with a high ee of 96% (entry 3, Table 33). Also the addition reactions to five and six-membered 3-substituted enones proceeded smoothly in most cases (entries 1–11, Table 33), providing the products with remarkable enantioselectivities. The only exceptions were ortho-substituted arylboronic acids, which did not react at all (entries 12 and 13, Table 33) [14].
Table 33:
Addition reactions of arylboronic acids to various enones catalysed by palladium bisoxazoline complex PdL14[14].
entry
substrate
Ar
yield (%)
ee (%)
1
A
Ph
93
93
2
A
4-Me-C6H4
68
90
3
B
Ph
100
96
4
B
3-Me-C6H4
89
97
5
B
4-Me-C6H4
96
97
6
B
4-F-C6H4
88
98
7
B
3-EtO-C6H4
44
93
8
B
3-Cl-C6H4
30a
98
9
B
3-Cl-4-MeO-C6H3
98
>99
10
B
4-MeO-C6H4
85
98
11
B
3,4-(CH2O2)-C6H3
98
96
12
B
2-Me-C6H4
0
–
13
B
ferrocenyl
0
–
14
C
Ph
80
94
15
D
Ph
91
99
16
E
Ph
0
–
17
F
Ph
28
69
18
G
Ph
57
88
a60 °C.
A substituent on the enone in position 3 significantly affected the reactivity (entries 3, 15, and 16, Table 33). In the case of dihydropyranone derivatives (entries 17 and 18, Table 33), the reactivity depended on the position of the oxygen in the ring. The tight geminal arrangement of oxygen with the reaction centre reduced the reactivity and enantioselectivity more than in the more distant arrangements. The substrate scope was expanded to 3-substituted linear enones, but the yields were only poor to good (up to 84%) and the enantioselectivities were low to moderate (up to 60% ee; Table 34) [14].
Table 34:
Addition reactions of arylboronic acids to linear enones catalysed by the bisoxazoline complex PdL14[14].
entry
substrate configuration
R
yield (%)
ee (%)
1
E
Ph
14
8
2
E
t-Bu
<10
–
3
E
t-BuO
84
23
4
E
BnO
81
25
5
Z
BnO
78
36
6
E
TBDPSO
38
60
7
E
TrO
53
51a
8
E
TIPSO
68
27a
aDetermined after ring opening of the ketal.
Another option to obtain the linear product is the ring opening of the addition product of the arylboronic acid to the dihydropyran-2-one derivative (Scheme 23) [14].
Scheme 23:
Ring opening of an enantioenriched tetrahydropyran-2-one derivative as alternative strategy to linear products [14].
Scheme 23:
Ring opening of an enantioenriched tetrahydropyran-2-one derivative as alternative strategy to line...
The Minnaard group next focused on the increase of the reactivity of ortho-substituted boronic acids [14,15]. An optimisation study showed that the presence of AgTFA (dehalogenation reagent) and NH4PF6 (Pd(II) stabilizing salt) in the reaction mixture was necessary. Additionally, the solvent was changed from a methanol/water mixture to a DCE/water biphasic system. It was also necessary to use a high excess of the starting enone (7 equiv). The results are summarised in Table 35 and it is clear that the yields for most of the cases were very low and exceeded 30% in only a few cases (mostly when a high catalyst amount was used). On the other hand, the enantioselectivities were excellent in almost every example (Table 35) [14,15].
Table 35:
Addition reactions of ortho-substituted arylboronic acids to five and six-membered enones [14,15].
entry
n
Ar
yield (%)
ee (%)
1
0
2-Me-C6H4
23
90
2
1
2-Me-C6H4
16
98
3
0
2-MeO-C6H4
45
80
4
1
2-MeO-C6H4
42a
96
5
0
2-F-C6H4
20
95
6
1
2-F-C6H4
23a
95
7
0
2-Cl-C6H4
12a
94
8
1
2-Cl-C6H4
<10
–
9
0
dibenzofuran-4-yl
51
94
10
1
dibenzofuran-4-yl
36
94
11
0
1-naphthyl
38a
85
12
1
1-naphthyl
26
95
13
0
2,3-diOMe-5-Me-C6H2
55a
92
14
1
2,3-diOMe-5-Me-C6H2
19
94
15
0
2,3-diMeO-C6H3
25
94
16
1
2,3-diMeO-C6H3
44
99
17
0
2-MeO-5-Me-C6H3
32a
80
18
1
2-MeO-5-Me-C6H3
28
91
19
0
2,5-diMeO-4-Me-C6H2
21a
74
20
1
2,5-diMeO-4-Me-C6H2
<10
84
21
0
2-MeO-4-Me-C6H3
<10
68
22
1
2-MeO-4-Me-C6H3
17
90
*8 mol % PdL14 used.
Selected addition products were used as intermediates in the total syntheses of various biologically active compounds (Scheme 24) [14-16].
Scheme 24:
Synthesis of biologically active compounds from addition products [14-16].
Scheme 24:
Synthesis of biologically active compounds from addition products [14-16].
Catalytic systems based on different groups of ligands
The use of the chiral 1,10-phenanthroline ligand L15 for the addition of phenylboronic acid to 2-cyclohexenone and chromone (Scheme 25) [61] was proposed by Tamura et al. in 2017. Excellent conversions and enantioselectivities (96–97%; 94–97% ee) were achieved for both studied substrates. However, a further use of this ligand has not been published yet.
Scheme 25:
Chiral 1,10-phenantroline derivative L15 as ligand for the Pd-catalysed addition reactions of phenylboronic acid to 2-cyclohexenone and chromone [61].
Scheme 25:
Chiral 1,10-phenantroline derivative L15 as ligand for the Pd-catalysed addition reactions of pheny...
Optically pure pyridine-hydrazones were successfully used for a number of various enantioselective transformations [62]. In 2019, Retamosa et al. used them for 1,4- and 1,6-addition reactions of boronic acids to cyclic (di)enones. Initial studies showed the best yields when DCE was used as a solvent upon the addition of 0.2 equiv of water [62]. Without the addition of water, no reproducible results were obtained. The addition of 1.1–1.5 equiv of water caused a minimal decrease of the enantioselectivity from 91 to 88% ee (entries 1 and 2, Table 36) [62].
Table 36:
Addition reactions of arylboronic acids to five and six-membered enones catalysed by L16/Pd(TFA)2[62].
entry
n
R
Ar
time (h)
yield (%)
ee (%)
1
1
Me
Ph
24
94
91
2
1
Me
Ph
24
90a
88a
3
1
Me
4-Me-C6H4
48
93
91
4
1
Me
4-F-C6H4
72
43
90
5
1
Me
4-Cl-C6H4
72
77
90
6
1
Me
4-MeO-C6H4
72
73
90
7
1
Me
4-CF3O-C6H4
72
65
90
8
1
Me
3,5-diMe-C6H3
24
75
92
9
1
Et
Ph
48
80
89
10
1
Ph
4-MeO-C6H4
72
0
–
11
1
H
Ph
48
76
87
12
0
Me
Ph
20
95
88
13
0
Me
2-MeO-C6H4
48
73
91
14
0
Me
4-Me-C6H4
48
97
88
15
0
Me
3,4-(CH2O2)C6H3
60
65b
86b
16
0
Me
2,5-diOMe-4-MeC6H2
72
38
93
a1.1 equiv of water used; bL16 9 mol % and Pd(TFA)2 7.5 mol % were used.
For the whole series of different substrates and boronic acids, there were enantioselectivities of about 90% ee and average to excellent yields of 43–97% (Table 36) [62]. This catalytic system worked for 3-unsubstituted enones but was much more powerful in the case of addition reactions to 3-substituted enones that lead to all-carbon quaternary stereogenic centres [62].
In the case of 1,6-additions, the amount of the starting dienones was increased to 4.17 equivalents relative to the boronic acids. Further, the boronic acid was gradually added over 12 hours and then the mixture was kept under the reaction conditions for another time period up to total 72 or 96 h. The prolonged reaction time increased the obtained yields but at the expense of reducing the enantioselectivity of the product (61 to 81%; 79 to 67% ee; entries 1 and 2, Table 37). This led to the conclusion that the ligand is not chemically stable in the reaction medium and undergoes decomposition over time. Only low to average conversions (up to 81%) and only average enantioselectivities (up to 80% ee; Table 37) were achieved for the studied substrates [62].
Table 37:
1,6-Addition reaction of arylboronic acids to dienones catalysed by L16/Pd(TFA)2[62].
entry
R
Ar
time (h)
yield (%)
ee (%)
1
Me
Ph
72
61
79
2
Me
Ph
96
81
67
3
Me
4-Me-C6H4
72
44
74
4
Me
4-Me-C6H4
96
78
68
5
Me
4-CF3O-C6H4
72
35
80
6
Me
4-CF3O-C6H4
96
47
72
7
n-Bu
Ph
72
31
52
One of the most recent contributions to this topic came from the group of Hong and Stoltz in 2020. Here, attention was focused on the development of a methodology for the enantioselective addition to 2-substituted chromones [63]. The original work from the Stoltz group using pyridine-oxazolines was very successful for addition reactions to 2-unsubstituted chromones (Table 23). However, in the attempted addition reaction of phenylboronic acid to 2-methylchromone, the expected product was not isolated (entry 6, Table 22) [51]. Therefore, a new optically pure substituted pyridine-dihydroisoquinoline L17 was developed (Table 38) [63]. The studied catalytic system of ligand L17 in combination with Pd(TFA)2 allowed the isolation of the desired products in excellent yields, especially for electron-rich boronic acids. The yields for the products from addition reactions with electron-poor boronic acids were only average. However, excellent enantioselectivities were achieved for all studied substrate combinations (90–99% ee; Table 38) [63].
Table 38:
Addition reactions of arylboronic acids to 2-substituted chromones catalysed by L17/Pd(TFA)2[63].
entry
R1
R2
Ar
yield (%)
ee (%)
1
Me
H
Ph
98
95
2
Me
H
4-Me-C6H4
80
96
3
Me
H
4-Et-C6H4
85
98
4
Me
H
4-MeO-C6H4
51
90
5
Me
H
4-t-Bu-C6H4
78
98
6
Me
H
3-MeO-C6H4
81
99
7
Me
H
3-Me-C6H4
82
99
8
Me
H
3,5-diMe-C6H3
77
97
9
Me
H
3,4-(CH2O2)-C6H3
47
96
10
Me
H
4-F-C6H4
80
98
11
Me
H
4-Cl-C6H4
86
99
12
Me
H
4-Br-C6H4
32
98
13
Me
H
4-CF3-C6H4
31
99
14
Me
H
3-F-C6H4
60
96
15
Me
H
3-Cl-C6H4
55
92
16
Et
H
Ph
93
98
17
iPr
H
Ph
47
97
18
Cy
H
Ph
48
98
19
Bn
H
Ph
52
98
20
Me
6-Me
Ph
89
98
21
Me
6-MeO
Ph
88
98
22
Me
7-MeO
Ph
92
98
23
Me
6-F
Ph
74
97
24
Me
6-Cl
Ph
90
96
25
Me
6-Br
Ph
64
99
Evaluation of current state and outlook
Asymmetric addition reactions to enones have so far been described in the literature in connection with catalysis. The catalyst is usually a complex of a transition metal with a suitable ligand. However, metal-free catalysis is also known [64]. Among the most successful transition-metal catalysts are those based on rhodium, as evidenced by the number of reports that deal with the issue. The rhodium-catalysed addition of various boronic acids to conjugated cyclic enones (the so-called Hayashi–Miyaura reaction) is a well-established method for 3-unsubstituted substrates as well as for 2-unsubstituted chromones [17-19,21-24]. On the other hand, there is only one example of the usage of a rhodium-based catalyst for the addition of arylboronic acid to 3-substituted enones. The olefino-oxazoline ligand L18 has been used for the rhodium-catalysed addition reaction of phenylboronic acid to 3-methyl-2-cyclohexenone and affording the product in a low yield and moderate enantioselectivity (36%; 85% ee; Scheme 26) [20]. Palladium-based catalysis provides better results in this area.
Scheme 26:
The Rh-catalysed addition reaction of phenylboronic acid to a 3-substituted enone [20].
Scheme 26:
The Rh-catalysed addition reaction of phenylboronic acid to a 3-substituted enone [20].
Up to now, asymmetric addition reactions to sterically hindered enones are still challenging. In Scheme 27, we present some underdeveloped methodologies.
We have so far tried to achieve asymmetric addition to some of these cyclic enones in our laboratory without success. Specifically, it was catalysis in a homogeneous medium, using ligand L9 and Pd (TFA)2. Also, continuous-flow reactions are currently a general challenge, especially for the pharmaceutical industry. The prerequisite for a successful continuous synthesis in the field of asymmetric addition reactions to enones is the mastery of recyclable heterogeneous catalysis. Very recently, we reported [57] the first heterogeneous polystyrene-supported recyclable catalyst for asymmetric conjugate addition reactions of arylboronic acids to five and six-membered enones. In our laboratory, we also attempted to perform this reaction under flow conditions. However, the change from batch to flow arrangement itself is another challenging task. Nevertheless, it should be noted at this point that in the case of rhodium complex catalysis, the asymmetric addition of phenylboronic acid to enones in continuous flow has been successful [24]. In 2021, Walhers et al. presented a theoretical study based on the Q2MM method about the asymmetric addition of arylboronic acids to conjugated cyclic enones, catalysed by a complex of L9 and Pd(TFA)2[68]. The authors prepared a training set from the data of currently known combinations of PyOx derivatives as ligands, boronic acids and enones (82 hits). They have calculated the predictions of enantioselectivities for Pd(TFA)2 complexes of 27 new PyOx-type ligands (for the reaction of 3-methyl-2-cyclohexenone with phenylboronic acid) and 59 new enones (in reactions with phenylboronic acid catalysed by L9/Pd(TFA)2). The calculation performed was related to a transition state and included steric and inductive effects. Although this approach may be suitable for predicting theoretically achievable enantioselectivity and is very promising, it is not engineered to predict reactivity. Besides, the reactivity (conversion or yield) depends on the reaction medium which is not included in the theoretical model. The experimental validation of the predicted results is therefore a challenge that has to be finished [68].
Conclusion
In this review, we focused on palladium-catalysed asymmetric 1,4-addition reactions of arylboronic acids to conjugated enones and chromones. The suitability of the ligand used, the reaction conditions, and additives in terms of the yield and enantioselectivity of the transformation have been discussed. The review is classified according to the type of ligand of the catalytic complex used. The yields and corresponding enantioselectivities from the relevant literature were summarised in clear tables. Based on the above results, we propose a flowchart facilitating the reader in selecting a suitable ligand for a given combination of enone and arylboronic acid (Scheme 28). However, the reader should be aware of its limitations because not all ligands have been studied on all substrates. Also, close to the end of the review, the catalysis by rhodium complexes has been mentioned. With these catalysts only reactions of 3-unsubstituted enone derivatives have been described. It can be said that, despite great efforts, some problems remain unresolved. Thus, palladium-based catalysts represent a more suitable alternative to the widely used rhodium complexes for these sterically hindered enone derivatives.
Scheme 28:
Flowchart for the selection of the proper catalytic system.
Scheme 28:
Flowchart for the selection of the proper catalytic system.
The authors thank the Czech Ministry of Education Youth and Sports (project number SGS_2021_004) for financial support.
References
Albuquerque de Oliveira Mendes, L.; Ponciano, C. S.; Depieri Cataneo, A. H.; Wowk, P. F.; Bordignon, J.; Silva, H.; Vieira de Almeida, M.; Ávila, E. P. Chem.-Biol. Interact.2020,331, 109218. doi:10.1016/j.cbi.2020.109218
Return to citation in text:
[1]
Tutunchi, H.; Naeini, F.; Ostadrahimi, A.; Hosseinzadeh‐Attar, M. J. Phytother. Res.2020,34, 3137–3147. doi:10.1002/ptr.6781
Return to citation in text:
[1]
Kobayashi, K.; Nishikata, T.; Yamamoto, Y.; Miyaura, N. Bull. Chem. Soc. Jpn.2008,81, 1019–1025. doi:10.1246/bcsj.81.1019
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
Bürki, C.; Whyte, A.; Arndt, S.; Hashmi, A. S. K.; Lautens, M. Org. Lett.2016,18, 5058–5061. doi:10.1021/acs.orglett.6b02505
Return to citation in text:
[1]
[2]
[3]
[4]
Wang, X.; Liu, B.; Searle, X.; Yeung, C.; Bogdan, A.; Greszler, S.; Singh, A.; Fan, Y.; Swensen, A. M.; Vortherms, T.; Balut, C.; Jia, Y.; Desino, K.; Gao, W.; Yong, H.; Tse, C.; Kym, P. J. Med. Chem.2018,61, 1436–1449. doi:10.1021/acs.jmedchem.7b01339
Return to citation in text:
[1]
[2]
[3]
Greszler, S. N.; Shelat, B.; Voight, E. A. Org. Lett.2019,21, 5725–5727. doi:10.1021/acs.orglett.9b02099
Return to citation in text:
[1]
[2]
[3]
Shockley, S. E.; Holder, J. C.; Stoltz, B. M. Org. Lett.2014,16, 6362–6365. doi:10.1021/ol5031537
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
Shaw, K.; Niyogi, S.; Nandi, R.; Bisai, V. Tetrahedron Lett.2020,61, 152169. doi:10.1016/j.tetlet.2020.152169
Return to citation in text:
[1]
[2]
[3]
Gottumukkala, A. L.; Matcha, K.; Lutz, M.; de Vries, J. G.; Minnaard, A. J. Chem. – Eur. J.2012,18, 6907–6914. doi:10.1002/chem.201200694
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
[12]
[13]
[14]
[15]
[16]
Buter, J.; Moezelaar, R.; Minnaard, A. J. Org. Biomol. Chem.2014,12, 5883–5890. doi:10.1039/c4ob01085j
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
Buter, J.; Heijnen, D.; Vila, C.; Hornillos, V.; Otten, E.; Giannerini, M.; Minnaard, A. J.; Feringa, B. L. Angew. Chem., Int. Ed.2016,55, 3620–3624. doi:10.1002/anie.201510328
Return to citation in text:
[1]
[2]
[3]
Defieber, C.; Paquin, J.-F.; Serna, S.; Carreira, E. M. Org. Lett.2004,6, 3873–3876. doi:10.1021/ol048240x
Return to citation in text:
[1]
[2]
Kurihara, K.; Sugishita, N.; Oshita, K.; Piao, D.; Yamamoto, Y.; Miyaura, N. J. Organomet. Chem.2007,692, 428–435. doi:10.1016/j.jorganchem.2006.04.042
Return to citation in text:
[1]
[2]
Shintani, R.; Tsutsumi, Y.; Nagaosa, M.; Nishimura, T.; Hayashi, T. J. Am. Chem. Soc.2009,131, 13588–13589. doi:10.1021/ja905432x
Return to citation in text:
[1]
[2]
Hahn, B. T.; Tewes, F.; Fröhlich, R.; Glorius, F. Angew. Chem., Int. Ed.2010,49, 1143–1146. doi:10.1002/anie.200905712
Return to citation in text:
[1]
[2]
[3]
Chen, G.; Gui, J.; Li, L.; Liao, J. Angew. Chem., Int. Ed.2011,50, 7681–7685. doi:10.1002/anie.201102586
Return to citation in text:
[1]
[2]
Thaler, T.; Guo, L.-N.; Steib, A. K.; Raducan, M.; Karaghiosoff, K.; Mayer, P.; Knochel, P. Org. Lett.2011,13, 3182–3185. doi:10.1021/ol200841x
Return to citation in text:
[1]
[2]
Yasukawa, T.; Miyamura, H.; Kobayashi, S. J. Am. Chem. Soc.2012,134, 16963–16966. doi:10.1021/ja307913e
Return to citation in text:
[1]
[2]
Shen, G.; Osako, T.; Nagaosa, M.; Uozumi, Y. J. Org. Chem.2018,83, 7380–7387. doi:10.1021/acs.joc.8b00178
Return to citation in text:
[1]
[2]
[3]
Morisaki, Y.; Imoto, H.; Hirano, K.; Hayashi, T.; Chujo, Y. J. Org. Chem.2011,76, 1795–1803. doi:10.1021/jo1024442
Return to citation in text:
[1]
[2]
[3]
Wong, J.; Gan, K.; Chen, H. J.; Pullarkat, S. A. Adv. Synth. Catal.2014,356, 3391–3400. doi:10.1002/adsc.201400473
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
Zhang, T.; Shi, M. Chem. – Eur. J.2008,14, 3759–3764. doi:10.1002/chem.200701982
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
Xu, Q.; Zhang, R.; Zhang, T.; Shi, M. J. Org. Chem.2010,75, 3935–3937. doi:10.1021/jo1006224
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
Mullick, A. B.; Jeletic, M. S.; Powers, A. R.; Ghiviriga, I.; Abboud, K. A.; Veige, A. S. Polyhedron2013,52, 810–819. doi:10.1016/j.poly.2012.07.046
Return to citation in text:
[1]
[2]
[3]
Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc.2011,133, 6902–6905. doi:10.1021/ja200664x
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
[12]
Holder, J. C.; Goodman, E. D.; Kikushima, K.; Gatti, M.; Marziale, A. N.; Stoltz, B. M. Tetrahedron2015,71, 5781–5792. doi:10.1016/j.tet.2014.11.048
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
[9]
[10]
[11]
[12]
Holder, J. C.; Marziale, A. N.; Gatti, M.; Mao, B.; Stoltz, B. M. Chem. – Eur. J.2013,19, 74–77. doi:10.1002/chem.201203643
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
Boeser, C. L.; Holder, J. C.; Taylor, B. L. H.; Houk, K. N.; Stoltz, B. M.; Zare, R. N. Chem. Sci.2015,6, 1917–1922. doi:10.1039/c4sc03337j
Return to citation in text:
[1]
[2]
Ochi, S.; Xia, Y.; Dong, G. Bull. Chem. Soc. Jpn.2020,93, 1213–1217. doi:10.1246/bcsj.20200147
Return to citation in text:
[1]
[2]
[3]
Van Zeeland, R.; Stanley, L. M. ACS Catal.2015,5, 5203–5206. doi:10.1021/acscatal.5b01272
Return to citation in text:
[1]
[2]
[3]
Kadam, A. A.; Ellern, A.; Stanley, L. M. Org. Lett.2017,19, 4062–4065. doi:10.1021/acs.orglett.7b01825
Return to citation in text:
[1]
[2]
[3]
[4]
Lestini, E.; Blackman, L. D.; Zammit, C. M.; Chen, T.; Williams, R. J.; Inam, M.; Couturaud, B.; O'Reilly, R. K. Polym. Chem.2018,9, 820–823. doi:10.1039/c7py02050c
Return to citation in text:
[1]
[2]
[3]
[4]
Bartáček, J.; Váňa, J.; Drabina, P.; Svoboda, J.; Kocúrik, M.; Sedlák, M. React. Funct. Polym.2020,153, 104615. doi:10.1016/j.reactfunctpolym.2020.104615
Return to citation in text:
[1]
[2]
[3]
[4]
Zhou, L.; Qiu, J.; Wang, M.; Xu, Z.; Wang, J.; Chen, T. J. Inorg. Organomet. Polym. Mater.2020,30, 4569–4577. doi:10.1007/s10904-020-01599-2
Return to citation in text:
[1]
[2]
[3]
[4]
Lamb, C. J. C.; Vilela, F.; Lee, A.-L. Org. Lett.2019,21, 8689–8694. doi:10.1021/acs.orglett.9b03293
Return to citation in text:
[1]
[2]
[3]
[4]
Lai, J.; Li, W.; Wei, S.; Li, S. Org. Chem. Front.2020,7, 2263–2268. doi:10.1039/d0qo00519c
Return to citation in text:
[1]
[2]
Tamura, M.; Ogata, H.; Ishida, Y.; Takahashi, Y. Tetrahedron Lett.2017,58, 3808–3813. doi:10.1016/j.tetlet.2017.08.041
Return to citation in text:
[1]
[2]
de Gracia Retamosa, M.; Álvarez‐Casao, Y.; Matador, E.; Gómez, Á.; Monge, D.; Fernández, R.; Lassaletta, J. M. Adv. Synth. Catal.2019,361, 176–184. doi:10.1002/adsc.201801021
Return to citation in text:
[1]
[2]
[3]
[4]
[5]
[6]
[7]
[8]
Baek, D.; Ryu, H.; Ryu, J. Y.; Lee, J.; Stoltz, B. M.; Hong, S. Chem. Sci.2020,11, 4602–4607. doi:10.1039/d0sc00412j
Return to citation in text:
[1]
[2]
[3]
[4]
Roscales, S.; Ortega, V.; Csákÿ, A. G. J. Org. Chem.2013,78, 12825–12830. doi:10.1021/jo402262m
Return to citation in text:
[1]
Gao, A.; Liu, X.-Y.; Ding, C.-H.; Hou, X.-L. Synlett2017,28, 2829–2832. doi:10.1055/s-0036-1590742
Return to citation in text:
[1]
Jordan-Hore, J. A.; Sanderson, J. N.; Lee, A.-L. Org. Lett.2012,14, 2508–2511. doi:10.1021/ol300794a
Return to citation in text:
[1]
Heintz, P. M.; Schumacher, B. P.; Chen, M.; Huang, W.; Stanley, L. M. ChemCatChem2019,11, 4286–4290. doi:10.1002/cctc.201900894
Return to citation in text:
[1]
Wahlers, J.; Maloney, M.; Salahi, F.; Rosales, A. R.; Helquist, P.; Norrby, P.-O.; Wiest, O. J. Org. Chem.2021,86, 5660–5667. doi:10.1021/acs.joc.1c00136
Return to citation in text:
[1]
[2]
References 47,49
47.
Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc.2011,133, 6902–6905. doi:10.1021/ja200664x
49.
Holder, J. C.; Goodman, E. D.; Kikushima, K.; Gatti, M.; Marziale, A. N.; Stoltz, B. M. Tetrahedron2015,71, 5781–5792. doi:10.1016/j.tet.2014.11.048
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
49.
Holder, J. C.; Goodman, E. D.; Kikushima, K.; Gatti, M.; Marziale, A. N.; Stoltz, B. M. Tetrahedron2015,71, 5781–5792. doi:10.1016/j.tet.2014.11.048
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
49.
Holder, J. C.; Goodman, E. D.; Kikushima, K.; Gatti, M.; Marziale, A. N.; Stoltz, B. M. Tetrahedron2015,71, 5781–5792. doi:10.1016/j.tet.2014.11.048
Wahlers, J.; Maloney, M.; Salahi, F.; Rosales, A. R.; Helquist, P.; Norrby, P.-O.; Wiest, O. J. Org. Chem.2021,86, 5660–5667. doi:10.1021/acs.joc.1c00136
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
51.
Holder, J. C.; Marziale, A. N.; Gatti, M.; Mao, B.; Stoltz, B. M. Chem. – Eur. J.2013,19, 74–77. doi:10.1002/chem.201203643
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Wahlers, J.; Maloney, M.; Salahi, F.; Rosales, A. R.; Helquist, P.; Norrby, P.-O.; Wiest, O. J. Org. Chem.2021,86, 5660–5667. doi:10.1021/acs.joc.1c00136
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
Albuquerque de Oliveira Mendes, L.; Ponciano, C. S.; Depieri Cataneo, A. H.; Wowk, P. F.; Bordignon, J.; Silva, H.; Vieira de Almeida, M.; Ávila, E. P. Chem.-Biol. Interact.2020,331, 109218. doi:10.1016/j.cbi.2020.109218
2.
Tutunchi, H.; Naeini, F.; Ostadrahimi, A.; Hosseinzadeh‐Attar, M. J. Phytother. Res.2020,34, 3137–3147. doi:10.1002/ptr.6781
Shockley, S. E.; Holder, J. C.; Stoltz, B. M. Org. Lett.2014,16, 6362–6365. doi:10.1021/ol5031537
27.
Shockley, S. E.; Holder, J. C.; Stoltz, B. M. Org. Process Res. Dev.2015,19, 974–981. doi:10.1021/acs.oprd.5b00169
47.
Kikushima, K.; Holder, J. C.; Gatti, M.; Stoltz, B. M. J. Am. Chem. Soc.2011,133, 6902–6905. doi:10.1021/ja200664x
48.
Holder, J. C.; Zou, L.; Marziale, A. N.; Liu, P.; Lan, Y.; Gatti, M.; Kikushima, K.; Houk, K. N.; Stoltz, B. M. J. Am. Chem. Soc.2013,135, 14996–15007. doi:10.1021/ja401713g
49.
Holder, J. C.; Goodman, E. D.; Kikushima, K.; Gatti, M.; Marziale, A. N.; Stoltz, B. M. Tetrahedron2015,71, 5781–5792. doi:10.1016/j.tet.2014.11.048
51.
Holder, J. C.; Marziale, A. N.; Gatti, M.; Mao, B.; Stoltz, B. M. Chem. – Eur. J.2013,19, 74–77. doi:10.1002/chem.201203643
52.
Boeser, C. L.; Holder, J. C.; Taylor, B. L. H.; Houk, K. N.; Stoltz, B. M.; Zare, R. N. Chem. Sci.2015,6, 1917–1922. doi:10.1039/c4sc03337j
Lestini, E.; Blackman, L. D.; Zammit, C. M.; Chen, T.; Williams, R. J.; Inam, M.; Couturaud, B.; O'Reilly, R. K. Polym. Chem.2018,9, 820–823. doi:10.1039/c7py02050c
Lestini, E.; Blackman, L. D.; Zammit, C. M.; Chen, T.; Williams, R. J.; Inam, M.; Couturaud, B.; O'Reilly, R. K. Polym. Chem.2018,9, 820–823. doi:10.1039/c7py02050c
58.
Zhou, L.; Qiu, J.; Wang, M.; Xu, Z.; Wang, J.; Chen, T. J. Inorg. Organomet. Polym. Mater.2020,30, 4569–4577. doi:10.1007/s10904-020-01599-2
Lestini, E.; Blackman, L. D.; Zammit, C. M.; Chen, T.; Williams, R. J.; Inam, M.; Couturaud, B.; O'Reilly, R. K. Polym. Chem.2018,9, 820–823. doi:10.1039/c7py02050c
Lestini, E.; Blackman, L. D.; Zammit, C. M.; Chen, T.; Williams, R. J.; Inam, M.; Couturaud, B.; O'Reilly, R. K. Polym. Chem.2018,9, 820–823. doi:10.1039/c7py02050c



![[Graphic 1]](/bjoc/content/inline/1860-5397-17-84-i29.svg?max-width=637&scale=1.0)
![[Graphic 2]](/bjoc/content/inline/1860-5397-17-84-i30.svg?max-width=637&scale=1.0)
![[Graphic 3]](/bjoc/content/inline/1860-5397-17-84-i31.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i1]](/bjoc/content/inline/1860-5397-17-84-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 4]](/bjoc/content/inline/1860-5397-17-84-i32.svg?max-width=637&scale=1.0)
![[Graphic 5]](/bjoc/content/inline/1860-5397-17-84-i33.svg?max-width=637&scale=1.0)
![[Graphic 6]](/bjoc/content/inline/1860-5397-17-84-i34.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i2]](/bjoc/content/inline/1860-5397-17-84-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i3]](/bjoc/content/inline/1860-5397-17-84-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 7]](/bjoc/content/inline/1860-5397-17-84-i35.svg?max-width=637&scale=1.0)
![[Graphic 8]](/bjoc/content/inline/1860-5397-17-84-i36.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i4]](/bjoc/content/inline/1860-5397-17-84-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i5]](/bjoc/content/inline/1860-5397-17-84-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 9]](/bjoc/content/inline/1860-5397-17-84-i37.svg?max-width=637&scale=1.0)
![[Graphic 10]](/bjoc/content/inline/1860-5397-17-84-i38.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i6]](/bjoc/content/inline/1860-5397-17-84-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i7]](/bjoc/content/inline/1860-5397-17-84-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 11]](/bjoc/content/inline/1860-5397-17-84-i39.svg?max-width=637&scale=1.0)
![[Graphic 12]](/bjoc/content/inline/1860-5397-17-84-i40.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i8]](/bjoc/content/inline/1860-5397-17-84-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 13]](/bjoc/content/inline/1860-5397-17-84-i41.svg?max-width=637&scale=1.0)
![[Graphic 14]](/bjoc/content/inline/1860-5397-17-84-i42.svg?max-width=637&scale=1.0)
![[Graphic 15]](/bjoc/content/inline/1860-5397-17-84-i43.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i9]](/bjoc/content/inline/1860-5397-17-84-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 16]](/bjoc/content/inline/1860-5397-17-84-i44.svg?max-width=637&scale=1.0)
![[Graphic 17]](/bjoc/content/inline/1860-5397-17-84-i45.svg?max-width=637&scale=1.0)
![[Graphic 18]](/bjoc/content/inline/1860-5397-17-84-i46.svg?max-width=637&scale=1.0)
![[Graphic 19]](/bjoc/content/inline/1860-5397-17-84-i47.svg?max-width=637&scale=1.0)
![[Graphic 20]](/bjoc/content/inline/1860-5397-17-84-i48.svg?max-width=637&scale=1.0)
![[Graphic 21]](/bjoc/content/inline/1860-5397-17-84-i49.svg?max-width=637&scale=1.0)
![[Graphic 22]](/bjoc/content/inline/1860-5397-17-84-i50.svg?max-width=637&scale=1.0)
![[Graphic 23]](/bjoc/content/inline/1860-5397-17-84-i51.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i10]](/bjoc/content/inline/1860-5397-17-84-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i11]](/bjoc/content/inline/1860-5397-17-84-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 24]](/bjoc/content/inline/1860-5397-17-84-i52.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i12]](/bjoc/content/inline/1860-5397-17-84-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i13]](/bjoc/content/inline/1860-5397-17-84-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 25]](/bjoc/content/inline/1860-5397-17-84-i53.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i14]](/bjoc/content/inline/1860-5397-17-84-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i15]](/bjoc/content/inline/1860-5397-17-84-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i16]](/bjoc/content/inline/1860-5397-17-84-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i17]](/bjoc/content/inline/1860-5397-17-84-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i18]](/bjoc/content/inline/1860-5397-17-84-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 26]](/bjoc/content/inline/1860-5397-17-84-i54.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i19]](/bjoc/content/inline/1860-5397-17-84-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 27]](/bjoc/content/inline/1860-5397-17-84-i55.svg?max-width=637&scale=1.0)
![[Graphic 28]](/bjoc/content/inline/1860-5397-17-84-i56.svg?max-width=637&scale=1.0)
![[Graphic 29]](/bjoc/content/inline/1860-5397-17-84-i57.svg?max-width=637&scale=1.0)
![[Graphic 30]](/bjoc/content/inline/1860-5397-17-84-i58.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i20]](/bjoc/content/inline/1860-5397-17-84-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 31]](/bjoc/content/inline/1860-5397-17-84-i59.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i21]](/bjoc/content/inline/1860-5397-17-84-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 32]](/bjoc/content/inline/1860-5397-17-84-i60.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i22]](/bjoc/content/inline/1860-5397-17-84-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 33]](/bjoc/content/inline/1860-5397-17-84-i61.svg?max-width=637&scale=1.0)
![[Graphic 34]](/bjoc/content/inline/1860-5397-17-84-i62.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i23]](/bjoc/content/inline/1860-5397-17-84-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 35]](/bjoc/content/inline/1860-5397-17-84-i63.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i24]](/bjoc/content/inline/1860-5397-17-84-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i25]](/bjoc/content/inline/1860-5397-17-84-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[Graphic 36]](/bjoc/content/inline/1860-5397-17-84-i64.svg?max-width=637&scale=1.0)
![[Graphic 37]](/bjoc/content/inline/1860-5397-17-84-i65.svg?max-width=637&scale=1.0)
![[Graphic 38]](/bjoc/content/inline/1860-5397-17-84-i66.svg?max-width=637&scale=1.0)
![[1860-5397-17-84-i26]](/bjoc/content/inline/1860-5397-17-84-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i27]](/bjoc/content/inline/1860-5397-17-84-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-17-84-i28]](/bjoc/content/inline/1860-5397-17-84-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)