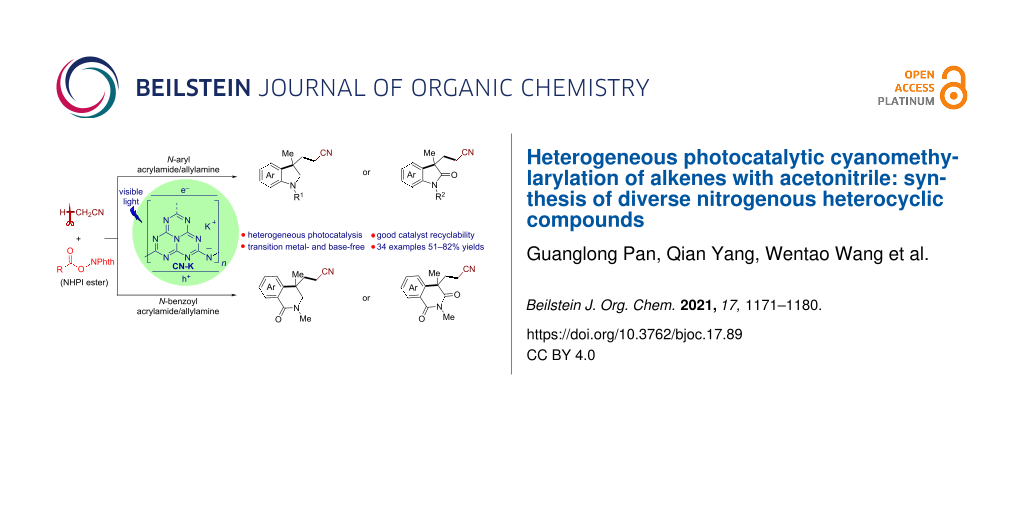
Abstract
A visible light-mediated heterogeneous photocatalytic cyanomethylarylation of alkenes with acetonitrile has been established using K-modified carbon nitride (CN-K) as a recyclable semiconductor photocatalyst. This protocol, employing readily accessible alkyl N-hydroxyphthalimide (NHPI) ester as a radical initiator, allows the efficient construction of a broad array of structural diverse nitrogenous heterocyclic compounds including indolines, oxindoles, isoquinolinones, and isoquinolinediones.

Graphical Abstract
Introduction
Nitrogenous heterocyclic compounds, such as indolines [1-4], oxindoles [5-7], isoquinolinones [8-10], and isoquinolinediones [11,12], are pivotal structural motifs in numerous pharmaceuticals, agrochemicals, and bioactive natural products. The oxidative cyanomethylarylation of N-aryl/benzoyl acrylamide or allylamine involving the key C–H functionalization of readily available acetonitrile is a straightforward and powerful method to access these useful structures [13-17]. Over the last decade, although numerous protocols have been disclosed for the direct cyanomethylarylation of alkenes, most of them rely on the use of transition metals (such as catalytic amounts of Pd, Cu, Fe catalyst or stoichiometric amounts of Ag, Mn salts) and strong oxidants (including PhI(OPiv)2, DTBP, and t-BuONO) in the presence of various base additives under high reaction temperatures or microwave stimulations [18-26]. Recently, visible light photoredox catalysis has emerged as a powerful and environment-friendly method in organic synthesis by activating organic molecules under mild reaction conditions [27,28]. In this context, the Li and Cai groups independently disclosed a photocatalytic cyanomethylarylation of N-aryl/benzoyl acrylamide for the synthesis of oxindoles and isoquinolinediones using diazonium salts and PIFA/1,3,5-trimethoxybenzene as radical initiators, respectively [29-31]. In this case, expensive Ru and 4CzIPN-based homogeneous photocatalysts are used; and the scope remains limited to activated alkenes. Therefore, it is still highly desirable to develop a general, efficient, and practical protocol for the cyanomethylarylation of unactivated alkenes from readily available radical initiators under mild and environmentally benign conditions.
Recently, we demonstrated K-modified carbon nitride (CN-K), a semiconductor material, exhibited a remarkably enhanced photocatalytic activity in the decarboxylative Giese reaction. The effect was due to its K-intercalated poly(heptazine)-based structure existing as small lamellar nanocrystallites, thus leading to an enhanced optical absorption, improved electron-hole separation and easy dispersion in polar solvents [32]. CN-K, which can be easily prepared by the direct KCl-induced structure remodeling of bulk g-C3N4 [33-36], showed great potentials for future industrial applications given its low cost, recyclability, and high chemical/thermal stability [37-39]. Meanwhile, CN-K has a band gap of 2.72 eV with suitable valence band and conduction band potentials, which allows its promising use in organic synthesis for oxidation and reduction of various substrates [40-43]. In the last few years, carbon nitride-based heterogeneous photocatalysts have also been utilized for several other radical-initiated synthetic transformations [44-56]. However, to the best of our knowledge, the application of a CN-based photocatalyst for the generation of cyanomethyl radicals from readily available acetonitrile has not been reported yet.
Herein, we disclose a CN-K-catalyzed cyanomethylarylation of both unactivated and activated alkenes with acetonitrile utilizing readily available alkyl N-hydroxyphthalimide (NHPI) esters as the radical initiator under mild conditions. A wide range of structurally diverse nitrogenous heterocyclic compounds including indolines, oxindoles, isoquinolinones, and isoquinolinediones have been accessed in high yields. This heterogeneous protocol features base- and transition metal-free, good catalyst recyclability, broad substrate scope, and high functional group tolerance (Scheme 1).
![[1860-5397-17-89-i1]](/bjoc/content/inline/1860-5397-17-89-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: CN-K-Catalyzed cyanomethylarylation of alkenes to access diverse heterocyclic compounds.
Scheme 1: CN-K-Catalyzed cyanomethylarylation of alkenes to access diverse heterocyclic compounds.
Results and Discussion
Our initial investigation focused on the CN-K photocatalyzed cascade alkyl radical addition/cyclization reaction of the N-arylallylamine 1a with tert-butyl N-hydroxyphthalimide (NHPI) ester (2a), a classical alkyl radical precursor [57-59], to construct indoline product 4. Surprisingly, the solvent acetonitrile incorporated indoline 3a was observed as the major product (21%, Table 1, entry 1). Stimulated by this result, we questioned whether it would be possible to develop a general and efficient CN-K-based heterogeneous protocol for the cyanomethylarylation of N-arylallylamines with acetonitrile. Pleasingly, after the systematic evaluation of various NHPI esters, which are easily prepared from readily available carboxylic acids, the yield of the product 3a could be increased to 75% when using the primary NHPI ester 2d as a radical initiator. The concentration of the reactant had a significant effect on the reaction. When the reaction was performed under higher concentrations (0.05 M), the target product was obtained in 62% yield after prolonged reaction time (Table 1, entry 5). Traditional g-C3N4 exhibited a low catalytic activity for this transformation (Table 1, entry 6). Switching from CN-K to a homogeneous organo photocatalyst such as eosin Y and 4CzIPN, led to lower yields of the desired product (Table 1, entries 7 and 8). The expensive Ru/Ir-based metal complexes gave similar results for the transformation (Table 1, entries 9 and 10). In the control experiments, no reaction was observed in the absence of the NHPI ester, CN-K, or light, which unambiguously manifested all of them were requisite for this transformation (Table 1, entries 11–13). In addition, no desired product was detected under air, further indicating a radical-initiated process was involved in the reaction (Table 1, entry 14).
Table 1: Optimization of the reaction conditions and control experimentsa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-17-89-i9.svg?max-width=637&scale=1.0)
|
|||
| entry | PC | NHPI ester 2 | yield (%)b |
| 1 | CN-K | 2a | 21 |
| 2 | CN–K | 2b | 13 |
| 3 | CN–K | 2c | 49 |
| 4 | CN–K | 2d | 75 (72)c |
| 5d | CN-K | 2d | 62 |
| 6 | g-C3N4 | 2d | 23 |
| 7 | eosin Y | 2d | 20 |
| 8 | 4CzIPN | 2d | 18 |
| 9e | [Ru(bpy)3]Cl2 | 2d | 73 |
| 10e | fac–Ir(ppy)3 | 2d | 60 |
| 11 | CN-K | – | 0 |
| 12 | – | 2d | 0 |
| 13f | CN-K | 2d | 0 |
| 14g | CN-K | 2d | 0 |
aReaction conditions: 1a (0.1 mmol, 1 equiv), 2 (0.2 mmol, 2 equiv), CH3CN (6 mL), PC (1 mg/mL), 2 × 24 W blue LEDs (460 ± 5 nm) without extra heating (at 40–45 °C) for 72 h. bDetermined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. cData in parentheses are the isolated yields. dPerformed with 0.05 M of 1a for 120 h. eUsing 2 mol % of PC. fIn the dark. gUnder air.
Under the optimal reaction conditions, we first evaluated the scope of N-arylallylamines for the synthesis of indolines. As shown in Scheme 2, substrates with various electron-donating groups (such as methyl or methoxy) and electron-withdrawing groups (chloro, bromo, or cyano) at the para-position of the aryl ring smoothly reacted with acetonitrile to afford the 5-substituted cyanomethylated indolines 3a–f in good yields (67–75%). When the benzene ring of the substrates was changed to naphthalene, the reaction successfully provided the benzoindoline 3g in 56% yield. Substrates with an N-Boc group also worked well to deliver the corresponding indoline 3h in 77% yield. Moreover, a scale-up reaction of 1a (2 mmol) was conducted, affording the corresponding product 3a (0.28 g) in a yield of 62%.
![[1860-5397-17-89-i2]](/bjoc/content/inline/1860-5397-17-89-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: CN-K-catalyzed cyanomethylarylation of N-arylallylamines for the synthesis of indolines. Reaction conditions: 1 (0.2 mmol, 1 equiv), 2d (0.4 mmol, 2 equiv), CH3CN (8 mL), CN-K (1 mg/mL), 2 × 24 W blue LEDs (460 ± 5 nm) without extra heating (at 40–45 °C). aPerformed with 0.017 M of 1.
Scheme 2: CN-K-catalyzed cyanomethylarylation of N-arylallylamines for the synthesis of indolines. Reaction c...
Next, we examined the efficiency of the CN-K-catalyzed cyanomethylarylation of N-benzoylallylamines for the synthesis of isoquinolinone derivatives. Under similar reaction conditions, various N-benzoylallylamines bearing either electron-donating or withdrawing substituents in the para-position of the phenyl ring (Scheme 3) were smoothly converted to the corresponding isoquinolinones 6a–e in moderate to good yields (51–69%, Scheme 3). The regioselectivity of this reaction was studied using the meta-substituted substrate 5f. The results indicated that the cyclization occurred at the less crowded positions, and the two regioisomers 6f and 6f’ were obtained in a 20:1 ratio.
![[1860-5397-17-89-i3]](/bjoc/content/inline/1860-5397-17-89-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: CN-K-catalyzed cyanomethylarylation of N-benzoylallylamines for the synthesis of isoquinolinones. Reaction conditions: 5 (0.2 mmol, 1 equiv), 2d (0.6 mmol, 3 equiv), CH3CN (8 mL), CN-K (1 mg/mL), 2 × 24 W blue LEDs (460 ± 5 nm), 60 °C.
Scheme 3: CN-K-catalyzed cyanomethylarylation of N-benzoylallylamines for the synthesis of isoquinolinones. R...
The established CN-K-catalyzed cyanomethylarylation protocol is not only effective for the above unactivated alkenes. Also activated alkenes including N-aryl and N-benzoyl acrylamides can be employed as substrates, allowing for the construction of valuable oxindole and isoquinolinedione derivatives. With respect to the generality of the N-aryl acrylamides 7 (Scheme 4), a range of frequently encountered functional groups were well tolerated affording the methoxy (8b), methyl (8c), the halogenated (8d–f), trifluromethyl (8g), cyano (8h), and carbonyl (8i and 8j) substituted products with 70–82% yield, which provide opportunities for further modification. The ortho-substituted substrate 7k gave the corresponding product 8k in a comparable yield (69%). Also the substrate bearing a meta-substituent exhibited good reactivity, delivering the corresponding regioisomers 8l and 8l’ in 62% with 1:1.6 ratio. Moreover, the naphthalene and tetrahydroisoquinoline-derived acrylamides were also compatible, giving the polycyclic products 8m and 8n in 77% and 70%, respectively. Additionally, protecting groups such as isopropyl, benzyl, or phenyl on the nitrogen atom did not considerably affect the yield (8o–8q, 68–76%). Substrates with benzyl and phenyl groups at the α-position afforded the products 8r and 8s in 74% and 24%, respectively. Substrates with a phenyl group at the β-position underwent arylation with the opposite regioselectivity to afford the six-membered product 8t in 22% yield. Regarding the scope of N-benzoyl acrylamides, the electronic property of a substituent on the phenyl ring also had little influence on the reaction, furnishing the desired isoquinolinediones 10a–c in moderate to high yields (Scheme 5, 60–70%).
![[1860-5397-17-89-i4]](/bjoc/content/inline/1860-5397-17-89-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: CN-K-catalyzed cyanomethylarylation of N-aryl acrylamides for the synthesis of oxindoles. Reaction conditions: 7 (0.2 mmol, 1 equiv), 2d (0.4 mmol, 2 equiv), CH3CN (8 mL), CN-K (0.5 mg/mL), 2 × 24 W blue LEDs (460 ± 5 nm), 80 °C. aConducted at 100 °C. bPerformed with 1 mg/mL of CN-K. cRatio = trans/cis.
Scheme 4: CN-K-catalyzed cyanomethylarylation of N-aryl acrylamides for the synthesis of oxindoles. Reaction ...
![[1860-5397-17-89-i5]](/bjoc/content/inline/1860-5397-17-89-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: CN-K-catalyzed cyanomethylarylation of N-benzoyl acrylamides for the synthesis of isoquinolinediones. Reaction conditions: 9 (0.2 mmol, 1 equiv), 2d (0.4 mmol, 2 equiv), CH3CN (8 mL), CN-K (1 mg/mL), 2 × 24 W blue LEDs (460 ± 5 nm), 80 °C.
Scheme 5: CN-K-catalyzed cyanomethylarylation of N-benzoyl acrylamides for the synthesis of isoquinolinedione...
To illustrate the practicability of this CN-K-based heterogeneous photocatalysis protocol, a recycling procedure was established. The CN-K was recovered via simple centrifugation after the reaction and subsequently reused. As shown in Figure 1, the CN-K catalyst could be recycled at least five times without the loss of photocatalytic activity when applied in the cyanomethylarylation of N-arylallylamine 1a with acetonitrile.
![[1860-5397-17-89-1]](/bjoc/content/figures/1860-5397-17-89-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Evaluation of catalyst recycling. Reaction conditions: 1a (0.1 mmol, 1 equiv), 2d (0.2 mmol, 2 equiv), CH3CN (6 mL), CN-K (1 mg/mL), 2 × 24 W blue LEDs (460 ± 5 nm) without extra heating (at 40–45 °C). After each reaction, the catalyst was recovered by centrifugation and reused in the next cycle.
Figure 1: Evaluation of catalyst recycling. Reaction conditions: 1a (0.1 mmol, 1 equiv), 2d (0.2 mmol, 2 equi...
We also utilized this strategy to test n-butyl nitrile under the standard conditions. As shown in Scheme 6a, the corresponding oxindole 11 was obtained in 45% yield. The synthetic utility was further demonstrated by a series of successful derivatizations of the cyano-substituted oxindole 8a. For instance, after the Ritter reaction, 8a was smoothly converted to N-tert-butylated acetamide 12 in 96% yield (Scheme 6b). A modified Witte–Seeliger reaction led to the formation of oxazoline 13 in 41% yield (Scheme 6c). Furthermore, the tricyclic indoline 14, a structural motif in diverse natural products [4], could be obtained in 60% yield through a reductive cyclization reaction (Scheme 6d).
![[1860-5397-17-89-i6]](/bjoc/content/inline/1860-5397-17-89-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Further survey of reaction scope and derivatization studies of 8a.
Scheme 6: Further survey of reaction scope and derivatization studies of 8a.
To investigate the mechanism of the cyanomethylarylation of alkenes, a series of control experiments was performed (Scheme 7). The cyanomethylarylation reaction of 7a gave the desired compound 8a as the major product in 70% yield, along with 23% yield of the byproduct 15. The latter compound was generated through a cascade alkyl radical addition/cyclization of the NHPI ester 2d to N-aryl acrylamide 7a (Scheme 7a). When the reaction was conducted in the presence of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO), both the desired product 8a and the byproduct 15 were not observed, which indicated that the reaction proceeds through a radical pathway (Scheme 7b). It is worth mentioning that when 7a was submitted to the reaction in a 1:1 mixture of CH3CN/CD3CN as the solvent, the product 8a was furnished in 44% yield and the corresponding deuterated product was not observed (Scheme 7c). In addition, when 7a was reacted in CD3CN, only the byproduct 15 was obtained in 34% yield (Scheme 7d). These results indicate a large primary isotope effect, which suggest that the C(sp3)–H bond cleavage of acetonitrile contributed to the rate-determining step.
![[1860-5397-17-89-i7]](/bjoc/content/inline/1860-5397-17-89-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Experiments for the mechanistic study.
Scheme 7: Experiments for the mechanistic study.
Based on the present results and the literature [18-26,31,60], the reaction pathway is proposed as shown in Scheme 8. As reported in our previous work [32], CN-K has a band gap of 2.72 eV with a conduction band potential of −1.88 V vs SCE, which is thermodynamically favored to induce a single-electron reduction of the NHPI ester (<−1.28 V vs SCE in CH3CN) [61] to generate the corresponding alkyl radical A. The subsequent hydrogen abstraction by radical A yields the key alkyl radical B. The addition of radical B or A to the alkene 7a following by an intramolecular cyclization provides the radical intermediates C or E, which are oxidized by the hole of CN-K via SET delivering the product 8a or byproduct 15 after deprotonation.
![[1860-5397-17-89-i8]](/bjoc/content/inline/1860-5397-17-89-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Plausible mechanism of the CN-K-catalyzed cyanomethylarylation of alkenes.
Scheme 8: Plausible mechanism of the CN-K-catalyzed cyanomethylarylation of alkenes.
Conclusion
We have demonstrated the application of a heterogeneous CN-K semiconducting photocatalyst in the cyanomethylarylation of alkenes with acetonitrile utilizing a readily available NHPI ester as radical initiator. This transition metal-free protocol tolerates a broad range of both unactivated and activated alkenes including N-aryl/benzoylallylamines and acrylamides, providing a facile access to a series of structural diverse indoline, oxindole, isoquinolinone, and isoquinolinedione derivatives with high efficiency. The CN-K catalyst can be easily recovered from the reaction mixture and reused several times, illustrating the practicability of this heterogeneous photocatalysis protocol. Further applying this sustainable and environmentally friendly CN-K heterogeneous photocatalyst to realize other synthetic useful transformations is undergoing.
Supporting Information
| Supporting Information File 1: Full experimental details, compound characterisation, and copies of NMR spectra. | ||
| Format: PDF | Size: 5.4 MB | Download |
Funding
The work was financially supported by the National Natural Science Foundation of China (Grant Nos. 21801030 and 21672210), the Basic and Frontier Research Project of Chongqing (cstc2018jcyjAX0413) and the Venture & Innovation Support Program for Chongqing Overseas Returnees (cx2018060). We thank Analytical and Testing Center of Chongqing University for assistance with NMR spectrum analysis.
References
-
Newhouse, T.; Baran, P. S. J. Am. Chem. Soc. 2008, 130, 10886–10887. doi:10.1021/ja8042307
Return to citation in text: [1] -
Mohn, T.; Plitzko, I.; Hamburger, M. Phytochemistry 2009, 70, 924–934. doi:10.1016/j.phytochem.2009.04.019
Return to citation in text: [1] -
Zhang, D.; Song, H.; Qin, Y. Acc. Chem. Res. 2011, 44, 447–457. doi:10.1021/ar200004w
Return to citation in text: [1] -
Matsuura, T.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6500–6503. doi:10.1021/ja980788+
Return to citation in text: [1] [2] -
Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050
Return to citation in text: [1] -
Jiang, T.; Kuhen, K. L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Tuntland, T.; Zhang, K.; Karanewsky, D.; He, Y. Bioorg. Med. Chem. Lett. 2006, 16, 2109–2112. doi:10.1016/j.bmcl.2006.01.066
Return to citation in text: [1] -
Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y
Return to citation in text: [1] -
Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669–1730. doi:10.1021/cr010212u
Return to citation in text: [1] -
Kaila, N.; Follows, B.; Leung, L.; Thomason, J.; Huang, A.; Moretto, A.; Janz, K.; Lowe, M.; Mansour, T. S.; Hubeau, C.; Page, K.; Morgan, P.; Fish, S.; Xu, X.; Williams, C.; Saiah, E. J. Med. Chem. 2014, 57, 1299–1322. doi:10.1021/jm401509e
Return to citation in text: [1] -
Hekal, M. H.; Abu El‐Azm, F. S. M.; Sallam, H. A. J. Heterocycl. Chem. 2019, 56, 795–803. doi:10.1002/jhet.3448
Return to citation in text: [1] -
Billamboz, M.; Bailly, F.; Barreca, M. L.; De Luca, L.; Mouscadet, J.-F.; Calmels, C.; Andréola, M.-L.; Witvrouw, M.; Christ, F.; Debyser, Z.; Cotelle, P. J. Med. Chem. 2008, 51, 7717–7730. doi:10.1021/jm8007085
Return to citation in text: [1] -
Billamboz, M.; Bailly, F.; Lion, C.; Touati, N.; Vezin, H.; Calmels, C.; Andréola, M.-L.; Christ, F.; Debyser, Z.; Cotelle, P. J. Med. Chem. 2011, 54, 1812–1824. doi:10.1021/jm1014692
Return to citation in text: [1] -
Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74–100. doi:10.1002/anie.201304268
Return to citation in text: [1] -
López, R.; Palomo, C. Angew. Chem., Int. Ed. 2015, 54, 13170–13184. doi:10.1002/anie.201502493
Return to citation in text: [1] -
Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H. Synthesis 2015, 47, 1195–1209. doi:10.1055/s-0034-1379903
Return to citation in text: [1] -
Li, C.-C.; Yang, S.-D. Org. Biomol. Chem. 2016, 14, 4365–4377. doi:10.1039/c6ob00554c
Return to citation in text: [1] -
Chu, X.-Q.; Ge, D.; Shen, Z.-L.; Loh, T.-P. ACS Catal. 2018, 8, 258–271. doi:10.1021/acscatal.7b03334
Return to citation in text: [1] -
Wu, T.; Mu, X.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 12578–12581. doi:10.1002/anie.201104575
Return to citation in text: [1] [2] -
Li, J.; Wang, Z.; Wu, N.; Gao, G.; You, J. Chem. Commun. 2014, 50, 15049–15051. doi:10.1039/c4cc07667b
Return to citation in text: [1] [2] -
Pan, C.; Zhang, H.; Zhu, C. Org. Biomol. Chem. 2015, 13, 361–364. doi:10.1039/c4ob02172j
Return to citation in text: [1] [2] -
Li, X.; Xu, J.; Gao, Y.; Fang, H.; Tang, G.; Zhao, Y. J. Org. Chem. 2015, 80, 2621–2626. doi:10.1021/jo502777b
Return to citation in text: [1] [2] -
Tang, S.; Li, S.-H.; Li, Z.-H.; Zhou, D.; Sheng, R.-L. Tetrahedron Lett. 2015, 56, 1423–1426. doi:10.1016/j.tetlet.2015.01.175
Return to citation in text: [1] [2] -
Ni, Z.; Huang, X.; Wang, J.; Pan, Y. RSC Adv. 2016, 6, 522–526. doi:10.1039/c5ra23471a
Return to citation in text: [1] [2] -
Liang, D.; Song, X.; Xu, L.; Sun, Y.; Dong, Y.; Wang, B.; Li, W. Tetrahedron 2019, 75, 3495–3503. doi:10.1016/j.tet.2019.05.018
Return to citation in text: [1] [2] -
Pan, C.; Wang, Y.; Wu, C.; Yu, J.-T. Catal. Commun. 2019, 131, 105802. doi:10.1016/j.catcom.2019.105802
Return to citation in text: [1] [2] -
Zhang, H.; Chen, P.; Liu, G. Synlett 2012, 23, 2749–2752. doi:10.1055/s-0032-1316555
Return to citation in text: [1] [2] -
Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044–2056. doi:10.1039/c5cs00655d
Return to citation in text: [1] -
Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. doi:10.1002/anie.201709766
Return to citation in text: [1] -
Liu, Y.; Zhang, J.-L.; Song, R.-J.; Li, J.-H. Org. Chem. Front. 2014, 1, 1289–1294. doi:10.1039/c4qo00251b
Return to citation in text: [1] -
Zhang, J.-L.; Liu, Y.; Song, R.-J.; Jiang, G.-F.; Li, J.-H. Synlett 2014, 25, 1031–1035. doi:10.1055/s-0033-1340956
Return to citation in text: [1] -
Lu, M.; Zhang, T.; Tan, D.; Chen, C.; Zhang, Y.; Huang, M.; Cai, S. Adv. Synth. Catal. 2019, 361, 4237–4242. doi:10.1002/adsc.201900712
Return to citation in text: [1] [2] -
Yang, Q.; Pan, G.; Wei, J.; Wang, W.; Tang, Y.; Cai, Y. ACS Sustainable Chem. Eng. 2021, 9, 2367–2377. doi:10.1021/acssuschemeng.0c08771
Return to citation in text: [1] [2] -
Liu, X.; Fechler, N.; Antonietti, M. Chem. Soc. Rev. 2013, 42, 8237–8265. doi:10.1039/c3cs60159e
Return to citation in text: [1] -
Wu, M.; Yan, J.-M.; Tang, X.-n.; Zhao, M.; Jiang, Q. ChemSusChem 2014, 7, 2654–2658. doi:10.1002/cssc.201402180
Return to citation in text: [1] -
Zhang, G.; Lin, L.; Li, G.; Zhang, Y.; Savateev, A.; Zafeiratos, S.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2018, 57, 9372–9376. doi:10.1002/anie.201804702
Return to citation in text: [1] -
Xu, Y.; He, X.; Zhong, H.; Singh, D. J.; Zhang, L.; Wang, R. Appl. Catal., B 2019, 246, 349–355. doi:10.1016/j.apcatb.2019.01.069
Return to citation in text: [1] -
Lang, X.; Chen, X.; Zhao, J. Chem. Soc. Rev. 2014, 43, 473–486. doi:10.1039/c3cs60188a
Return to citation in text: [1] -
Friedmann, D.; Hakki, A.; Kim, H.; Choi, W.; Bahnemann, D. Green Chem. 2016, 18, 5391–5411. doi:10.1039/c6gc01582d
Return to citation in text: [1] -
Chen, J.; Cen, J.; Xu, X.; Li, X. Catal. Sci. Technol. 2016, 6, 349–362. doi:10.1039/c5cy01289a
Return to citation in text: [1] -
Savateev, A.; Antonietti, M. ACS Catal. 2018, 8, 9790–9808. doi:10.1021/acscatal.8b02595
Return to citation in text: [1] -
Savateev, A.; Ghosh, I.; König, B.; Antonietti, M. Angew. Chem., Int. Ed. 2018, 57, 15936–15947. doi:10.1002/anie.201802472
Return to citation in text: [1] -
Zheng, Y.; Lin, L.; Wang, B.; Wang, X. Angew. Chem., Int. Ed. 2015, 54, 12868–12884. doi:10.1002/anie.201501788
Return to citation in text: [1] -
Markushyna, Y.; Smith, C. A.; Savateev, A. Eur. J. Org. Chem. 2020, 1294–1309. doi:10.1002/ejoc.201901112
Return to citation in text: [1] -
Cai, Y.; Tang, Y.; Fan, L.; Lefebvre, Q.; Hou, H.; Rueping, M. ACS Catal. 2018, 8, 9471–9476. doi:10.1021/acscatal.8b02937
Return to citation in text: [1] -
Geng, P.; Tang, Y.; Pan, G.; Wang, W.; Hu, J.; Cai, Y. Green Chem. 2019, 21, 6116–6122. doi:10.1039/c9gc02870f
Return to citation in text: [1] -
Kurpil, B.; Otte, K.; Mishchenko, A.; Lamagni, P.; Lipiński, W.; Lock, N.; Antonietti, M.; Savateev, A. Nat. Commun. 2019, 10, 945. doi:10.1038/s41467-019-08652-w
Return to citation in text: [1] -
Markushyna, Y.; Teutloff, C.; Kurpil, B.; Cruz, D.; Lauermann, I.; Zhao, Y.; Antonietti, M.; Savateev, A. Appl. Catal., B 2019, 248, 211–217. doi:10.1016/j.apcatb.2019.02.016
Return to citation in text: [1] -
Mazzanti, S.; Kurpil, B.; Pieber, B.; Antonietti, M.; Savateev, A. Nat. Commun. 2020, 11, 1387. doi:10.1038/s41467-020-15131-0
Return to citation in text: [1] -
Yang, C.; Li, R.; Zhang, K. A. I.; Lin, W.; Landfester, K.; Wang, X. Nat. Commun. 2020, 11, 1239. doi:10.1038/s41467-020-14983-w
Return to citation in text: [1] -
Wang, J.; Xue, L.; Hong, M.; Ni, B.; Niu, T. Green Chem. 2020, 22, 411–416. doi:10.1039/c9gc03679b
Return to citation in text: [1] -
Wen, J.; Yang, X.; Sun, Z.; Yang, J.; Han, P.; Liu, Q.; Dong, H.; Gu, M.; Huang, L.; Wang, H. Green Chem. 2020, 22, 230–237. doi:10.1039/c9gc03580j
Return to citation in text: [1] -
Ghosh, I.; Khamrai, J.; Savateev, A.; Shlapakov, N.; Antonietti, M.; König, B. Science 2019, 365, 360–366. doi:10.1126/science.aaw3254
Return to citation in text: [1] -
Pieber, B.; Malik, J. A.; Cavedon, C.; Gisbertz, S.; Savateev, A.; Cruz, D.; Heil, T.; Zhang, G.; Seeberger, P. H. Angew. Chem., Int. Ed. 2019, 58, 9575–9580. doi:10.1002/anie.201902785
Return to citation in text: [1] -
Cavedon, C.; Madani, A.; Seeberger, P. H.; Pieber, B. Org. Lett. 2019, 21, 5331–5334. doi:10.1021/acs.orglett.9b01957
Return to citation in text: [1] -
Khamrai, J.; Ghosh, I.; Savateev, A.; Antonietti, M.; König, B. ACS Catal. 2020, 10, 3526–3532. doi:10.1021/acscatal.9b05598
Return to citation in text: [1] -
Qin, Y.; Martindale, B. C. M.; Sun, R.; Rieth, A. J.; Nocera, D. G. Chem. Sci. 2020, 11, 7456–7461. doi:10.1039/d0sc02131h
Return to citation in text: [1] -
Slutskyy, Y.; Overman, L. E. Org. Lett. 2016, 18, 2564–2567. doi:10.1021/acs.orglett.6b00895
Return to citation in text: [1] -
Jin, Y.; Yang, H.; Fu, H. Org. Lett. 2016, 18, 6400–6403. doi:10.1021/acs.orglett.6b03300
Return to citation in text: [1] -
Wang, G.-Z.; Shang, R.; Fu, Y. Org. Lett. 2018, 20, 888–891. doi:10.1021/acs.orglett.8b00023
Return to citation in text: [1] -
Tang, Q.; Liu, X.; Liu, S.; Xie, H.; Liu, W.; Zeng, J.; Cheng, P. RSC Adv. 2015, 5, 89009–89014. doi:10.1039/c5ra17292f
Return to citation in text: [1] -
Kachkovskyi, G.; Faderl, C.; Reiser, O. Adv. Synth. Catal. 2013, 355, 2240–2248. doi:10.1002/adsc.201300221
Return to citation in text: [1]
| 32. | Yang, Q.; Pan, G.; Wei, J.; Wang, W.; Tang, Y.; Cai, Y. ACS Sustainable Chem. Eng. 2021, 9, 2367–2377. doi:10.1021/acssuschemeng.0c08771 |
| 61. | Kachkovskyi, G.; Faderl, C.; Reiser, O. Adv. Synth. Catal. 2013, 355, 2240–2248. doi:10.1002/adsc.201300221 |
| 1. | Newhouse, T.; Baran, P. S. J. Am. Chem. Soc. 2008, 130, 10886–10887. doi:10.1021/ja8042307 |
| 2. | Mohn, T.; Plitzko, I.; Hamburger, M. Phytochemistry 2009, 70, 924–934. doi:10.1016/j.phytochem.2009.04.019 |
| 3. | Zhang, D.; Song, H.; Qin, Y. Acc. Chem. Res. 2011, 44, 447–457. doi:10.1021/ar200004w |
| 4. | Matsuura, T.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6500–6503. doi:10.1021/ja980788+ |
| 13. | Girard, S. A.; Knauber, T.; Li, C.-J. Angew. Chem., Int. Ed. 2014, 53, 74–100. doi:10.1002/anie.201304268 |
| 14. | López, R.; Palomo, C. Angew. Chem., Int. Ed. 2015, 54, 13170–13184. doi:10.1002/anie.201502493 |
| 15. | Song, R.-J.; Liu, Y.; Xie, Y.-X.; Li, J.-H. Synthesis 2015, 47, 1195–1209. doi:10.1055/s-0034-1379903 |
| 16. | Li, C.-C.; Yang, S.-D. Org. Biomol. Chem. 2016, 14, 4365–4377. doi:10.1039/c6ob00554c |
| 17. | Chu, X.-Q.; Ge, D.; Shen, Z.-L.; Loh, T.-P. ACS Catal. 2018, 8, 258–271. doi:10.1021/acscatal.7b03334 |
| 4. | Matsuura, T.; Overman, L. E.; Poon, D. J. J. Am. Chem. Soc. 1998, 120, 6500–6503. doi:10.1021/ja980788+ |
| 11. | Billamboz, M.; Bailly, F.; Barreca, M. L.; De Luca, L.; Mouscadet, J.-F.; Calmels, C.; Andréola, M.-L.; Witvrouw, M.; Christ, F.; Debyser, Z.; Cotelle, P. J. Med. Chem. 2008, 51, 7717–7730. doi:10.1021/jm8007085 |
| 12. | Billamboz, M.; Bailly, F.; Lion, C.; Touati, N.; Vezin, H.; Calmels, C.; Andréola, M.-L.; Christ, F.; Debyser, Z.; Cotelle, P. J. Med. Chem. 2011, 54, 1812–1824. doi:10.1021/jm1014692 |
| 18. | Wu, T.; Mu, X.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 12578–12581. doi:10.1002/anie.201104575 |
| 19. | Li, J.; Wang, Z.; Wu, N.; Gao, G.; You, J. Chem. Commun. 2014, 50, 15049–15051. doi:10.1039/c4cc07667b |
| 20. | Pan, C.; Zhang, H.; Zhu, C. Org. Biomol. Chem. 2015, 13, 361–364. doi:10.1039/c4ob02172j |
| 21. | Li, X.; Xu, J.; Gao, Y.; Fang, H.; Tang, G.; Zhao, Y. J. Org. Chem. 2015, 80, 2621–2626. doi:10.1021/jo502777b |
| 22. | Tang, S.; Li, S.-H.; Li, Z.-H.; Zhou, D.; Sheng, R.-L. Tetrahedron Lett. 2015, 56, 1423–1426. doi:10.1016/j.tetlet.2015.01.175 |
| 23. | Ni, Z.; Huang, X.; Wang, J.; Pan, Y. RSC Adv. 2016, 6, 522–526. doi:10.1039/c5ra23471a |
| 24. | Liang, D.; Song, X.; Xu, L.; Sun, Y.; Dong, Y.; Wang, B.; Li, W. Tetrahedron 2019, 75, 3495–3503. doi:10.1016/j.tet.2019.05.018 |
| 25. | Pan, C.; Wang, Y.; Wu, C.; Yu, J.-T. Catal. Commun. 2019, 131, 105802. doi:10.1016/j.catcom.2019.105802 |
| 26. | Zhang, H.; Chen, P.; Liu, G. Synlett 2012, 23, 2749–2752. doi:10.1055/s-0032-1316555 |
| 31. | Lu, M.; Zhang, T.; Tan, D.; Chen, C.; Zhang, Y.; Huang, M.; Cai, S. Adv. Synth. Catal. 2019, 361, 4237–4242. doi:10.1002/adsc.201900712 |
| 60. | Tang, Q.; Liu, X.; Liu, S.; Xie, H.; Liu, W.; Zeng, J.; Cheng, P. RSC Adv. 2015, 5, 89009–89014. doi:10.1039/c5ra17292f |
| 8. | Scott, J. D.; Williams, R. M. Chem. Rev. 2002, 102, 1669–1730. doi:10.1021/cr010212u |
| 9. | Kaila, N.; Follows, B.; Leung, L.; Thomason, J.; Huang, A.; Moretto, A.; Janz, K.; Lowe, M.; Mansour, T. S.; Hubeau, C.; Page, K.; Morgan, P.; Fish, S.; Xu, X.; Williams, C.; Saiah, E. J. Med. Chem. 2014, 57, 1299–1322. doi:10.1021/jm401509e |
| 10. | Hekal, M. H.; Abu El‐Azm, F. S. M.; Sallam, H. A. J. Heterocycl. Chem. 2019, 56, 795–803. doi:10.1002/jhet.3448 |
| 44. | Cai, Y.; Tang, Y.; Fan, L.; Lefebvre, Q.; Hou, H.; Rueping, M. ACS Catal. 2018, 8, 9471–9476. doi:10.1021/acscatal.8b02937 |
| 45. | Geng, P.; Tang, Y.; Pan, G.; Wang, W.; Hu, J.; Cai, Y. Green Chem. 2019, 21, 6116–6122. doi:10.1039/c9gc02870f |
| 46. | Kurpil, B.; Otte, K.; Mishchenko, A.; Lamagni, P.; Lipiński, W.; Lock, N.; Antonietti, M.; Savateev, A. Nat. Commun. 2019, 10, 945. doi:10.1038/s41467-019-08652-w |
| 47. | Markushyna, Y.; Teutloff, C.; Kurpil, B.; Cruz, D.; Lauermann, I.; Zhao, Y.; Antonietti, M.; Savateev, A. Appl. Catal., B 2019, 248, 211–217. doi:10.1016/j.apcatb.2019.02.016 |
| 48. | Mazzanti, S.; Kurpil, B.; Pieber, B.; Antonietti, M.; Savateev, A. Nat. Commun. 2020, 11, 1387. doi:10.1038/s41467-020-15131-0 |
| 49. | Yang, C.; Li, R.; Zhang, K. A. I.; Lin, W.; Landfester, K.; Wang, X. Nat. Commun. 2020, 11, 1239. doi:10.1038/s41467-020-14983-w |
| 50. | Wang, J.; Xue, L.; Hong, M.; Ni, B.; Niu, T. Green Chem. 2020, 22, 411–416. doi:10.1039/c9gc03679b |
| 51. | Wen, J.; Yang, X.; Sun, Z.; Yang, J.; Han, P.; Liu, Q.; Dong, H.; Gu, M.; Huang, L.; Wang, H. Green Chem. 2020, 22, 230–237. doi:10.1039/c9gc03580j |
| 52. | Ghosh, I.; Khamrai, J.; Savateev, A.; Shlapakov, N.; Antonietti, M.; König, B. Science 2019, 365, 360–366. doi:10.1126/science.aaw3254 |
| 53. | Pieber, B.; Malik, J. A.; Cavedon, C.; Gisbertz, S.; Savateev, A.; Cruz, D.; Heil, T.; Zhang, G.; Seeberger, P. H. Angew. Chem., Int. Ed. 2019, 58, 9575–9580. doi:10.1002/anie.201902785 |
| 54. | Cavedon, C.; Madani, A.; Seeberger, P. H.; Pieber, B. Org. Lett. 2019, 21, 5331–5334. doi:10.1021/acs.orglett.9b01957 |
| 55. | Khamrai, J.; Ghosh, I.; Savateev, A.; Antonietti, M.; König, B. ACS Catal. 2020, 10, 3526–3532. doi:10.1021/acscatal.9b05598 |
| 56. | Qin, Y.; Martindale, B. C. M.; Sun, R.; Rieth, A. J.; Nocera, D. G. Chem. Sci. 2020, 11, 7456–7461. doi:10.1039/d0sc02131h |
| 5. | Marti, C.; Carreira, E. M. Eur. J. Org. Chem. 2003, 2209–2219. doi:10.1002/ejoc.200300050 |
| 6. | Jiang, T.; Kuhen, K. L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Tuntland, T.; Zhang, K.; Karanewsky, D.; He, Y. Bioorg. Med. Chem. Lett. 2006, 16, 2109–2112. doi:10.1016/j.bmcl.2006.01.066 |
| 7. | Singh, G. S.; Desta, Z. Y. Chem. Rev. 2012, 112, 6104–6155. doi:10.1021/cr300135y |
| 57. | Slutskyy, Y.; Overman, L. E. Org. Lett. 2016, 18, 2564–2567. doi:10.1021/acs.orglett.6b00895 |
| 58. | Jin, Y.; Yang, H.; Fu, H. Org. Lett. 2016, 18, 6400–6403. doi:10.1021/acs.orglett.6b03300 |
| 59. | Wang, G.-Z.; Shang, R.; Fu, Y. Org. Lett. 2018, 20, 888–891. doi:10.1021/acs.orglett.8b00023 |
| 32. | Yang, Q.; Pan, G.; Wei, J.; Wang, W.; Tang, Y.; Cai, Y. ACS Sustainable Chem. Eng. 2021, 9, 2367–2377. doi:10.1021/acssuschemeng.0c08771 |
| 37. | Lang, X.; Chen, X.; Zhao, J. Chem. Soc. Rev. 2014, 43, 473–486. doi:10.1039/c3cs60188a |
| 38. | Friedmann, D.; Hakki, A.; Kim, H.; Choi, W.; Bahnemann, D. Green Chem. 2016, 18, 5391–5411. doi:10.1039/c6gc01582d |
| 39. | Chen, J.; Cen, J.; Xu, X.; Li, X. Catal. Sci. Technol. 2016, 6, 349–362. doi:10.1039/c5cy01289a |
| 29. | Liu, Y.; Zhang, J.-L.; Song, R.-J.; Li, J.-H. Org. Chem. Front. 2014, 1, 1289–1294. doi:10.1039/c4qo00251b |
| 30. | Zhang, J.-L.; Liu, Y.; Song, R.-J.; Jiang, G.-F.; Li, J.-H. Synlett 2014, 25, 1031–1035. doi:10.1055/s-0033-1340956 |
| 31. | Lu, M.; Zhang, T.; Tan, D.; Chen, C.; Zhang, Y.; Huang, M.; Cai, S. Adv. Synth. Catal. 2019, 361, 4237–4242. doi:10.1002/adsc.201900712 |
| 40. | Savateev, A.; Antonietti, M. ACS Catal. 2018, 8, 9790–9808. doi:10.1021/acscatal.8b02595 |
| 41. | Savateev, A.; Ghosh, I.; König, B.; Antonietti, M. Angew. Chem., Int. Ed. 2018, 57, 15936–15947. doi:10.1002/anie.201802472 |
| 42. | Zheng, Y.; Lin, L.; Wang, B.; Wang, X. Angew. Chem., Int. Ed. 2015, 54, 12868–12884. doi:10.1002/anie.201501788 |
| 43. | Markushyna, Y.; Smith, C. A.; Savateev, A. Eur. J. Org. Chem. 2020, 1294–1309. doi:10.1002/ejoc.201901112 |
| 27. | Chen, J.-R.; Hu, X.-Q.; Lu, L.-Q.; Xiao, W.-J. Chem. Soc. Rev. 2016, 45, 2044–2056. doi:10.1039/c5cs00655d |
| 28. | Marzo, L.; Pagire, S. K.; Reiser, O.; König, B. Angew. Chem., Int. Ed. 2018, 57, 10034–10072. doi:10.1002/anie.201709766 |
| 18. | Wu, T.; Mu, X.; Liu, G. Angew. Chem., Int. Ed. 2011, 50, 12578–12581. doi:10.1002/anie.201104575 |
| 19. | Li, J.; Wang, Z.; Wu, N.; Gao, G.; You, J. Chem. Commun. 2014, 50, 15049–15051. doi:10.1039/c4cc07667b |
| 20. | Pan, C.; Zhang, H.; Zhu, C. Org. Biomol. Chem. 2015, 13, 361–364. doi:10.1039/c4ob02172j |
| 21. | Li, X.; Xu, J.; Gao, Y.; Fang, H.; Tang, G.; Zhao, Y. J. Org. Chem. 2015, 80, 2621–2626. doi:10.1021/jo502777b |
| 22. | Tang, S.; Li, S.-H.; Li, Z.-H.; Zhou, D.; Sheng, R.-L. Tetrahedron Lett. 2015, 56, 1423–1426. doi:10.1016/j.tetlet.2015.01.175 |
| 23. | Ni, Z.; Huang, X.; Wang, J.; Pan, Y. RSC Adv. 2016, 6, 522–526. doi:10.1039/c5ra23471a |
| 24. | Liang, D.; Song, X.; Xu, L.; Sun, Y.; Dong, Y.; Wang, B.; Li, W. Tetrahedron 2019, 75, 3495–3503. doi:10.1016/j.tet.2019.05.018 |
| 25. | Pan, C.; Wang, Y.; Wu, C.; Yu, J.-T. Catal. Commun. 2019, 131, 105802. doi:10.1016/j.catcom.2019.105802 |
| 26. | Zhang, H.; Chen, P.; Liu, G. Synlett 2012, 23, 2749–2752. doi:10.1055/s-0032-1316555 |
| 33. | Liu, X.; Fechler, N.; Antonietti, M. Chem. Soc. Rev. 2013, 42, 8237–8265. doi:10.1039/c3cs60159e |
| 34. | Wu, M.; Yan, J.-M.; Tang, X.-n.; Zhao, M.; Jiang, Q. ChemSusChem 2014, 7, 2654–2658. doi:10.1002/cssc.201402180 |
| 35. | Zhang, G.; Lin, L.; Li, G.; Zhang, Y.; Savateev, A.; Zafeiratos, S.; Wang, X.; Antonietti, M. Angew. Chem., Int. Ed. 2018, 57, 9372–9376. doi:10.1002/anie.201804702 |
| 36. | Xu, Y.; He, X.; Zhong, H.; Singh, D. J.; Zhang, L.; Wang, R. Appl. Catal., B 2019, 246, 349–355. doi:10.1016/j.apcatb.2019.01.069 |
© 2021 Pan et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the author(s) and source are credited and that individual graphics may be subject to special legal provisions.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc/terms)