Abstract
A convenient and efficient synthesis of novel achiral and chiral heterocyclic amino acid-like building blocks was developed. Regioisomeric methyl 5-(N-Boc-cycloaminyl)-1,2-oxazole-4-carboxylates were prepared by the reaction of β-enamino ketoesters (including azetidine, pyrrolidine or piperidine enamines) with hydroxylamine hydrochloride. Unambiguous structural assignments were based on chiral HPLC analysis, 1H, 13C, and 15N NMR spectroscopy, HRMS, and single-crystal X-ray diffraction data.

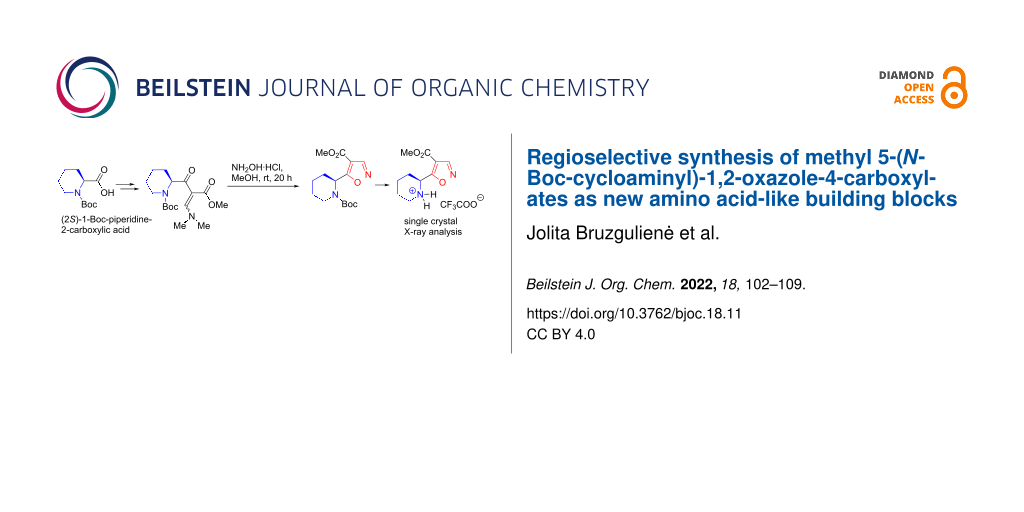
Graphical Abstract
Introduction
1,2-Oxazoles (isoxazoles) constitute an important class of heterocyclic compounds that plays a fundamental role in drug discovery [1-7]. Many amino-functionalized 1,2-oxazole derivatives are biologically active substances that include naturally occurring and synthetic neuroactive compounds. Specifically, natural products such as muscimol (I) and ibotenic acid (II) (Figure 1) have been isolated from several fungal species and are active on the γ-aminobutyric acid (GABA) and glutamate receptors of the central nervous system (CNS), respectively [8,9]. Various unnatural amino acids bearing a 1,2-oxazole moiety, such as nonproteinogenic α-amino acids, have been used as excitatory amino acid receptor agonists [10-13]. For example, (S)-AMPA (III) and (S)-ACPA (IV) are specific agonists of an AMPA receptor that mimic the effects of the neurotransmitter glutamate [14-16].
![[1860-5397-18-11-1]](/bjoc/content/figures/1860-5397-18-11-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Examples of amino-functionalized 1,2-oxazole derivatives I–VIII.
Figure 1: Examples of amino-functionalized 1,2-oxazole derivatives I–VIII.
Unnatural heteroarene amino acids have also been widely used as building blocks to prepare various heterocyclic peptides [17-22]. In particular, 1,2-oxazole amino acid derivatives, such as compounds V [20], VI [21], and VII, VIII [22], can be easily synthesized and are suitable for insertion with the corresponding heterocycle into a peptide-like structure.
Heterocyclic amino acids and related compounds have been used to prepare synthetic DNA-encoded compound libraries for the discovery of small molecule protein ligands [23-25]. Recently, a highly specific and potent p38α kinase inhibitor containing a 3-amino-1-phenyl-1H-pyrazole-4-carboxylic acid residue was identified directly from the 12.6-million-member DNA-encoded small molecule library using yoctoReactor technology [26]. We have developed efficient protocols that provide easy access to highly functional heterocyclic compounds as novel amino acid-like building blocks by combining thiazole, selenazole, pyrazole, indazole, and indole moieties with both carboxyl functional groups and cycloaminyl units [27-31].
In recent decades, various methods of constructing 1,2-oxazole ring systems have been developed [1-7,32]. The two primary pathways to 1,2-oxazoles are: the 1,3-dipolar cycloaddition of alkenes and alkynes with nitrile oxides, and the reaction of a three-carbon atom component, such as a α,β-unsaturated ketone or a 1,3-diketone with hydroxylamine hydrochloride [33]. Recently, Rosa et al. reported a useful procedure for the synthesis of various regioisomeric 1,2-oxazole derivatives. Accordingly, the synthetic route starts from the condensation of 1,3-diketones with N,N-dimethylformamide dimethylacetal to form β-enamino ketoester. The latter undergoes a subsequent cycloaddition reaction with hydroxylamine to form regioisomerically substituted 1,2-oxazoles [34,35].
This study aimed to develop and synthesize methyl 5-(cycloaminyl)-1,2-oxazole-4-carboxylates, as new amino acid-like building blocks. This type of functionalized heterocycles could exhibit not only useful biological properties, but also find application as building blocks for the generation of DNA-encoded chemical libraries.
Results and Discussion
The synthetic strategy for the synthesis of novel functionalized 1,2-oxazole derivatives is outlined in Scheme 1. The synthetic sequence began with preparing β-keto esters 2a–h by treating N-Boc-protected cyclic amino acids 1a–h with Meldrum’s acid in the presence of EDC·HCl and DMAP, followed by methanolysis of the corresponding adducts [27,28,31,36-38]. Reaction of the resulting β-keto esters 2a–h with N,N-dimethylformamide dimethylacetal afforded cycloaminyl β-enamino ketoesters 3a–h. After isolation of compounds 3a–h from the corresponding reaction mixtures, they were identified using LC–MS analysis, and were immediately treated with hydroxylamine hydrochloride in an appropriate solvent to obtain the target 1,2-oxazoles 4a–h. A representative β-enamino ketoester 3a was subjected to a detailed NMR analysis (Figure 2a). The 1H NMR spectrum of compound 3a showed the appearance of a new downfield enamine proton signal which resonated at δ 7.80 ppm. The connectivity of the β-enamino ketoester moiety and the N-Boc-protected azetidine fragment were easily confirmed based on long-range 1H,13C correlations, obtained from gs-HMBC spectra. The aforementioned enamine proton and protons 2’(4’)-H (δ 4.00-4.09 ppm) from the azetidine ring system shared the HMBC cross-peak with the ketone carbonyl carbon (δ 195.4 ppm). Finally, in the 1H,15N-HMBC spectrum of 3a, an expected long-range correlation between the enamine proton (δ 7.80 ppm) and the dimethylamino nitrogen (δ −269.7 ppm) was observed, thus allowing to prove the formation of β-enamino ketoester.
![[1860-5397-18-11-i1]](/bjoc/content/inline/1860-5397-18-11-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Conversion of cyclic amino acids to 1,2-oxazole derivatives.
Scheme 1: Conversion of cyclic amino acids to 1,2-oxazole derivatives.
Analysis of the possible resulting reaction showed that the condensation reaction between β-enamino ketoester precursors 3a–h and hydroxylamine could lead to the formation of two isomeric 1,2-oxazoles (Scheme 2) [34,35]. In the first route, enaminone 3a–h and hydroxylamine gives intermediate A, which then removes one molecule of dimethylamine to form intermediate B. This is followed by intramolecular cyclization to intermediate C, and subsequent dehydration to generate the final products 4a–h. Alternatively, nucleophilic attack of hydroxylamine to the carbonyl carbon atom of the enone moiety forms intermediate D. The dehydration of the latter provides oxime E, which through the formation of intermediate F, should be capable to form final product VII. However, as demonstrated with substrate 3a, the reaction only gave product 4a with a yield of 65% (Scheme 1). LC–MS analysis and flash column chromatography did not allow to detect the formation of the second possible isomeric product methyl 3-(N-Boc-azetidin-3-yl)-1,2-oxazole-4-carboxylate (VII) (Figure 1) [22].
![[1860-5397-18-11-i2]](/bjoc/content/inline/1860-5397-18-11-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Plausible mechanisms for the formation of 1,2-oxazoles 4a–h and VII from β-enamino ketoesters 3a–h with hydroxylamine.
Scheme 2: Plausible mechanisms for the formation of 1,2-oxazoles 4a–h and VII from β-enamino ketoesters 3a–h ...
The structural assignment of regiospecific compound 4a was readily deduced via detailed spectral data analysis. The IR spectrum of 4a contained characteristic absorption bands such as 1723 (C=O, ester), and 1687 (C=O, Boc) cm−1. The 1H NMR spectrum of compound 4a revealed a characteristic resonance for the Boc group protons, a singlet in the δ 1.45 ppm region, while the signal of the protons of the COOMe group appeared as a singlet at approximately δ 3.85 ppm (Figure 2b). The azetidine methylene protons (CHaHb-2’ and CHaHb-4’) exhibited a doublet of doublets at δ 4.24 ppm (J = 8.7, 6.5 Hz) and a triplet at δ 4.32 ppm (J = 8.8 Hz), while the methine proton (3’-H) yielded a triplet of triplets at δ 4.48 ppm (J = 8.9, 6.5 Hz). A comparison between the DEPT-90, DEPT-135 and 13C NMR spectra of compound 4a clearly indicated the presence of methine carbons C-3 (δ 150.6 ppm) and C-3’ (δ 26.1 ppm), respectively. The 1H,13C-HMBC spectrum of compound 4a revealed that the methylene protons from the azetidine moiety and the 1,2-oxazole methine proton H-3, exhibited long-range correlations with quaternary carbon C-5 (Figure 2c). The aforementioned protonated carbons C-3 and C-3’ showed correlations in the 1,1-ADEQUATE spectrum thus allowing to assign a neighboring quaternary carbons C-4 (δ 109.9 ppm) and C-5 (δ 175.4 ppm), H–C(3)–C(4) and H–C(3’)–C(5), respectively. The 1H,15N-HMBC experiment revealed an expected long-range correlation between the 1,2-oxazole methine H-3 proton (δ 8.50 ppm) and nitrogen N-2 which resonated at δ −0.6 ppm, while the azetidine ring protons showed a sole correlation with the azetidine nitrogen N-1’ (δ −311.7 ppm) [29,39].
![[1860-5397-18-11-2]](/bjoc/content/figures/1860-5397-18-11-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: (a) 1H NMR (italics), 13C NMR (normal), and 15N NMR (bold) chemical shifts (ppm) of compound 3a in CDCl3; (b) 1H NMR (italics), 13C NMR (normal), and 15N NMR (bold) chemical shifts (ppm) of compound 4a in CDCl3; (c) relevant 1H,13C HMBC, 1H,15N HMBC and 1,1-ADEQUATE correlations of compound 4a.
Figure 2: (a) 1H NMR (italics), 13C NMR (normal), and 15N NMR (bold) chemical shifts (ppm) of compound 3a in ...
Furthermore, 15N-labeled methyl 5-(N-Boc-azetidin-3-yl)-1,2-oxazole-4-carboxylate (5) was synthesized by analogy to 4a, by the reaction of β-enamino ketoester 3a with 15N-hydroxylamine hydrochloride (Scheme 3). Incorporation of a 15N atom in azaheterocycles is an important method for studying molecular structures, which significantly expands the possibilities of using NMR methods [40]. The 15N-labeled aromatic heterocyclic structures usually have well-resolved 1H,15N (JHN) and 13C,15N (JCN) coupling constants, as well as additional splitting of the corresponding signals in the standard proton decoupled 1D 13C NMR and 1D 1H NMR spectra [41,42]. The 1H,15N coupling constants in the azoles, especially the rather large 2JHN values, which are in the range of 13–15 Hz is widely accepted in the structure assignments and are even considered as diagnostic for this class of compounds, because the 3JHN is typically in the range of 1–3 Hz. For example, the 1H,15N coupling constants were measured for a series of 1,2-oxazoles, which provided the 2JHN values in the range of 14.4–14.7 Hz, while the 3JHN values were in the range of 1–3 Hz [42,43]. The coupling constants 13C,15N of a series of 15N-labeled pyrazoles gave 1JCN values of 8–11 Hz, while 2JCN were less than 2 Hz [44]. The 15N-labeled 1,2-thiazole moiety was readily determined by measuring the corresponding direct 13C,15N coupling constant 1JC3-N2 of 6.9 Hz [44].
![[1860-5397-18-11-i3]](/bjoc/content/inline/1860-5397-18-11-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of compound 15N-1,2-oxazole 5. The coupling constants of JHN and JCN from 15N2 are indicated by arrows.
Scheme 3: Synthesis of compound 15N-1,2-oxazole 5. The coupling constants of JHN and JCN from 15N2 are indica...
Therefore, the 1H NMR spectrum of 15N-labeled methyl 5-(N-Boc-azetidin-3-yl)-1,2-oxazole-4-carboxylate (5) showed a 1H,15N coupling constant (2JH3–N2 = 14.36 Hz), while in the 13C NMR spectrum, the 13C,15N interaction was observed for signals C-3 (1JC3-N2 = 4.55 Hz), C-4 (2JC4-N2 = 1.30 Hz), and C-5 (2JC5-N2 = 1.96 Hz), which unambiguously indicates the presence of a 1,2-oxazole ring in the target compound.
Also, chiral methyl 5-(N-Boc-cycloaminyl)-1,2-oxazole-4-carboxylates 4b–g were obtained from β-enamino ketoesters 3b–g with hydroxylamine hydrochloride in methanol (Scheme 1). Synthesized compounds 4b–g exhibited optical activity, and the corresponding (R)- or (S)-enantiomers rotated the plane of plane-polarized light in opposite directions. The enantiomeric purity of chiral compounds 4b–g was assessed via chiral HPLC analysis of enantiomeric samples, as shown in Figure 3 (4b,c, ee 100%), Supporting Information File 1, Figure S51 (4d,e, ee 98%) and Supporting Information File 1, Figure S52 (4f,g, ee 97%).
![[1860-5397-18-11-3]](/bjoc/content/figures/1860-5397-18-11-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Stacked chromatogram view of pairs of enantiomers with area, %: (R)-4b, ee 100% (tR = 10.1 min) and (S)-4c, ee 100% (tR = 9.6 min); conditions: CHIRAL ART Cellulose-SB (100 × 4.6 mm I.D., S-3 µm, chiral selector cellulose tris(3,5-dimethylphenylcarbamate), YMC); mobile phase: ACN/(H2O + 0.1% HCOOH (30:70 isocratic mode); T = 36 °C; flow rate 1.0 mL/min. Samples were prepared in methanol. The injection volume was 10 μL, λdet = 245 nm.
Figure 3: Stacked chromatogram view of pairs of enantiomers with area, %: (R)-4b, ee 100% (tR = 10.1 min) and...
The structure of the newly synthesized chiral 1,2-oxazole derivatives 4b–g was described and confirmed via NMR spectroscopy data (Supporting Information File 1 in Figures S21–S38). In 1H NMR and 13C NMR spectra of compounds 4b and 4c, double sets of signals with different intensities were observed. It is possible that these signals are associated with the presence of equilibrating conformers arising from the rotation of the tert-butoxycarbonyl (Boc) moiety around a C–N single bond (Figure 4a). It is known that some N-Boc-substituted heterocyclic compounds, including oxazolidines, consist of NMR spectra showing two sets of signals due to the dynamic equilibrium between the two conformers formed by the rotation of the Boc moiety, and their syn- and anti-orientation is present in the molecule [45,46].
![[1860-5397-18-11-4]](/bjoc/content/figures/1860-5397-18-11-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: (a) Structure of 4b with syn- and anti-conformers; (b) superimposed 1H NMR and 1D gradient NOE spectra with a selective irradiation of signal at δ 1.44 ppm.
Figure 4: (a) Structure of 4b with syn- and anti-conformers; (b) superimposed 1H NMR and 1D gradient NOE spec...
Ley et al. demonstrated that selective chemical exchange NMR experiments are very useful in the determination of equilibrating rotamers, including chiral N-Boc amino acid derivatives, from non-equilibrating diastereomers [47]. Chemical exchange NMR experiments such as saturation transfer have been widely used in such cases (e.g., 1D selective NOESY). In this case, the 1H NMR spectrum of 4b revealed singlets of tert-butyl protons at δ 1.24 ppm (major rotamer) and δ 1.44 ppm (minor rotamer). When the signal at δ 1.44 ppm was irradiated, two negative signals of the same phase at δ 1.44 ppm and δ 1.24 ppm were observed (Figure 4b; Supporting Information File 1 in Figure S53a). Additionally, when the pyrrolidine ring proton 2-H (δ 5.61–5.65 ppm) was irradiated, two negative signals of the same phase at δ 5.51–5.56 ppm and δ 5.61–5.65 ppm appeared, implying chemical exchange and therefore the presence of rotamers (Supporting Information File 1 in Figure S53b).
The synthesis of methyl 5-(N-Boc-piperidin-4-yl)-1,2-oxazole-4-carboxylate (4h), a nonchiral amino acid-like building block, was obtained by the reaction of β-enamino ketoester 3h with hydroxylamine hydrochloride (Scheme 1). The 1H NMR spectrum of compound 4h showed a characteristic resonance for the Boc-group methyl protons, a singlet at δ 1.47 ppm, while the 1,2-oxazole methine proton appeared as a singlet at δ 8.46 ppm. The 13C NMR spectrum of 4h contained the characteristic signals of the 1,2-oxazole ring skeleton carbons at δ 108.3 (C-4), 150.2 (C-3), and 179.5 (C-5) ppm. The 15N NMR spectrum of 4h exhibited characteristic resonances of nitrogen atoms at δ −294.6 (piperidine) and δ −3.1 (1,2-oxazole) ppm, respectively.
Next, we studied the reaction of compounds 4b and 4g with trifluoroacetic acid. Removal of the Boc protection in the presence of CF3COOH in dichloromethane yielded trifluoroacetates 6a and 6b as white solids (Scheme 4). A single crystal of 6b was prepared via recrystallization from dichloromethane for X-ray diffraction analysis [48].
![[1860-5397-18-11-i4]](/bjoc/content/inline/1860-5397-18-11-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Synthesis of 2-[4-(methoxycarbonyl)-1,2-oxazol-5-yl]cycloaminyl-1-ium trifluoroacetates 6a,b.
Scheme 4: Synthesis of 2-[4-(methoxycarbonyl)-1,2-oxazol-5-yl]cycloaminyl-1-ium trifluoroacetates 6a,b.
The asymmetric unit of the crystal of 6b consists of two (2S)-2-[4-(methoxycarbonyl)-1,2-oxazol-5-yl]piperidin-1-ium cations and two 2,2,2-trifluoroacetate anions (2C10H15N2O3+·2C2F3O2−) (Figure 5; Table 1, Supporting Information File 1 in Tables S1–S3). The substituted piperidinium moieties are in chair conformation. The 1,2-oxazole rings occupy equatorial positions at the 2nd atom of the piperidinium ring, and the dihedral angle H(10)–C(10)–C(5)–O(1) is 158°. The methoxycarbonyl group is in the same plane as the 1,2-oxazole ring.
![[1860-5397-18-11-5]](/bjoc/content/figures/1860-5397-18-11-5.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 5: ORTEP diagram of the asymmetric unit consisting of two cations 6b(A) and 6b(B) and triflate anions.
Figure 5: ORTEP diagram of the asymmetric unit consisting of two cations 6b(A) and 6b(B) and triflate anions.
Table 1: Bond lengths and angles of 1,2-oxazole fragments in an asymmetric unit of 6b.
| Molecule 6b(A) | d, (Å) | Φ, deg | |
| O(1)–N(2) | 1.420(2) | O(1)–N(2)–C(3) | 105.00(16) |
| N(2)–C(3) | 1.298(3) | N(2)–C(3)–C(4) | 112.61(17) |
| C(3)–C(4) | 1.424(3) | C(3)–C(4)–C(5) | 104.01(16) |
| C(4)–C(5) | 1.361(2) | C(4)–C(5)–O(1) | 109.44(16) |
| C(5)–O(1) | 1.351(2) | C(5)–O(1)–N(2) | 108.95(14) |
| Molecule 6b(B) | d, (Å) | Φ, deg | |
| O(21)–N(22) | 1.419(2) | O(21)–N(22)–C(23) | 104.71(16) |
| N(22)–C(23) | 1.305(3) | N(22)–C(23)–C(24) | 112.48(17) |
| C(23)–C(24) | 1.425(3) | C(23)–C(24)–C(25) | 103.89(16) |
| C(24)–C(25) | 1.360(2) | C(24)–C(25)–O(21) | 109.68(16) |
| C(25)–O(21) | 1.345(2) | C(25)–O(21)–N(22) | 109.23(14) |
The bond lengths and angles of 1,2-oxazole rings are shown in Table 1 which correlates with the literature data [49]. In addition, there were some marginal differences between the bond lengths in the two 1,2-oxazole rings.
Conclusion
A series of novel 1,2-oxazole-4-carboxylate derivatives possessing Boc-protected 4-, 5- and 6-membered saturated nitrogen heterocycles were synthesized as amino-acid-like building blocks. Construction of 5-cycloaminyl-1,2-oxazole compounds was based on the reaction of β-enamino ketoester precursors with hydroxylamine hydrochloride in moderate yields. When using the starting chiral saturated N-heterocyclic carboxylic acids, the target adducts were obtained with up to 97–100% ee. Discrimination between the regioisomeric compounds methyl 5-(N-Boc-azetidin-3-yl)-1,2-oxazole-4-carboxylate and methyl 3-(N-Boc-azetidin-3-yl)-1,2-oxazole-4-carboxylate was based on data from 1H, 13C and 15N NMR experiments.
In the NMR spectra of chiral 1,2-oxazoles, two sets of signals with different intensities were observed due to the existence of two Boc-group rotational conformers. The X-ray structure of (2S)-2-[4-(methoxycarbonyl)-1,2-oxazol-5-yl]piperidin-1-ium trifluoroacetate (6b) finally supported this structure analysis.
Supporting Information
| Supporting Information File 1: General information, synthesis procedures, and spectral data. | ||
| Format: PDF | Size: 4.6 MB | Download |
References
-
Sysak, A.; Obmińska-Mrukowicz, B. Eur. J. Med. Chem. 2017, 137, 292–309. doi:10.1016/j.ejmech.2017.06.002
Return to citation in text: [1] [2] -
Zhu, J.; Mo, J.; Lin, H.-z.; Chen, Y.; Sun, H.-p. Bioorg. Med. Chem. 2018, 26, 3065–3075. doi:10.1016/j.bmc.2018.05.013
Return to citation in text: [1] [2] -
Li, J.; Lin, Z.; Wu, W.; Jiang, H. Org. Chem. Front. 2020, 7, 2325–2348. doi:10.1039/d0qo00609b
Return to citation in text: [1] [2] -
Hu, F.; Szostak, M. Adv. Synth. Catal. 2015, 357, 2583–2614. doi:10.1002/adsc.201500319
Return to citation in text: [1] [2] -
Zimecki, M.; Bąchor, U.; Mączyński, M. Molecules 2018, 23, 2724. doi:10.3390/molecules23102724
Return to citation in text: [1] [2] -
Agrawal, N.; Mishra, P. Med. Chem. Res. 2018, 27, 1309–1344. doi:10.1007/s00044-018-2152-6
Return to citation in text: [1] [2] -
Morita, T.; Yugandar, S.; Fuse, S.; Nakamura, H. Tetrahedron Lett. 2018, 59, 1159–1171. doi:10.1016/j.tetlet.2018.02.020
Return to citation in text: [1] [2] -
Krogsgaard-Larsen, P.; Brehm, L.; Schaumburg, K. Acta Chem. Scand., Ser. B 1981, 35, 311–324. doi:10.3891/acta.chem.scand.35b-0311
Return to citation in text: [1] -
Obermaier, S.; Müller, M. Angew. Chem., Int. Ed. 2020, 59, 12432–12435. doi:10.1002/anie.202001870
Return to citation in text: [1] -
Madsen, U.; Stensbol, T. B.; Krogsgaard-Larsen, P. Curr. Med. Chem. 2001, 8, 1291–1301. doi:10.2174/0929867013372210
Return to citation in text: [1] -
Krogsgaard-Larsen, P.; Ebert, B.; Lund, T. M.; Bräuner-Osborne, H.; Sløk, F. A.; Johansen, T. N.; Brehm, L.; Madsen, U. Eur. J. Med. Chem. 1996, 31, 515–537. doi:10.1016/0223-5234(96)89549-3
Return to citation in text: [1] -
Kim, J.-H.; Marton, J.; Ametamey, S. M.; Cumming, P. Molecules 2020, 25, 4749. doi:10.3390/molecules25204749
Return to citation in text: [1] -
Sløk, F. A.; Ebert, B.; Lang, Y.; Krogsgaard-Larsen, P.; Lenz, S. M.; Madsen, U. Eur. J. Med. Chem. 1997, 32, 329–338. doi:10.1016/s0223-5234(97)89085-x
Return to citation in text: [1] -
Bleakman, D.; Lodge, D. Neuropharmacology 1998, 37, 1187–1204. doi:10.1016/s0028-3908(98)00139-7
Return to citation in text: [1] -
Johansen, T. N.; Stensbøl, T. B.; Nielsen, B.; Vogensen, S. B.; Frydenvang, K.; Sløk, F. A.; Brünumuner-Osborne, H.; Madsen, U.; Krogsgaard-Larsen, P. Chirality 2001, 13, 523–532. doi:10.1002/chir.1172
Return to citation in text: [1] -
Burkhart, D. J.; Twamley, B.; Natale, N. R. Tetrahedron Lett. 2001, 42, 8415–8418. doi:10.1016/s0040-4039(01)01796-8
Return to citation in text: [1] -
Cerminara, I.; Chiummiento, L.; Funicello, M.; Guarnaccio, A.; Lupattelli, P. Pharmaceuticals 2012, 5, 297–316. doi:10.3390/ph5030297
Return to citation in text: [1] -
Bruyat, P.; Jean, L.; Renard, P.-Y. Eur. J. Org. Chem. 2019, 3134–3141. doi:10.1002/ejoc.201900378
Return to citation in text: [1] -
Sekirnik née Measures, A. R.; Hewings, D. S.; Theodoulou, N. H.; Jursins, L.; Lewendon, K. R.; Jennings, L. E.; Rooney, T. P. C.; Heightman, T. D.; Conway, S. J. Angew. Chem., Int. Ed. 2016, 55, 8353–8357. doi:10.1002/anie.201602908
Return to citation in text: [1] -
Jones, R. C. F.; Hollis, S. J.; Iley, J. N. Tetrahedron: Asymmetry 2000, 11, 3273–3276. doi:10.1016/s0957-4166(00)00318-9
Return to citation in text: [1] [2] -
Chennakrishnareddy, G.; Vasantha, B.; Narendra, N.; Sureshbabu, V. V. Int. J. Pept. Res. Ther. 2011, 17, 185–191. doi:10.1007/s10989-011-9256-x
Return to citation in text: [1] [2] -
Chalyk, B. A.; Kandaurova, I. Y.; Hrebeniuk, K. V.; Manoilenko, O. V.; Kulik, I. B.; Iminov, R. T.; Kubyshkin, V.; Tverdokhlebov, A. V.; Ablialimov, O. K.; Mykhailiuk, P. K. RSC Adv. 2016, 6, 25713–25723. doi:10.1039/c6ra02365g
Return to citation in text: [1] [2] [3] -
Kunig, V.; Potowski, M.; Gohla, A.; Brunschweiger, A. Biol. Chem. 2018, 399, 691–710. doi:10.1515/hsz-2018-0119
Return to citation in text: [1] -
Madsen, D.; Azevedo, C.; Micco, I.; Petersen, L. K.; Hansen, N. J. V. Prog. Med. Chem. 2020, 59, 181–249. doi:10.1016/bs.pmch.2020.03.001
Return to citation in text: [1] -
Blakskjaer, P.; Heitner, T.; Hansen, N. J. V. Curr. Opin. Chem. Biol. 2015, 26, 62–71. doi:10.1016/j.cbpa.2015.02.003
Return to citation in text: [1] -
Petersen, L. K.; Blakskjær, P.; Chaikuad, A.; Christensen, A. B.; Dietvorst, J.; Holmkvist, J.; Knapp, S.; Kořínek, M.; Larsen, L. K.; Pedersen, A. E.; Röhm, S.; Sløk, F. A.; Hansen, N. J. V. Med. Chem. Commun. 2016, 7, 1332–1339. doi:10.1039/c6md00241b
Return to citation in text: [1] -
Malinauskienė, V.; Kveselytė, A.; Dzedulionytė, K.; Bieliauskas, A.; Burinskas, S.; Sløk, F. A.; Šačkus, A. Chem. Heterocycl. Compd. 2018, 54, 469–473. doi:10.1007/s10593-018-2291-1
Return to citation in text: [1] [2] -
Matulevičiūtė, G.; Arbačiauskienė, E.; Kleizienė, N.; Kederienė, V.; Ragaitė, G.; Dagilienė, M.; Bieliauskas, A.; Milišiūnaitė, V.; Sløk, F. A.; Šačkus, A. Molecules 2021, 26, 3808. doi:10.3390/molecules26133808
Return to citation in text: [1] [2] -
Iškauskienė, M.; Ragaitė, G.; Sløk, F. A.; Šačkus, A. Mol. Diversity 2020, 24, 1235–1251. doi:10.1007/s11030-019-09987-8
Return to citation in text: [1] [2] -
Sackus, A.; Kveselyte, A.; Malinauskiene, V.; Dzedulionyte, K.; Bieliauskas, A.; Burinskas, S.; Krikstolaityte, S.; Sløk, A. F. Heterocyclic amino acids as scaffolds for the synthesis of functionalized chiral 1,3-thiazole and 1,3-selenazole derivatives. In 256th National Meeting and Exposition of the American Chemical Society (ACS), Boston, MA, USA, Aug 19–23, 2018; American Chemical Society, 2018; Abstract 586.
Return to citation in text: [1] -
Dzedulionytė, K.; Voznikaitė, P.; Bieliauskas, A.; Malinauskienė, V.; Sløk, F. A.; Šačkus, A. Molbank 2021, M1207. doi:10.3390/m1207
Return to citation in text: [1] [2] -
Plumet, J. ChemPlusChem 2020, 85, 2252–2271. doi:10.1002/cplu.202000448
Return to citation in text: [1] -
Pinho e Melo, T. M. V. D. Curr. Org. Chem. 2005, 9, 925–958. doi:10.2174/1385272054368420
Return to citation in text: [1] -
Silva, R. G. M.; da Silva, M. J. V.; Jacomini, A. P.; Moura, S.; Back, D. F.; Basso, E. A.; Rosa, F. A. RSC Adv. 2018, 8, 4773–4778. doi:10.1039/c7ra13343j
Return to citation in text: [1] [2] -
Mittersteiner, M.; Andrade, V. P.; Bonacorso, H. G.; Martins, M. A. P.; Zanatta, N. Eur. J. Org. Chem. 2020, 6405–6417. doi:10.1002/ejoc.202001039
Return to citation in text: [1] [2] -
Gao, W.; Lau, T.; Pan, S.; Phillips, D. P.; Wang, X. Compounds and compositions as TGR5 agonists. WO Pat. Appl. 2012/082947A1, June 21, 2012.
Return to citation in text: [1] -
Brooks, D. W.; Lu, L. D.-L.; Masamune, S. Angew. Chem., Int. Ed. Engl. 1979, 18, 72–74. doi:10.1002/anie.197900722
Return to citation in text: [1] -
Greck, C.; Thomassigny, C.; Le Bouc, G. ARKIVOC 2012, No. viii, 231–249. doi:10.3998/ark.5550190.0013.821
Return to citation in text: [1] -
Schofield, M. H.; Sorel, M.-A.; Manalansan, R. J.; Richardson, D. P.; Markgraf, J. H. Magn. Reson. Chem. 2006, 44, 851–855. doi:10.1002/mrc.1860
Return to citation in text: [1] -
Deev, S. L.; Khalymbadzha, I. A.; Shestakova, T. S.; Charushin, V. N.; Chupakhin, O. N. RSC Adv. 2019, 9, 26856–26879. doi:10.1039/c9ra04825a
Return to citation in text: [1] -
Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876
Return to citation in text: [1] -
Chen, B. C.; Von Philipsborn, W.; Nagarajan, K. Helv. Chim. Acta 1983, 66, 1537–1555. doi:10.1002/hlca.19830660522
Return to citation in text: [1] [2] -
Maslova, M. M.; Pol'shakov, V. I.; Anisimova, O. S.; Marchenko, N. B.; Glushkov, R. G. Pharm. Chem. J. 1993, 26, 889–893. doi:10.1007/bf00767668
Return to citation in text: [1] -
Akiba, K.; Kashiwagi, K.; Ohyama, Y.; Yamamoto, Y.; Ohkata, K. J. Am. Chem. Soc. 1985, 107, 2721–2730. doi:10.1021/ja00295a026
Return to citation in text: [1] [2] -
Avenoza, A.; Busto, J. H.; Corzana, F.; Jiménez-Osés, G.; Peregrina, J. M. Tetrahedron 2003, 59, 5713–5718. doi:10.1016/s0040-4020(03)00852-4
Return to citation in text: [1] -
Garner, P.; Park, J. M. J. Org. Chem. 1987, 52, 2361–2364. doi:10.1021/jo00388a004
Return to citation in text: [1] -
Hu, D. X.; Grice, P.; Ley, S. V. J. Org. Chem. 2012, 77, 5198–5202. doi:10.1021/jo300734r
Return to citation in text: [1] -
The CCDC deposition number of (2S)-2-[4-(methoxycarbonyl)-1,2-oxazol-5-yl]piperidin-1-ium trifluoroacetate (6b) is 2003749; formula C10H15N2O3·C2F3O2; unit cell parameters: a = 9.1009(1), b = 17.8668(1), c = 9.8366(1) Å, β = 117.186 (2)°, space group P21.
Return to citation in text: [1] -
Allen, F. H.; Kennard, O.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. J. Chem. Soc., Perkin Trans. 2 1987, S1–S19. doi:10.1039/p298700000s1
Return to citation in text: [1]
| 1. | Sysak, A.; Obmińska-Mrukowicz, B. Eur. J. Med. Chem. 2017, 137, 292–309. doi:10.1016/j.ejmech.2017.06.002 |
| 2. | Zhu, J.; Mo, J.; Lin, H.-z.; Chen, Y.; Sun, H.-p. Bioorg. Med. Chem. 2018, 26, 3065–3075. doi:10.1016/j.bmc.2018.05.013 |
| 3. | Li, J.; Lin, Z.; Wu, W.; Jiang, H. Org. Chem. Front. 2020, 7, 2325–2348. doi:10.1039/d0qo00609b |
| 4. | Hu, F.; Szostak, M. Adv. Synth. Catal. 2015, 357, 2583–2614. doi:10.1002/adsc.201500319 |
| 5. | Zimecki, M.; Bąchor, U.; Mączyński, M. Molecules 2018, 23, 2724. doi:10.3390/molecules23102724 |
| 6. | Agrawal, N.; Mishra, P. Med. Chem. Res. 2018, 27, 1309–1344. doi:10.1007/s00044-018-2152-6 |
| 7. | Morita, T.; Yugandar, S.; Fuse, S.; Nakamura, H. Tetrahedron Lett. 2018, 59, 1159–1171. doi:10.1016/j.tetlet.2018.02.020 |
| 17. | Cerminara, I.; Chiummiento, L.; Funicello, M.; Guarnaccio, A.; Lupattelli, P. Pharmaceuticals 2012, 5, 297–316. doi:10.3390/ph5030297 |
| 18. | Bruyat, P.; Jean, L.; Renard, P.-Y. Eur. J. Org. Chem. 2019, 3134–3141. doi:10.1002/ejoc.201900378 |
| 19. | Sekirnik née Measures, A. R.; Hewings, D. S.; Theodoulou, N. H.; Jursins, L.; Lewendon, K. R.; Jennings, L. E.; Rooney, T. P. C.; Heightman, T. D.; Conway, S. J. Angew. Chem., Int. Ed. 2016, 55, 8353–8357. doi:10.1002/anie.201602908 |
| 20. | Jones, R. C. F.; Hollis, S. J.; Iley, J. N. Tetrahedron: Asymmetry 2000, 11, 3273–3276. doi:10.1016/s0957-4166(00)00318-9 |
| 21. | Chennakrishnareddy, G.; Vasantha, B.; Narendra, N.; Sureshbabu, V. V. Int. J. Pept. Res. Ther. 2011, 17, 185–191. doi:10.1007/s10989-011-9256-x |
| 22. | Chalyk, B. A.; Kandaurova, I. Y.; Hrebeniuk, K. V.; Manoilenko, O. V.; Kulik, I. B.; Iminov, R. T.; Kubyshkin, V.; Tverdokhlebov, A. V.; Ablialimov, O. K.; Mykhailiuk, P. K. RSC Adv. 2016, 6, 25713–25723. doi:10.1039/c6ra02365g |
| 27. | Malinauskienė, V.; Kveselytė, A.; Dzedulionytė, K.; Bieliauskas, A.; Burinskas, S.; Sløk, F. A.; Šačkus, A. Chem. Heterocycl. Compd. 2018, 54, 469–473. doi:10.1007/s10593-018-2291-1 |
| 28. | Matulevičiūtė, G.; Arbačiauskienė, E.; Kleizienė, N.; Kederienė, V.; Ragaitė, G.; Dagilienė, M.; Bieliauskas, A.; Milišiūnaitė, V.; Sløk, F. A.; Šačkus, A. Molecules 2021, 26, 3808. doi:10.3390/molecules26133808 |
| 31. | Dzedulionytė, K.; Voznikaitė, P.; Bieliauskas, A.; Malinauskienė, V.; Sløk, F. A.; Šačkus, A. Molbank 2021, M1207. doi:10.3390/m1207 |
| 36. | Gao, W.; Lau, T.; Pan, S.; Phillips, D. P.; Wang, X. Compounds and compositions as TGR5 agonists. WO Pat. Appl. 2012/082947A1, June 21, 2012. |
| 37. | Brooks, D. W.; Lu, L. D.-L.; Masamune, S. Angew. Chem., Int. Ed. Engl. 1979, 18, 72–74. doi:10.1002/anie.197900722 |
| 38. | Greck, C.; Thomassigny, C.; Le Bouc, G. ARKIVOC 2012, No. viii, 231–249. doi:10.3998/ark.5550190.0013.821 |
| 14. | Bleakman, D.; Lodge, D. Neuropharmacology 1998, 37, 1187–1204. doi:10.1016/s0028-3908(98)00139-7 |
| 15. | Johansen, T. N.; Stensbøl, T. B.; Nielsen, B.; Vogensen, S. B.; Frydenvang, K.; Sløk, F. A.; Brünumuner-Osborne, H.; Madsen, U.; Krogsgaard-Larsen, P. Chirality 2001, 13, 523–532. doi:10.1002/chir.1172 |
| 16. | Burkhart, D. J.; Twamley, B.; Natale, N. R. Tetrahedron Lett. 2001, 42, 8415–8418. doi:10.1016/s0040-4039(01)01796-8 |
| 34. | Silva, R. G. M.; da Silva, M. J. V.; Jacomini, A. P.; Moura, S.; Back, D. F.; Basso, E. A.; Rosa, F. A. RSC Adv. 2018, 8, 4773–4778. doi:10.1039/c7ra13343j |
| 35. | Mittersteiner, M.; Andrade, V. P.; Bonacorso, H. G.; Martins, M. A. P.; Zanatta, N. Eur. J. Org. Chem. 2020, 6405–6417. doi:10.1002/ejoc.202001039 |
| 10. | Madsen, U.; Stensbol, T. B.; Krogsgaard-Larsen, P. Curr. Med. Chem. 2001, 8, 1291–1301. doi:10.2174/0929867013372210 |
| 11. | Krogsgaard-Larsen, P.; Ebert, B.; Lund, T. M.; Bräuner-Osborne, H.; Sløk, F. A.; Johansen, T. N.; Brehm, L.; Madsen, U. Eur. J. Med. Chem. 1996, 31, 515–537. doi:10.1016/0223-5234(96)89549-3 |
| 12. | Kim, J.-H.; Marton, J.; Ametamey, S. M.; Cumming, P. Molecules 2020, 25, 4749. doi:10.3390/molecules25204749 |
| 13. | Sløk, F. A.; Ebert, B.; Lang, Y.; Krogsgaard-Larsen, P.; Lenz, S. M.; Madsen, U. Eur. J. Med. Chem. 1997, 32, 329–338. doi:10.1016/s0223-5234(97)89085-x |
| 33. | Pinho e Melo, T. M. V. D. Curr. Org. Chem. 2005, 9, 925–958. doi:10.2174/1385272054368420 |
| 8. | Krogsgaard-Larsen, P.; Brehm, L.; Schaumburg, K. Acta Chem. Scand., Ser. B 1981, 35, 311–324. doi:10.3891/acta.chem.scand.35b-0311 |
| 9. | Obermaier, S.; Müller, M. Angew. Chem., Int. Ed. 2020, 59, 12432–12435. doi:10.1002/anie.202001870 |
| 34. | Silva, R. G. M.; da Silva, M. J. V.; Jacomini, A. P.; Moura, S.; Back, D. F.; Basso, E. A.; Rosa, F. A. RSC Adv. 2018, 8, 4773–4778. doi:10.1039/c7ra13343j |
| 35. | Mittersteiner, M.; Andrade, V. P.; Bonacorso, H. G.; Martins, M. A. P.; Zanatta, N. Eur. J. Org. Chem. 2020, 6405–6417. doi:10.1002/ejoc.202001039 |
| 23. | Kunig, V.; Potowski, M.; Gohla, A.; Brunschweiger, A. Biol. Chem. 2018, 399, 691–710. doi:10.1515/hsz-2018-0119 |
| 24. | Madsen, D.; Azevedo, C.; Micco, I.; Petersen, L. K.; Hansen, N. J. V. Prog. Med. Chem. 2020, 59, 181–249. doi:10.1016/bs.pmch.2020.03.001 |
| 25. | Blakskjaer, P.; Heitner, T.; Hansen, N. J. V. Curr. Opin. Chem. Biol. 2015, 26, 62–71. doi:10.1016/j.cbpa.2015.02.003 |
| 27. | Malinauskienė, V.; Kveselytė, A.; Dzedulionytė, K.; Bieliauskas, A.; Burinskas, S.; Sløk, F. A.; Šačkus, A. Chem. Heterocycl. Compd. 2018, 54, 469–473. doi:10.1007/s10593-018-2291-1 |
| 28. | Matulevičiūtė, G.; Arbačiauskienė, E.; Kleizienė, N.; Kederienė, V.; Ragaitė, G.; Dagilienė, M.; Bieliauskas, A.; Milišiūnaitė, V.; Sløk, F. A.; Šačkus, A. Molecules 2021, 26, 3808. doi:10.3390/molecules26133808 |
| 29. | Iškauskienė, M.; Ragaitė, G.; Sløk, F. A.; Šačkus, A. Mol. Diversity 2020, 24, 1235–1251. doi:10.1007/s11030-019-09987-8 |
| 30. | Sackus, A.; Kveselyte, A.; Malinauskiene, V.; Dzedulionyte, K.; Bieliauskas, A.; Burinskas, S.; Krikstolaityte, S.; Sløk, A. F. Heterocyclic amino acids as scaffolds for the synthesis of functionalized chiral 1,3-thiazole and 1,3-selenazole derivatives. In 256th National Meeting and Exposition of the American Chemical Society (ACS), Boston, MA, USA, Aug 19–23, 2018; American Chemical Society, 2018; Abstract 586. |
| 31. | Dzedulionytė, K.; Voznikaitė, P.; Bieliauskas, A.; Malinauskienė, V.; Sløk, F. A.; Šačkus, A. Molbank 2021, M1207. doi:10.3390/m1207 |
| 22. | Chalyk, B. A.; Kandaurova, I. Y.; Hrebeniuk, K. V.; Manoilenko, O. V.; Kulik, I. B.; Iminov, R. T.; Kubyshkin, V.; Tverdokhlebov, A. V.; Ablialimov, O. K.; Mykhailiuk, P. K. RSC Adv. 2016, 6, 25713–25723. doi:10.1039/c6ra02365g |
| 1. | Sysak, A.; Obmińska-Mrukowicz, B. Eur. J. Med. Chem. 2017, 137, 292–309. doi:10.1016/j.ejmech.2017.06.002 |
| 2. | Zhu, J.; Mo, J.; Lin, H.-z.; Chen, Y.; Sun, H.-p. Bioorg. Med. Chem. 2018, 26, 3065–3075. doi:10.1016/j.bmc.2018.05.013 |
| 3. | Li, J.; Lin, Z.; Wu, W.; Jiang, H. Org. Chem. Front. 2020, 7, 2325–2348. doi:10.1039/d0qo00609b |
| 4. | Hu, F.; Szostak, M. Adv. Synth. Catal. 2015, 357, 2583–2614. doi:10.1002/adsc.201500319 |
| 5. | Zimecki, M.; Bąchor, U.; Mączyński, M. Molecules 2018, 23, 2724. doi:10.3390/molecules23102724 |
| 6. | Agrawal, N.; Mishra, P. Med. Chem. Res. 2018, 27, 1309–1344. doi:10.1007/s00044-018-2152-6 |
| 7. | Morita, T.; Yugandar, S.; Fuse, S.; Nakamura, H. Tetrahedron Lett. 2018, 59, 1159–1171. doi:10.1016/j.tetlet.2018.02.020 |
| 32. | Plumet, J. ChemPlusChem 2020, 85, 2252–2271. doi:10.1002/cplu.202000448 |
| 21. | Chennakrishnareddy, G.; Vasantha, B.; Narendra, N.; Sureshbabu, V. V. Int. J. Pept. Res. Ther. 2011, 17, 185–191. doi:10.1007/s10989-011-9256-x |
| 20. | Jones, R. C. F.; Hollis, S. J.; Iley, J. N. Tetrahedron: Asymmetry 2000, 11, 3273–3276. doi:10.1016/s0957-4166(00)00318-9 |
| 26. | Petersen, L. K.; Blakskjær, P.; Chaikuad, A.; Christensen, A. B.; Dietvorst, J.; Holmkvist, J.; Knapp, S.; Kořínek, M.; Larsen, L. K.; Pedersen, A. E.; Röhm, S.; Sløk, F. A.; Hansen, N. J. V. Med. Chem. Commun. 2016, 7, 1332–1339. doi:10.1039/c6md00241b |
| 40. | Deev, S. L.; Khalymbadzha, I. A.; Shestakova, T. S.; Charushin, V. N.; Chupakhin, O. N. RSC Adv. 2019, 9, 26856–26879. doi:10.1039/c9ra04825a |
| 22. | Chalyk, B. A.; Kandaurova, I. Y.; Hrebeniuk, K. V.; Manoilenko, O. V.; Kulik, I. B.; Iminov, R. T.; Kubyshkin, V.; Tverdokhlebov, A. V.; Ablialimov, O. K.; Mykhailiuk, P. K. RSC Adv. 2016, 6, 25713–25723. doi:10.1039/c6ra02365g |
| 29. | Iškauskienė, M.; Ragaitė, G.; Sløk, F. A.; Šačkus, A. Mol. Diversity 2020, 24, 1235–1251. doi:10.1007/s11030-019-09987-8 |
| 39. | Schofield, M. H.; Sorel, M.-A.; Manalansan, R. J.; Richardson, D. P.; Markgraf, J. H. Magn. Reson. Chem. 2006, 44, 851–855. doi:10.1002/mrc.1860 |
| 48. | The CCDC deposition number of (2S)-2-[4-(methoxycarbonyl)-1,2-oxazol-5-yl]piperidin-1-ium trifluoroacetate (6b) is 2003749; formula C10H15N2O3·C2F3O2; unit cell parameters: a = 9.1009(1), b = 17.8668(1), c = 9.8366(1) Å, β = 117.186 (2)°, space group P21. |
| 49. | Allen, F. H.; Kennard, O.; Watson, D. G.; Brammer, L.; Orpen, A. G.; Taylor, R. J. Chem. Soc., Perkin Trans. 2 1987, S1–S19. doi:10.1039/p298700000s1 |
| 45. | Avenoza, A.; Busto, J. H.; Corzana, F.; Jiménez-Osés, G.; Peregrina, J. M. Tetrahedron 2003, 59, 5713–5718. doi:10.1016/s0040-4020(03)00852-4 |
| 46. | Garner, P.; Park, J. M. J. Org. Chem. 1987, 52, 2361–2364. doi:10.1021/jo00388a004 |
| 47. | Hu, D. X.; Grice, P.; Ley, S. V. J. Org. Chem. 2012, 77, 5198–5202. doi:10.1021/jo300734r |
| 44. | Akiba, K.; Kashiwagi, K.; Ohyama, Y.; Yamamoto, Y.; Ohkata, K. J. Am. Chem. Soc. 1985, 107, 2721–2730. doi:10.1021/ja00295a026 |
| 44. | Akiba, K.; Kashiwagi, K.; Ohyama, Y.; Yamamoto, Y.; Ohkata, K. J. Am. Chem. Soc. 1985, 107, 2721–2730. doi:10.1021/ja00295a026 |
| 41. | Deev, S. L.; Shenkarev, Z. O.; Shestakova, T. S.; Chupakhin, O. N.; Rusinov, V. L.; Arseniev, A. S. J. Org. Chem. 2010, 75, 8487–8497. doi:10.1021/jo1017876 |
| 42. | Chen, B. C.; Von Philipsborn, W.; Nagarajan, K. Helv. Chim. Acta 1983, 66, 1537–1555. doi:10.1002/hlca.19830660522 |
| 42. | Chen, B. C.; Von Philipsborn, W.; Nagarajan, K. Helv. Chim. Acta 1983, 66, 1537–1555. doi:10.1002/hlca.19830660522 |
| 43. | Maslova, M. M.; Pol'shakov, V. I.; Anisimova, O. S.; Marchenko, N. B.; Glushkov, R. G. Pharm. Chem. J. 1993, 26, 889–893. doi:10.1007/bf00767668 |
© 2022 Bruzgulienė et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.