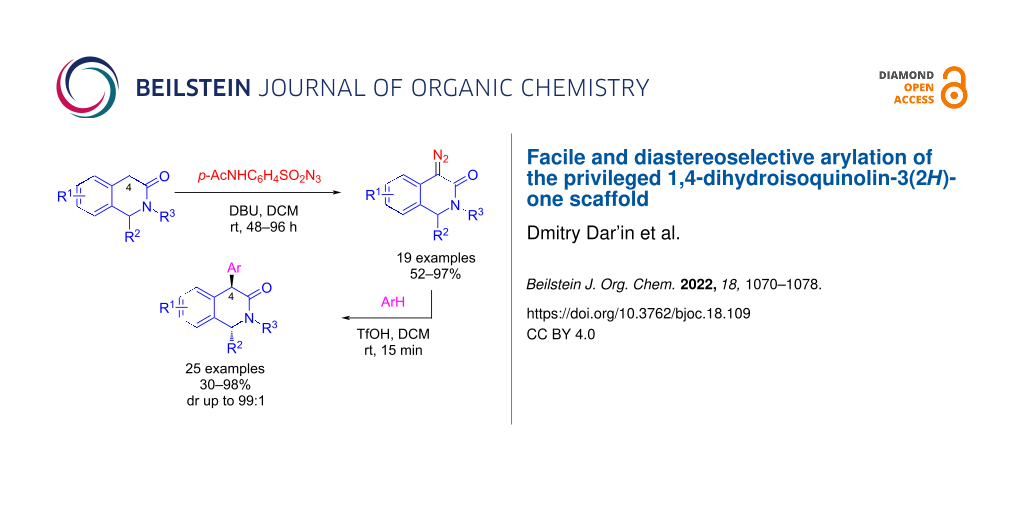
Abstract
A practically convenient and streamlined protocol for the trans-diastereoselective introduction of an aryl substituent at position 4 of the 1,4-dihydroisoquinol-3-one (1,4-DHIQ) scaffold is presented. The protocol involves direct Regitz diazo transfer onto readily available 3(2H)-isoquinolones followed by TfOH-promoted hydroarylation by an arene molecule. Screening of the novel 1,2,4-trisubstituted 1,4-DHIQs against cancer cell lines confirmed high cytotoxicity of selected analogs, which validates this new chemotype for further investigations as anticancer cytotoxic agents.

Graphical Abstract
Introduction
Besides being derivatives of (or a precursor to) the 1,2,3,4-tetrahydroisoquinoline core which itself bears a special significance from the standpoint of associated biological activities and relevance to the naturally occurring alkaloids [1], 1,4-dihydro-3(2H)-isoquinolones (1,4-DHIQs) undoubtedly represent a privileged scaffold [2] for drug design considering such diversely bioactive compounds documented in the literature as ligand for serotonin 5-HT1A receptors 1 [3], AChE and BACE-1 inhibitor 2 [4], inhibitor of oncogenic p53-MDM2 protein–protein interaction 3 [5], positive allosteric modulator of ionotropic glutamate receptor NMDA-1 4 [6], insulin-like growth factor 1 receptor inhibitor 5 [7], and metabotropic glutamate receptor 7 modulator 6 [8] (Figure 1). The privileged 1,4-DHIQs would be a highly suitable platform for a stereodefined presentation of three different diversity vectors of the lactam moiety. However, the methods for the preparation of 1,4-DHIQs with convenient and independent variation of the three lactam substituents are absent in the literature. While pondering possible solutions to fill this void, we drew inspiration in our recent success achieving direct Brønsted acid-catalyzed C-arylation of 4-diazo-isoquinoline-1,3-diones 7 [9] which are, in turn, obtainable via the Regitz diazo transfer reaction onto readily available homophthalimides 8 [10-12]. We reasoned that a similar strategy could be adopted for the preparation of 1,2,4-trisubstituted 1,4-DHIQs 9 if access to their diazo precursors 10 was gained. N-Sulfonyl analogs of compounds 10 have recently been synthesized via an innovative Dimroth rearrangement of 4-diazoisochroman-3-imines [13] and employed in several acid- and metal-promoted transformations by Lu, Wang et al. [14-18]. However, preparation of precursor 10 by direct diazo transfer onto the methylene group of readily available [19,20] 3(2H)-isoquinolones 11 has not been described in the literature. Lured by the prospects of applying a diazo chemistry route to an expedited synthesis of hitherto undescribed 1,2,4-trisubstituted 1,4-DHIQs 9 from 11, we set off to investigate the obtainability of 10 from 11 by the Regitz [21] diazo transfer and the usage of 10 in acid-promoted direct arylation by aromatic hydrocarbons (Figure 2) [13]. Herein, we describe the results obtained in the course of this investigation.
![[1860-5397-18-109-1]](/bjoc/content/figures/1860-5397-18-109-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Diverse bioactive compounds based on the privileged 1,4-DHIQ scaffold.
Figure 1: Diverse bioactive compounds based on the privileged 1,4-DHIQ scaffold.
![[1860-5397-18-109-2]](/bjoc/content/figures/1860-5397-18-109-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Strategy investigated in this work.
Figure 2: Strategy investigated in this work.
Results and Discussion
We began our investigation by synthesizing a sufficiently broad range of 3(2H)-isoquinolones 11 from phenylacetyl chlorides 12 and Schiff bases 13 using conditions described in the literature [19,20]. It was soon established that room-temperature TfOH-promoted cyclocondensation (method A) [19] and AlCl3-promoted reaction conducted at elevated temperature (method B) can be employed interchangeably, with yields registered for 3(2H)-isoquinolones 11 mostly from good to excellent. While the scope of the 3(2H)-isoquinolone synthesis was substantially expanded by these findings compared to the previously reported results [21], certain limitations were also noted. For example, attempts to involve imines derived from ethyl glyoxylate, cyclohexanone, and cinnamaldehyde gave no desired product (11t–v). Quite surprisingly, the Schiff base derived from methyl o-formylbenzoate also gave no desired product while cyclization led to isobenzofuran-1(3H)-one 14 (Scheme 1).
![[1860-5397-18-109-i1]](/bjoc/content/inline/1860-5397-18-109-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Preparation of 3(2H)-isoquinolones 11. aObtained as a 10:1 mixture of regioisomers; purified by crystallization. bEmployed in the next step without purification (not characterized). cCompound 14 was identified as the reaction product.
Scheme 1: Preparation of 3(2H)-isoquinolones 11. aObtained as a 10:1 mixture of regioisomers; purified by cry...
Having secured a supply of diversely substituted 3(2H)-isoquinolones 11a–s, we proceeded to investigate their suitability as substrates in the Regitz diazo transfer. We reasoned that if the C–H acidity of the methylene group in 11 would turn out to be insufficient for the Regitz protocol to be directly applied, these substrates could have been additionally activated by trifluoroacetylation (Danheiser method [22]) or ethoxalylation [23]. Fortunately, all of the substrates 11a–s were converted cleanly and smoothly over 2–5 days into their diazo derivatives 10a–s using p-(acetamido)benzenesulfonyl azide (p-ABSA) as the diazo group donor [24] and DBU as the base. The yields of diazo compounds 10 were generally good to excellent throughout. The notable exception is the evident drop in the isolated yield in the case of substrates bearing a nitrophenyl group at position 1 (cf. compounds 10j and 10n). This lowering of the yield, however, likely has to do with the combination of the nitrophenyl substituent and an N-alkyl group (considering that N-aryl nitrophenyl-substituted compound 10o was obtained in a respectable 91% yield). The structures of compounds 10a–s were unequivocally confirmed by 1H and 13C NMR spectroscopy (paying a particular attention to the appearance of the C=N2 signal in the spectrum), mass spectrometry and, in the case of compound 10c, by single-crystal X-ray crystallography (Scheme 2).
![[1860-5397-18-109-i2]](/bjoc/content/inline/1860-5397-18-109-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Preparation of 4-diazo-3(2H)-isoquinolones 10. aConfirmed by single-crystal X-ray crystallography (see Supporting Information File 1).
Scheme 2: Preparation of 4-diazo-3(2H)-isoquinolones 10. aConfirmed by single-crystal X-ray crystallography (...
Using compound 10a as the model substrate, we proceeded to screen for suitable reaction conditions that would allow involving this kind of diazo compounds in the TfOH-promoted benzene C-arylation reaction (Table 1) [13]. The excess of benzene was varied in the range 10 to 60 equiv and the yield of product 9a was found to improve from 63% to 83% which also allowed lowering the excess of triflic acid to 1.5 equiv. Product 9a was in all cases obtained after only 15 min reaction with high diastereoselectivity and the principal diastereomer was rightly deemed to be trans-configured (vide infra). The order of mixing the reagents was found to be crucial for the successful arylation of compound 10a. Specifically, the arylation was conducted on adding a DCM solution of substrate 10a to a vigorously stirred mixture of benzene and triflic acid. On the contrary, when adding TfOH to a solution of 10a in benzene, the formation of a complex mixture of unidentifiable products was observed, with only a trace amount of 9a detectable by 1H NMR.
Table 1: Conditions screening for the TfOH-promoted arylation of diazo substrate 10a.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-109-i6.svg?max-width=637&scale=1.0)
|
||||
| Entry |
PhH
(equiv) |
TfOH
(equiv) |
% Yield of 9a (dr) | % Yield of 15a |
| 1 | 10 | 2.0 | 63% (98:2) | 26% |
| 2 | 20 | 2.0 | 73% (98:2) | 20% |
| 3 | 60 | 2.0 | 80% (98:2) | 8% |
| 4 | 60 | 1.5 | 83% (98:2) | 13% |
| 5 | 60 | 1.0 | 65% (96:4) | 19% |
| 6 | 0 | 1.1 | – | 51% |
aConcentration: 0.15 M, scale: 0.3 mmol.
The formation of product 9a was unavoidably accompanied by a varying amount of byproduct 15a (vide infra for the mechanistic reasoning for its formation) which demonstrated, along with other analogs of 15 which were isolated and characterized, limited chemical stability and deteriorated on prolonged standing as a solution in CDCl3 at ambient temperature. Compound 15a was the exclusive isolable product when benzene was eliminated from the reaction mixture (Table 1, entry 6). Interestingly, the formation of byproduct 15 (which could be, in principle, obtained by DDQ oxidation of 11 [25]) via the elimination of a diazo group has not been reported.
Having identified the optimum conditions for the C-arylation of diazo substrates 10 (60 equiv ArH, 1.5 equiv TfOH, DCM, rt, 15 min), we proceeded to investigate the scope of this transformation for 4-diazo-3(2H)-isoquinolones 10a–s as well as various arenes (Scheme 3). In all cases, this (generally high-yielding) transformation resulted in the highly diastereoselective formation of C-arylation products 10 which in some cases was accompanied by a regioisomer formation (with respect to the entering arene moiety) and the formation of 3-isoquinolones 15 (isolated and characterized in several instances). Not unexpectedly, less electron-rich arenes furnished higher amounts of the 3-isoquinolone 15 byproduct compared to their electron-rich counterparts (cf. 9o vs 9p).
![[1860-5397-18-109-i3]](/bjoc/content/inline/1860-5397-18-109-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: TfOH-promoted arylation of diazo substrates 10. aStructure confirmed by single-crystal X-ray analysis. bYields in parentheses estimated by 1H NMR. cRun on 5 × scale (1.5 mmol) with 88% yield. d5.0 equiv of arene was used.
Scheme 3: TfOH-promoted arylation of diazo substrates 10. aStructure confirmed by single-crystal X-ray analys...
Likewise, electron-donating substituents in the 4-diazo-3(2H)-isoquinolone benzene ring increased the tendency of 3-isoquinolones 15 to form (cf. 9(15)f–h). Using more reactive (electron-rich) arenes results in lower diastereoselectivity and regiospecificity of the reaction (cf. 9q, 9v, and 9w). The structure and the initially anticipated trans-configuration of the products 9 was unequivocally confirmed by 1H and 13C NMR spectroscopy as well as, in the case of compound 9a, by single-crystal X-ray analysis.
A curious and somewhat unexpected result was obtained when trying to employ N-formyl-N-methylaniline as an arene in the TfOH-promoted arylation of 10a. Instead of the anticipated product 9aa, 73% yield of predominantly trans-configured formate ester 16 was obtained and its structure was confirmed by single-crystal X-ray analysis. Presumably, the formation of ester 16 can be justified by the trapping of the carbocation intermediate 17 (vide infra) by the formamide carbonyl oxygen atom followed by hydrolysis of the iminium moiety (Scheme 4).
![[1860-5397-18-109-i4]](/bjoc/content/inline/1860-5397-18-109-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Unexpected outcome of the TfOH-promoted arylation of 10a with N-formyl-N-methylaniline giving rise to ester 16.
Scheme 4: Unexpected outcome of the TfOH-promoted arylation of 10a with N-formyl-N-methylaniline giving rise ...
Mechanistically, the arylation of diazo substrates 10 likely proceeds via the protonation of the diazo moiety and elimination of a nitrogen molecule, whereby carbocation 17 is generated. The latter can either be deprotonated to give byproduct 15 or be intercepted by an arene molecule in SEAr fashion to give arylation product 9. The trans-diastereoselectivity in the latter process is reasonably justified by the approach of the arene molecule to carbocation 17 from the sterically less hindered side (Scheme 5).
![[1860-5397-18-109-i5]](/bjoc/content/inline/1860-5397-18-109-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Plausible mechanism for the conversion of diazo substrates 10 to 4-aryl products 9 (shown for ArH = benzene) and 3-isoquinoline byproducts 15.
Scheme 5: Plausible mechanism for the conversion of diazo substrates 10 to 4-aryl products 9 (shown for ArH =...
Compounds 9 have a pronounced three-dimensional character which makes this novel chemotype a promising probe for protein–protein interactions, including oncogenic ones [26]. As the first step towards biological characterization of compounds 9, they were screened for cytotoxicity against the NCI-H460 lung carcinoma cell line. The most potent cytotoxic agent (9j) reduced the number of viable cells by >95% at 30 μM concentration. Dose–response testing of this compound against both NCI-H460 and A549 lung carcinoma cell lines resulted in the determination of IC50 values for compound 9j as 31.4 ± 0.48 μM and 13.6 ± 3.34 μM, respectively (see Supporting Information File 1 for details).
Conclusion
In summary, we have presented a practically convenient and streamlined protocol for the trans-diastereoselective introduction of an aryl substituent at position 4 of the 1,4-dihydroisoquinol-3-one (1,4-DHIQ) scaffold. The protocol relies on hitherto undescribed direct Regitz diazo transfer onto readily available 3(2H)-isoquinolones followed by TfOH-promoted arylation. The generally high-yielding two-step sequence was shown to be applicable to a wide range of substrates. To a varying degree, the arylation step was accompanied by the elimination of the nitrogen molecule and deprotonation to furnish 3-isoquinolone byproducts (formed exclusively in the absence of the arene). The extent of this side reaction was found to be dependent on the electronic character of the 1,4-DHIQs and the carbocation-intercepting arene molecule. Considering the pronounced three-dimensional character of 1,2,4-trisubstituted 1,4-DHIQ adducts synthesized in this work, they were deemed efficient probes for the perturbation of vital cellular targets. Screening of these compounds against lung carcinoma cancer cell lines confirmed high cytotoxicity of selected analogs, which validates this new chemotype for further investigation as anticancer cytotoxic agents.
Supporting Information
Deposition Numbers 2158046 (for 9a), 2170881 (for 10c) and 2170877 (for 16) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre (http://www.ccdc.cam.ac.uk/structures) and Fachinformationszentrum Karlsruhe Access Structures service.
| Supporting Information File 1: General experimental information, X-ray crystallographic data, synthetic procedures, analytical data and NMR spectra for the reported compounds. | ||
| Format: PDF | Size: 12.5 MB | Download |
References
-
Faheem; Karan Kumar, B.; Chandra Sekhar, K. V. G.; Chander, S.; Kunjiappan, S.; Murugesan, S. RSC Adv. 2021, 11, 12254–12287. doi:10.1039/d1ra01480c
Return to citation in text: [1] -
Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347–361. doi:10.1016/j.cbpa.2010.02.018
Return to citation in text: [1] -
Mokrosz, J. L.; Bojarski, A. J.; Mackowiak, M.; Bielecka, Z.; Boksa, J. Pharmazie 1994, 49, 328–333.
Return to citation in text: [1] -
Zhao, X.-j.; Gong, D.-m.; Jiang, Y.-r.; Guo, D.; Zhu, Y.; Deng, Y.-c. Eur. J. Med. Chem. 2017, 138, 738–747. doi:10.1016/j.ejmech.2017.07.006
Return to citation in text: [1] -
Gessier, F.; Kallen, J.; Jacoby, E.; Chène, P.; Stachyra-Valat, T.; Ruetz, S.; Jeay, S.; Holzer, P.; Masuya, K.; Furet, P. Bioorg. Med. Chem. Lett. 2015, 25, 3621–3625. doi:10.1016/j.bmcl.2015.06.058
Return to citation in text: [1] -
Epplin, M. P.; Mohan, A.; Harris, L. D.; Zhu, Z.; Strong, K. L.; Bacsa, J.; Le, P.; Menaldino, D. S.; Traynelis, S. F.; Liotta, D. C. J. Med. Chem. 2020, 63, 7569–7600. doi:10.1021/acs.jmedchem.9b01733
Return to citation in text: [1] -
Gunzinger, J.; Leander, K. Isoquinolines as IGF-1R inhibitors. WO Pat. Appl. WO 2007/029106 A1, March 15, 2007.
Return to citation in text: [1] -
Teall, M.; White, K.; Mack, S.; Liwicki, G.; Stephenson, A.; Dickson, L. MGLUR7 Agonist compounds for treating MGLUR7 – regulated diseases, disorders, or conditions. WO Patent WO/2018/092921, May 24, 2018.
Return to citation in text: [1] -
Golushko, A.; Dar’in, D.; Kantin, G.; Guranova, N.; Vasilyev, A. V.; Krasavin, M. Synthesis 2019, 51, 3815–3824. doi:10.1055/s-0037-1611882
Return to citation in text: [1] -
Kantin, G.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2018, 4857–4859. doi:10.1002/ejoc.201800955
Return to citation in text: [1] -
Dar’in, D.; Kantin, G.; Krasavin, M. Chem. Commun. 2019, 55, 5239–5242. doi:10.1039/c9cc02042j
Return to citation in text: [1] -
Dar'in, D.; Kantin, G.; Chupakhin, E.; Sharoyko, V.; Krasavin, M. Chem. – Eur. J. 2021, 27, 8221–8227. doi:10.1002/chem.202100880
Return to citation in text: [1] -
Li, Z.; Chen, J.; Wu, L.; Ren, A.; Lu, P.; Wang, Y. Org. Lett. 2020, 22, 26–30. doi:10.1021/acs.orglett.9b03708
Return to citation in text: [1] [2] [3] -
Li, Z.; Xie, J.; Lu, P.; Wang, Y. J. Org. Chem. 2020, 85, 5525–5535. doi:10.1021/acs.joc.0c00283
Return to citation in text: [1] -
Chen, J.; Li, Z.; Suleman, M.; Wang, Z.; Lu, P.; Wang, Y. Org. Biomol. Chem. 2020, 18, 7671–7676. doi:10.1039/d0ob01748e
Return to citation in text: [1] -
Wu, L.; Chen, J.; Xie, J.; Lu, P.; Wang, Y. Tetrahedron 2021, 84, 132019. doi:10.1016/j.tet.2021.132019
Return to citation in text: [1] -
Xie, J.; Suleman, M.; Wang, Z.; Mao, X.; Mao, B.; Fan, J.; Lu, P.; Wang, Y. Org. Biomol. Chem. 2021, 19, 6341–6345. doi:10.1039/d1ob00859e
Return to citation in text: [1] -
Qi, M.; Suleman, M.; Xie, J.; Lu, P.; Wang, Y. J. Org. Chem. 2022, 87, 4088–4096. doi:10.1021/acs.joc.1c02905
Return to citation in text: [1] -
Zhang, Y.; Kindelin, P. J.; DeSchepper, D. J.; Zheng, C.; Klumpp, D. A. Synthesis 2006, 1775–1780. doi:10.1055/s-2006-942387
Return to citation in text: [1] [2] [3] -
Venkov, A. P.; Mollov, N. M. Synthesis 1982, 216–217. doi:10.1055/s-1982-29752
Return to citation in text: [1] [2] -
Regitz, M. Tetrahedron Lett. 1964, 5, 1403–1407. doi:10.1016/s0040-4039(00)90489-1
Return to citation in text: [1] [2] -
Danheiser, R. L.; Miller, R. F.; Brisbois, R. G.; Park, S. Z. J. Org. Chem. 1990, 55, 1959–1964. doi:10.1021/jo00293a053
Return to citation in text: [1] -
Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133
Return to citation in text: [1] -
Baum, J. S.; Shook, D. A.; Davies, H. M. L.; Smith, H. D. Synth. Commun. 1987, 17, 1709–1716. doi:10.1080/00397918708063988
Return to citation in text: [1] -
Kaiser, D.; de la Torre, A.; Shaaban, S.; Maulide, N. Angew. Chem., Int. Ed. 2017, 56, 5921–5925. doi:10.1002/anie.201701538
Return to citation in text: [1] -
Kuenemann, M. A.; Bourbon, L. M. L.; Labbé, C. M.; Villoutreix, B. O.; Sperandio, O. J. Chem. Inf. Model. 2014, 54, 3067–3079. doi:10.1021/ci500487q
Return to citation in text: [1]
| 22. | Danheiser, R. L.; Miller, R. F.; Brisbois, R. G.; Park, S. Z. J. Org. Chem. 1990, 55, 1959–1964. doi:10.1021/jo00293a053 |
| 19. | Zhang, Y.; Kindelin, P. J.; DeSchepper, D. J.; Zheng, C.; Klumpp, D. A. Synthesis 2006, 1775–1780. doi:10.1055/s-2006-942387 |
| 21. | Regitz, M. Tetrahedron Lett. 1964, 5, 1403–1407. doi:10.1016/s0040-4039(00)90489-1 |
| 1. | Faheem; Karan Kumar, B.; Chandra Sekhar, K. V. G.; Chander, S.; Kunjiappan, S.; Murugesan, S. RSC Adv. 2021, 11, 12254–12287. doi:10.1039/d1ra01480c |
| 5. | Gessier, F.; Kallen, J.; Jacoby, E.; Chène, P.; Stachyra-Valat, T.; Ruetz, S.; Jeay, S.; Holzer, P.; Masuya, K.; Furet, P. Bioorg. Med. Chem. Lett. 2015, 25, 3621–3625. doi:10.1016/j.bmcl.2015.06.058 |
| 13. | Li, Z.; Chen, J.; Wu, L.; Ren, A.; Lu, P.; Wang, Y. Org. Lett. 2020, 22, 26–30. doi:10.1021/acs.orglett.9b03708 |
| 4. | Zhao, X.-j.; Gong, D.-m.; Jiang, Y.-r.; Guo, D.; Zhu, Y.; Deng, Y.-c. Eur. J. Med. Chem. 2017, 138, 738–747. doi:10.1016/j.ejmech.2017.07.006 |
| 19. | Zhang, Y.; Kindelin, P. J.; DeSchepper, D. J.; Zheng, C.; Klumpp, D. A. Synthesis 2006, 1775–1780. doi:10.1055/s-2006-942387 |
| 20. | Venkov, A. P.; Mollov, N. M. Synthesis 1982, 216–217. doi:10.1055/s-1982-29752 |
| 3. | Mokrosz, J. L.; Bojarski, A. J.; Mackowiak, M.; Bielecka, Z.; Boksa, J. Pharmazie 1994, 49, 328–333. |
| 19. | Zhang, Y.; Kindelin, P. J.; DeSchepper, D. J.; Zheng, C.; Klumpp, D. A. Synthesis 2006, 1775–1780. doi:10.1055/s-2006-942387 |
| 20. | Venkov, A. P.; Mollov, N. M. Synthesis 1982, 216–217. doi:10.1055/s-1982-29752 |
| 26. | Kuenemann, M. A.; Bourbon, L. M. L.; Labbé, C. M.; Villoutreix, B. O.; Sperandio, O. J. Chem. Inf. Model. 2014, 54, 3067–3079. doi:10.1021/ci500487q |
| 2. | Welsch, M. E.; Snyder, S. A.; Stockwell, B. R. Curr. Opin. Chem. Biol. 2010, 14, 347–361. doi:10.1016/j.cbpa.2010.02.018 |
| 21. | Regitz, M. Tetrahedron Lett. 1964, 5, 1403–1407. doi:10.1016/s0040-4039(00)90489-1 |
| 9. | Golushko, A.; Dar’in, D.; Kantin, G.; Guranova, N.; Vasilyev, A. V.; Krasavin, M. Synthesis 2019, 51, 3815–3824. doi:10.1055/s-0037-1611882 |
| 13. | Li, Z.; Chen, J.; Wu, L.; Ren, A.; Lu, P.; Wang, Y. Org. Lett. 2020, 22, 26–30. doi:10.1021/acs.orglett.9b03708 |
| 13. | Li, Z.; Chen, J.; Wu, L.; Ren, A.; Lu, P.; Wang, Y. Org. Lett. 2020, 22, 26–30. doi:10.1021/acs.orglett.9b03708 |
| 8. | Teall, M.; White, K.; Mack, S.; Liwicki, G.; Stephenson, A.; Dickson, L. MGLUR7 Agonist compounds for treating MGLUR7 – regulated diseases, disorders, or conditions. WO Patent WO/2018/092921, May 24, 2018. |
| 14. | Li, Z.; Xie, J.; Lu, P.; Wang, Y. J. Org. Chem. 2020, 85, 5525–5535. doi:10.1021/acs.joc.0c00283 |
| 15. | Chen, J.; Li, Z.; Suleman, M.; Wang, Z.; Lu, P.; Wang, Y. Org. Biomol. Chem. 2020, 18, 7671–7676. doi:10.1039/d0ob01748e |
| 16. | Wu, L.; Chen, J.; Xie, J.; Lu, P.; Wang, Y. Tetrahedron 2021, 84, 132019. doi:10.1016/j.tet.2021.132019 |
| 17. | Xie, J.; Suleman, M.; Wang, Z.; Mao, X.; Mao, B.; Fan, J.; Lu, P.; Wang, Y. Org. Biomol. Chem. 2021, 19, 6341–6345. doi:10.1039/d1ob00859e |
| 18. | Qi, M.; Suleman, M.; Xie, J.; Lu, P.; Wang, Y. J. Org. Chem. 2022, 87, 4088–4096. doi:10.1021/acs.joc.1c02905 |
| 25. | Kaiser, D.; de la Torre, A.; Shaaban, S.; Maulide, N. Angew. Chem., Int. Ed. 2017, 56, 5921–5925. doi:10.1002/anie.201701538 |
| 7. | Gunzinger, J.; Leander, K. Isoquinolines as IGF-1R inhibitors. WO Pat. Appl. WO 2007/029106 A1, March 15, 2007. |
| 23. | Zhukovsky, D.; Dar'in, D.; Kantin, G.; Krasavin, M. Eur. J. Org. Chem. 2019, 2397–2400. doi:10.1002/ejoc.201900133 |
| 6. | Epplin, M. P.; Mohan, A.; Harris, L. D.; Zhu, Z.; Strong, K. L.; Bacsa, J.; Le, P.; Menaldino, D. S.; Traynelis, S. F.; Liotta, D. C. J. Med. Chem. 2020, 63, 7569–7600. doi:10.1021/acs.jmedchem.9b01733 |
| 10. | Kantin, G.; Dar'in, D.; Krasavin, M. Eur. J. Org. Chem. 2018, 4857–4859. doi:10.1002/ejoc.201800955 |
| 11. | Dar’in, D.; Kantin, G.; Krasavin, M. Chem. Commun. 2019, 55, 5239–5242. doi:10.1039/c9cc02042j |
| 12. | Dar'in, D.; Kantin, G.; Chupakhin, E.; Sharoyko, V.; Krasavin, M. Chem. – Eur. J. 2021, 27, 8221–8227. doi:10.1002/chem.202100880 |
| 24. | Baum, J. S.; Shook, D. A.; Davies, H. M. L.; Smith, H. D. Synth. Commun. 1987, 17, 1709–1716. doi:10.1080/00397918708063988 |
© 2022 Dar’in et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.