Abstract
The conversion of tetrazolo[1,5-a]quinoxalines to 1,2,3-triazoloquinoxalines and triazoloimidazoquinoxalines under typical conditions of a CuAAC reaction has been investigated. Derivatives of the novel compound class of triazoloimidazoquinoxalines (TIQ) and rhenium(I) triazoloquinoxaline complexes as well as a new TIQ rhenium complex were synthesized. As a result, a small 1,2,3-triazoloquinoxaline library was obtained and the method could be expanded towards 4-substituted tetrazoloquinoxalines. The compatibility of various aliphatic and aromatic alkynes towards the reaction was investigated and the denitrogenative annulation towards imidazoloquinoxalines could be observed as a competing reaction depending on the alkyne concentration and the substitutions at the quinoxaline.

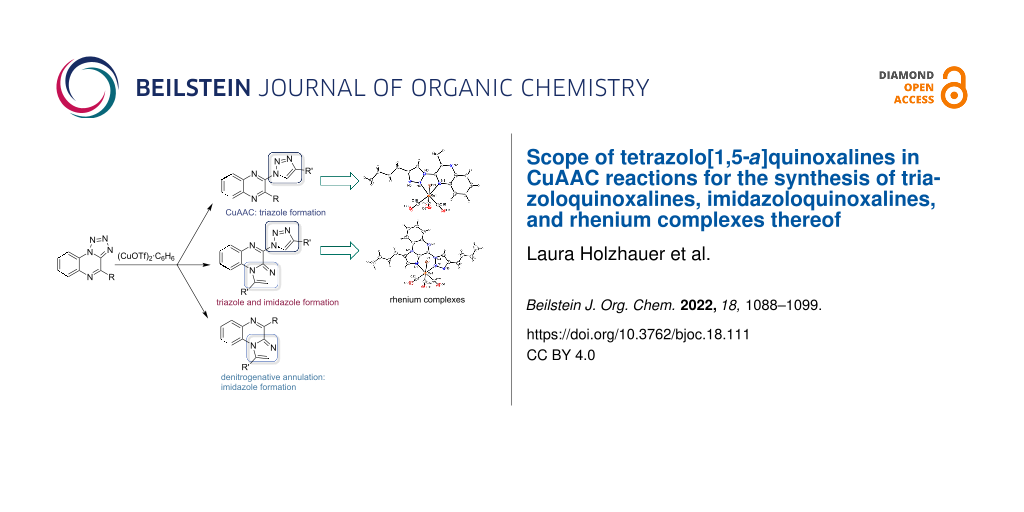
Graphical Abstract
Introduction
Quinoxalines are amongst the most versatile N-heterocyclic compounds, combining a straightforward synthesis with a diverse set of possible functionalizations and a wide range of applications in drug development and materials sciences [1]. Different quinoxaline derivatives possess antibacterial [2], antifungal [3], and antiviral properties [4] and form the core structure of commercially available drugs like brimonidine, varenicline, and quinacillin [5]. Quinoxalines can also be used in organic solar cell polymers [1,6] and have been described as donor moieties in many TADF and OLED compounds [7-9]. Amongst many other possible ways to modify and extend the core structure of quinoxalines, the conversion of tetrazolo[1,5-a]quinoxalines offers several advantages, as tetrazolo[1,5-a]quinoxalines can be used as quinoxaline-azide precursor, serving as a precursor for new nitrogen-enriched quinoxaline-based structures. Literature-known procedures for such a quinoxaline modification starting from tetrazolo[1,5-a]quinoxalines 1 are the synthesis of 1,2,3-triazoloquinoxalines 3 via copper-catalyzed azide–alkyne cycloaddition (CuAAC) [10] and the synthesis of imidazo[1,2-a]quinoxalines 2, which was recently reported for the first time using tetraphenylporphyrin iron(III) chloride as a catalyst (Scheme 1) [11].
![[1860-5397-18-111-i1]](/bjoc/content/inline/1860-5397-18-111-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Reactions of tetrazoloquinoxalines 1 to 1,2,3-triazoloquinoxalines 3 via CuAAC and denitrogenative annulation to imidazo[1,2-a]quinoxalines 2 catalyzed by an iron porphyrin catalyst 5 in combination with Zn. The scheme includes all quinoxaline-based derivatives that were obtained by these procedures so far [10-12].
Scheme 1: Reactions of tetrazoloquinoxalines 1 to 1,2,3-triazoloquinoxalines 3 via CuAAC and denitrogenative ...
While the target compounds, 1,2,3-triazoloquinoxalines 3 and imidazo[1,2-a]quinoxalines 2, offer a wide range of possible applications, the current knowledge on their formation from tetrazolo[1,5-a]quinoxalines 1 is still limited. Triazole-linked N-heterocycles like pyridotriazoles and quinolinotriazoles exert a variety of favorable biological properties like anticancer and antimicrobial activities as well as protein kinase inhibition [10,13-15]. Moreover, a vast diversity of metal complexes incorporating 1,2,3-triazoles as ligands have been reported [16-18]. Triazole ligands with N-heterocycles such as Pyta (4-(2-pyridyl)-1,2,3-triazole) and related structures were employed to obtain novel metal complexes as catalysts [19,20] and imaging probes [21], as well as metallosupramolecular assemblies [22]. The so-called inverse constellation of the triazole bound to the heterocycle via the nitrogen has been shown to possess interesting properties compared to the “regular” form [23,24], underlining the importance of accessing the desired triazole-heterocycle products from ring-fused 1,2,3,4-tetrazoles. Although some triazoloquinoxalines with a spacer moiety have been reported in the past [25,26], only three successfully synthesized derivatives of 1,2,3-triazoloquinoxalines 3 without a spacer are known [10,12]. To date, only one study describes the formation of a metal complex with an inverse triazoloquinoxaline ligand [12].
Imidazo[1,2-a]quinoxalines have been reported to possess anticancer and antitumor properties [27,28] and show activity as adenosine receptor antagonists [29] as well as PDE4 inhibitors [30]. The reaction of ring-fused tetrazoles to imidazole-fused products via denitrogenative annulation leading to 2 is, compared to the ever-present CuAAC, less known and was only shown with one example so far [11].
The study described herein intends to investigate the reactivity of tetrazolo[1,5-a]quinoxalines 1 concerning the competing formation of 1,2,3-triazoloquinoxalines 3 and imidazo[1,2-a]quinoxalines 2 under conditions known for copper-catalyzed azide–alkyne cycloaddition (CuAAC) [10]. The currently published porphyrin-catalyzed process requires glovebox conditions and the use of an expensive catalyst [11]. We intend to elucidate the conditions that favor the triazole formation or the imidazole, giving indications for alternative strategies to access imidazo[1,2-a]quinoxalines.
Results
All tetrazolo[1,5-a]quinoxaline precursors were synthesized in three to five steps from commercially available o-1,2-phenylenediamine (8, Scheme 2). Condensation to the corresponding quinoxalinone and subsequent chlorination was followed by introduction of the tetrazole moiety into the molecule via sodium azide to yield 11a–e. Alternatively, 4-chlorotetrazolo[1,5-a]quinoxaline (11f) was obtained after reaction of 2,3-dichloroquinoxaline (10f) with hydrazine and sodium nitrite. Further derivation of 11f led to compounds 11g–l which include different substitution patterns for R2. The tetrazolo[1,5-a]quinoxaline products 11a–l were obtained in yields of 36% to 81% for all steps (see Supporting Information File 1 for the entire scheme).
![[1860-5397-18-111-i2]](/bjoc/content/inline/1860-5397-18-111-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis of tetrazolo[1,5-a]quinoxalines. Reaction conditions: (a) 9, THF or 4 M HCl, 70–110 °C, 2–3 h; (b) POCl3, 100 °C, 2–4 h, yields over two steps are given above; (c) NaN3, DMF, 60–80 °C, 2–26 h; (d) H2NNH2·H2O, EtOH, 25 °C, 21 h; (e) NaNO2, AcOH/H2O, 0 °C, 3 h; (f) diverse conditions, see Supporting Information File 1 for details.
Scheme 2: Synthesis of tetrazolo[1,5-a]quinoxalines. Reaction conditions: (a) 9, THF or 4 M HCl, 70–110 °C, 2...
Starting from 11a, a small library of 1,2,3-triazole-substituted quinoxalines was synthesized applying the method of Chattopadhyay et al. [10] with minor adjustments. Altogether, a series of 21 different aliphatic and aromatic terminal alkynes were reacted with tetrazolo[1,5-a]quinoxaline and Cu(I) triflate as a catalyst at 100 °C in dry toluene, using DIPEA as an additional base. The use of DIPEA resulted in faster conversions and slightly higher yields (see Table S1, Supporting Information File 1). In total, 14 novel triazoloquinoxalines could be obtained successfully with yields ranging from 11% to 89%, showing the compatibility of the conversion with a diverse set of alkynes. Reduction of the starting material 11a to quinoxalin-2-amine as a side product was observed in some cases (see Supporting Information File 1 for details). The wide range of tolerated alkynes allows the installation of functional groups for further modification of the triazoloquinoxalines. For example, the alkyne-bearing compound 14f can be used for further CuAAC reactions and compounds including leaving groups, such as in 14j, can be easily converted by nucleophilic substitutions. In addition, compounds with alkene- (14m) or hydroxy- (14o) functionality can also be applied for various other reactions. Possible modifications of compounds 14 were exemplarily shown for 14j, which was converted to the amine-substituted product 14j* via nucleophilic substitution with a yield of 77% (see Scheme 3). However, alkynes 4 with reactive and electron-withdrawing functional groups, such as carboxylic acids, were not tolerated in the reaction of 11 to 14, or led to lower yields (for not successful reactions, please see Supporting Information File 1). The highest yields could be observed for the compounds 14j–l (Scheme 3).
![[1860-5397-18-111-i3]](/bjoc/content/inline/1860-5397-18-111-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Synthesis of 1,2,3-triazole-substituted quinoxalines via CuAAC from tetrazolo[1,5-a]quinoxaline (11a). aSynthesis of 14j* from 14j = Et2NH, K2CO3, DMF, 70 °C, 1 d.
Scheme 3: Synthesis of 1,2,3-triazole-substituted quinoxalines via CuAAC from tetrazolo[1,5-a]quinoxaline (11a...
To extend the scope of the reaction of tetrazolo[1,5-a]quinoxalines with alkynes under CuAAC conditions, different substituted quinoxalines 11 were reacted with hexyne (4k) as a model system (Table 1). A variation of the experimental setting for the substituted derivates found that the reaction gives better yields in the absence of DIPEA (see Table S2, Supporting Information File 1). Therefore, no base was used in the following experiments to convert substituted tetrazolo[1,5-a]quinoxalines with alkynes. Under these conditions, in addition to the reaction to the expected 1,2,3-triazoloquinoxalines, denitrogenative annulation was observed as a competing reaction, leading to imidazole product 16. This competing reaction was also observed for an aromatic alkyne (see Supporting Information File 1), but did not occur in any of the previous experiments with unsubstituted tetrazolo[1,5-a]quinoxalines. Moreover, the denitrogenative reduction to quinoxaline-2-amines 17 was noticed as a side reaction. Depending on the residue in 4-position (R, Table 1) on the pyrazine ring of the tetrazolo[1,5-a]quinoxaline, the formation of either the triazole or the imidazole product or both products occurred. For groups with electron-donating properties or a positive mesomeric effect combined with a low steric demand, such as methyl and methoxy groups, the triazole product was preferably formed. Increased steric demand of the groups such as for isopropyl residues led to the formation of the imidazole product instead. When using starting materials that incorporate functional groups with strong electron-withdrawing effects such as trifluoromethyl or chlorine, the imidazole product 16 was formed without any detectable amount of the triazole compound 15.
Table 1: Results of the reaction of different tetrazolo[1,5-a]quinoxalines 11 with hexyne (4k) under CuAAC conditions.a
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-111-i9.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Starting material | R | Equiv of hexyne (4k) | Yield [%] | ||
| 15a | 16a | 17a | ||||
| 1 | 11b | Me | 31 | 0 | 18 | |
| 2 | 11b | Me | 2 | 17 | 0 | nd |
| 3 | 11b | Me | 1.1 | 15 | 0 | 33b |
| 15b | 16b | 17b | ||||
| 4 | 11c | iPr | 5 | 8 | 17 | 11 |
| 5 | 11c | iPr | 2.5 | 0 | 13 | 34 |
| 6 | 11c | iPr | 1.1 | 0 | 22 | 41 |
| 15c | 16c | 17c | ||||
| 7 | 11d | CF3 | 8 | 0 | 0 | 41 |
| 8 | 11d | CF3 | 2 | 0 | 17 | 66 |
| 15d | 16d | 17d | ||||
| 9 | 11e | Ph | 5 | 11 | 0 | 11 |
| 10 | 11e | Ph | 2 | 11 | 0 | 24 |
| 11 | 11e | Ph | 1.1 | 9 | 0 | 31 |
| 15e | 16e | 17e | ||||
| 12 | 11f | Cl | 5 | 0 | 4 | 23 |
| 15f | 16f | 17f | ||||
| 13 | 11g | OMe | 2 | 49 | 0 | 0 |
| 15g | 16g | 17g | ||||
| 14 | 11j | NHC6H4COCH3 | 2.5 | 8 | 0 | 9 |
| 15h | 16h | 17h | ||||
| 15 | 11k | O(CH2)2(CF2)7CF3 | 15 | 62 | 13 | 0 |
| 16 | 11k | O(CH2)2(CF2)7CF3 | 5 | 50 | 15 | 21 |
| 17 | 11k | O(CH2)2(CF2)7CF3 | 2 | 10 | 19 | 55 |
| 18 | 11k | O(CH2)2(CF2)7CF3 | 1.1 | 0 | 22 | 29 |
a1.1–5 equiv hexyne, 10 mol % (CuOTf)2·C6H6 (7), toluene, 100 °C, 3 d. Full results including also not successful conversions are available in Supporting Information File 1; bobtained with impurities, nd = not determined.
In the cases when both products were observed, the ratio of the gained products depended strongly on the amount of alkyne used in the reaction. To investigate this effect, the perfluoro-substituted compound 11k was used as a model substrate as it showed the formation of both products under standard conditions with two equivalents of hexyne. When the amount of alkyne was reduced to 1.1 equivalents, no more triazole product could be isolated; the yield of the imidazole product was only slightly affected. In contrast, an increase in the alkyne amount led to a noticeable improvement of the yield from 10% up to 62%. In parallel, the imidazole formation decreased from 22% to 13% under the same conditions. The experiments were thus repeated with the methyl-, isopropyl- and phenyl-substituted compounds 11b, 11c, and 11e; again, increasing the amount of alkyne led to increased formation of the triazole product, especially for 11b and 11c.
These observations match with the general mechanism of CuAAC reactions and denitrogenative annulation according to Roy et al. [11]. Copper-catalyzed azide–alkyne cycloadditions are initiated via the (dual) complexation of the alkyne, whereas denitrogenative annulation on 1,2,3,4-tetrazoles is assumed to start via complexation of the open-form azide 18 (see Scheme 4). Increasing the amount of alkyne 4 increases the probability of the alkyne being coordinated in contrast to the tetrazole, which leads to launching of the CuAAC cycle. The probability of coordination on the tetrazole should also be indirectly impacted by this. However, the imidazole formation is only slightly decreased when the alkyne concentration is raised for compounds 11c and 11k. In contrast to that, no imidazole formation could be observed for compound 11d when 8 equivalents of alkyne were used. Therefore, further investigations will be necessary to determine why the imidazole formation is not completely suppressed in some cases when increasing the alkyne concentration drastically.
![[1860-5397-18-111-i4]](/bjoc/content/inline/1860-5397-18-111-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Mechanism of CuAAC vs denitrogenative annulation.
Scheme 4: Mechanism of CuAAC vs denitrogenative annulation.
The denitrogenative annulation reaction was then further explored using derivate 11d regarding the influences of different catalysts and additives (for details and results see Supporting Information File 1, Tables S3 and S4). Improving this route provides an alternative to the literature-known method [11] that requires both a special porphyrin complex and glovebox conditions. Using neither silver(I) triflate nor copper(I) iodide yielded the imidazole product, indicating that the use of copper(I) triflate is crucial for the reaction to take place. The increase of the amount of catalyst did not significantly improve the yield, while the addition of a base (DIPEA) or Lewis acid (AlCl3) resulted in suppression of imidazole formation and almost complete conversion to the amine 17. Addition of Zn(OTf)2 reduced the yield of the desired product 16 whereas addition of zinc powder seems to have different effects depending on the derivative (see Supporting Information File 1).
We could then show that the conversion of tetrazoles to both triazoles and imidazoles can occur together in the same molecule. When bis(tetrazolo)[1,5-a:5',1'-c]quinoxaline (24) was reacted with alkynes under Cu(I) triflate catalysis (see Scheme 5), CuAAC and denitrogenative annulation were observed in parallel to form triazoloimidazoquinoxalines (TIQs) as the main product, which have not been described in the literature yet. It remains unclear if one of the reactions takes place first and is required for the second reaction or whether both reactions occur independently of each other. Single crystals for 25b were obtained from slow evaporation of methanol under ambient pressure and the assumed structure of the TIQ product could unambiguously be confirmed via single crystal X-ray crystallography. Several other byproducts, such as the bistriazolo product were isolated (see Supporting Information File 1).
![[1860-5397-18-111-i5]](/bjoc/content/inline/1860-5397-18-111-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of bis(tetrazolo)[1,5-a:5',1'-c]quinoxaline (24) and conversion to triazoloimidazoquinoxalines (TIQs): 2.5 equiv hexyne, 10 mol % (CuOTf)2·C6H6 (7), toluene, 100 °C, 4 h to 3 d. ORTEP diagram of triazoloimidazoquinoxaline 25b with the thermal ellipsoids shown at 50% probability.
Scheme 5: Synthesis of bis(tetrazolo)[1,5-a:5',1'-c]quinoxaline (24) and conversion to triazoloimidazoquinoxa...
The obtained triazoloquinoxaline and TIQ products are promising ligands for complexation with different metals. The formation of organometallic complexes is a well-established method to obtain interesting materials for catalysis [31-33] and optoelectronics [34,35], as well as for biological applications [36,37]. Therefore, the obtained triazole and TIQ products were employed to act as ligands in rhenium tricarbonyl complexes. These are especially used as CO2 reduction catalysts [38-40] and noninvasive imaging probes [12,41]; examples for the application in organic light-emitting diodes [35] and as photoactive CO-releasing molecule [42,43] have been reported as well.
For the complexation experiments, compounds with three different residues on the triazole moiety (14a, 14k and 14j*) were selected. Moreover, the two substituted ligands 15a and 15d were employed to obtain novel substituted rhenium triazoloquinoxaline complexes and the TIQ compound 25b was tested for use as a ligand in rhenium tricarbonyl complexes. The complexes were prepared by reaction of the ligands with rhenium pentacarbonyl bromide (26) in toluene at 110 °C (see Scheme 6 and Scheme 7) as reported in the literature [12]. The structures of all obtained complexes could be confirmed via single crystal X-ray crystallography, verifying unambiguously the formation of the obtained products. Single crystals for complexes 27a–d were obtained via slow evaporation of a solution in either methylene chloride, ethyl acetate, or deuterated chloroform under ambient conditions. The rhenium atom is coordinated to three carbonyl groups, the bromine atom and two nitrogens of the 1,2,3-triazoloquinoxaline ligand in a distorted octahedral coordination geometry in all cases. The obtained data for the alkyl-chain complex 27a corresponds to similar published results [12].
![[1860-5397-18-111-i6]](/bjoc/content/inline/1860-5397-18-111-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of rhenium tricarbonyl complexes 27a–d and ORTEP diagrams of the resulting molecular structures with the thermal ellipsoids shown at 50% probability.
Scheme 6: Synthesis of rhenium tricarbonyl complexes 27a–d and ORTEP diagrams of the resulting molecular stru...
For complex 29, single crystals were formed from slow evaporation of a methylene chloride solution under ambient conditions. The crystal structure confirmed that rhenium is coordinated to three carbonyl groups, the bromine atom and two nitrogens of the 1,2,3-triazoloquinoxaline ligand. However, in this case, instead of coordination via the quinoxaline nitrogen and the 2-nitrogen of the triazole ring, the complex is formed via complexation of the 3-nitrogen of the triazole ring and the nitrogen of the amine side chain. The complex has a yellow color in contrast to the red complexes 27a–d.
![[1860-5397-18-111-i7]](/bjoc/content/inline/1860-5397-18-111-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Synthesis of rhenium tricarbonyl complex 29 and ORTEP diagram of the resulting molecular structure with the thermal ellipsoids shown at 50% probability.
Scheme 7: Synthesis of rhenium tricarbonyl complex 29 and ORTEP diagram of the resulting molecular structure ...
Using TIQ ligand 25b for a complexation attempt with Re(CO)5Br, an orange complex (30) was successfully isolated in 79% yield. Single crystals were obtained from slow evaporation of a solution of 25b in acetonitrile under ambient conditions. Crystal structure analysis of compound 30 confirmed that the rhenium complexation happens via the nitrogen of the imidazole and the 2-nitrogen of the triazole group in addition to three carbonyl groups and one bromine atom (see Scheme 8).
![[1860-5397-18-111-i8]](/bjoc/content/inline/1860-5397-18-111-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of a TIQ rhenium complex and ORTEP diagram of the obtained product 30 with the thermal ellipsoids shown at 50% probability.
Scheme 8: Synthesis of a TIQ rhenium complex and ORTEP diagram of the obtained product 30 with the thermal el...
UV–vis absorption spectra of all obtained rhenium complexes (Figure 1) and those of the free ligands (Figure S4, Supporting Information File 1) were measured in acetonitrile. The molar extinction coefficients ε of the complexes were calculated from the obtained quantitative data (see Table 2). Complexes 27a–d show similar properties to the literature [12] containing a low-energy broad absorption band with a maximum at 424–432 nm (see Table 2) and an absorption maximum at around 356 nm with a shoulder peak at around 344 nm for 27a, 27b, and 27c. Complex 29 displays different absorption properties due to the different complexation; it possesses a peak with a center at around 340 nm but no noticeable absorption in the range of 420–430 nm. The TIQ complex 30 shows two minor peaks at 332 nm and 350 nm and an intense broad peak at 386 nm, thus being blue-shifted compared to the triazoloquinoxaline complexes 27a–d.
![[1860-5397-18-111-1]](/bjoc/content/figures/1860-5397-18-111-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: UV–vis absorption spectra of the obtained metal complexes (18 µM solutions) in acetonitrile at 20 °C.
Figure 1: UV–vis absorption spectra of the obtained metal complexes (18 µM solutions) in acetonitrile at 20 °...
Table 2: Absorption maxima (λmax) and molar extinction coefficient ε at the absorption maximum [44].
| Compound | λmax [nm] | Log(ε) [M−1·cm−1] |
| 27a | 256 | 4.39 |
| 27b | 260 | 4.54 |
| 27c | 248 | 4.39 |
| 27d | 254 | 4.45 |
| 29 | 256 | 4.40 |
| 30 | 260 | 4.37 |
To characterize the electrochemical properties of the obtained complexes, cyclic voltammetry measurements were performed. For complexes 27a–d, irreversible oxidation previously assigned to the Re(I)/Re(II) couple [38,45] can be observed at 1.6 V vs SCE (see Table 3 and Figure 2); for complexes 29 and 30, this peak is shifted towards 1.4 V, indicating the stronger electron-donating nature of the ligands [38]. Moreover, an additional oxidation state at 1.91 V is present for complex 30 (see Supporting Information File 1 for full trace). For the other compounds, this oxidation state is hardly recognizable as it is almost hidden beneath the increase of the curve related to oxidation of the solvent.
![[1860-5397-18-111-2]](/bjoc/content/figures/1860-5397-18-111-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Cyclic voltammetry traces for rhenium complexes 27a–d, 29 and 30: 0.5 mM in MeCN solution with 0.1 M Bu4NPF6 under nitrogen at 25 °C, recorded at 0.1 V/s at a glassy carbon electrode and referenced to the saturated calomel electrode (SCE) using Fc/Fc+ as an internal standard (0.46 V vs SCE [12]).
Figure 2: Cyclic voltammetry traces for rhenium complexes 27a–d, 29 and 30: 0.5 mM in MeCN solution with 0.1 ...
Table 3: Electrochemical data for rhenium complexes 27a–d, 29 and 30. For full scan range (−2.0 V to 2.5 V), please refer to the Supporting Information File 1 (Figures S5, S6, and S7).
| Entry | Compound | Eox [V] | ERed [V] |
| 1 | 27a | 1.60 | −0.72, −1.18 |
| 2 | 27b | 1.09a, 1.62 | −0.68, −1.02 |
| 3 | 27c | 1.59 | −0.74, −0.96 |
| 4 | 27d | 1.60 | −0.71 |
| 5 | 29 | 1.47 | −0.92, −1.27 |
| 6 | 30 | 1.43, 1.91 | −1.17, −1.9 |
aMinor features.
Scanning towards negative potentials, two reduction waves can be observed between −0.6 V and −1.5 V for complexes 27a–d that can be assigned to reduction of the ligand [45]. For 29 and 30, reduction features of the ligands are anodically shifted. The reduction of complex 30 seems to be reversible (for further experiments please see Supporting Information File 1). The anodic shift shows that the more electron-rich nature of the TIQ ligand compared to the triazoloquinoxaline ligand has a visible influence on the reduction behavior of the complex.
Conclusion
New derivatives of 1,2,3-triazoloquinoxalines have been synthesized starting from tetrazolo[1,5-a]quinoxalines via CuAAC by varying the alkyne and the residues on the quinoxaline building blocks. During the investigation of the formation of 1,2,3-triazoloquinoxalines, denitrogenative annulation towards imidazole derivatives could be identified as a competing reaction for some substituted quinoxalines. Following the proposed mechanism, a dependency of obtained product ratio on the alkyne concentration was observed. These results expand the scope of accessible 1,2,3-triazoloquinoxalines and provide an alternative synthesis route from tetrazolo[1,5-a]quinoxalines to imidazo[1,2-a]quinoxalines.
For bis(tetrazolo)[1,5-a:5',1'-c]quinoxalines, the formation of triazoloimidazoquinoxalines was shown with two derivatives. Five rhenium complexes with 1,2,3-triazoloquinoxalines and a novel TIQ rhenium complex were synthesized, and their structures were confirmed via X-ray crystallography. All complexes were characterized and compared regarding their absorption and electrochemical properties. The TIQ complex could be confirmed to possess rather different properties than the triazoloquinoxaline complexes in these measurements, including a blue-shift in the absorption spectrum and anodically shifted features in cyclic voltammetry measurements.
Abbreviations
CuAAC, copper-catalyzed azide–alkyne cycloaddition; DIPEA, N,N-diisopropylethylamine; OLED, organic-light emitting diode; SCE, saturated calomel electrode; TADF, thermally activated delayed fluorescence; TEMPO, 2,2,6,6-tetramethylpiperidinyloxyl; TIQ, triazoloimidazoquinoxaline.
Supporting Information
The Supporting Information covers detailed material on the conducted experiments and their results, including unsuccessful experiments. All experimental details, including the analytical description of the obtained target compounds and byproducts, are available in Supporting Information File 1. Information on the availability of the data and the physical material of the target compounds is added to the Supporting Information File 2. Data that refers to the herein described experiments were submitted to the repository chemotion (https://www.chemotion-repository.net/). All DOIs minted for the data are linked in Supporting Information File 1. New data obtained in this study is assigned to the collection embargo numbers LSH_2021-02-02 and CML_2020-12-18. The material that was obtained in this study (target compounds, please see Supporting Information File 2) was submitted to the Molecule Archive at KIT and can be requested from there (https://compound-platform.eu/home).
| Supporting Information File 1: Experimental part. | ||
| Format: PDF | Size: 2.6 MB | Download |
| Supporting Information File 2: NMR spectra. | ||
| Format: PDF | Size: 10.1 MB | Download |
| Supporting Information File 3: Information on the availability of the data and the physical material of the target compounds. | ||
| Format: XLSX | Size: 481.0 KB | Download |
Acknowledgements
We are very thankful to Jérôme Klein for providing three precursor compounds and synthetic procedures for other tetrazole precursors. We thank André Jung for the deciding hint regarding the imidazole structure and the Soft Matter Synthesis Laboratory for the opportunity to use their UV–vis spectrophotometer.
Funding
L.H. acknowledges funding by the Landesgraduiertenförderung Baden-Württemberg. C.L. acknowledges funding by the ERASMUS program and the regional international mobility scholarship of Lyon. We acknowledge the support by the Joint Lab VirtMat within the Helmholtz research area Information. This work was supported by the Helmholtz program Information. We acknowledge support by Deutsche Forschungsgemeinschaft for the DFG-core facility Molecule Archive, to which all target compounds were registered for further re-use (DFG project number: 284178167).
References
-
Gedefaw, D.; Prosa, M.; Bolognesi, M.; Seri, M.; Andersson, M. R. Adv. Energy Mater. 2017, 7, 1700575. doi:10.1002/aenm.201700575
Return to citation in text: [1] [2] -
Xia, R.; Guo, T.; He, J.; Chen, M.; Su, S.; Jiang, S.; Tang, X.; Chen, Y.; Xue, W. Monatsh. Chem. 2019, 150, 1325–1334. doi:10.1007/s00706-019-02449-9
Return to citation in text: [1] -
Ajani, O. O.; Obafemi, C. A.; Nwinyi, O. C.; Akinpelu, D. A. Bioorg. Med. Chem. 2010, 18, 214–221. doi:10.1016/j.bmc.2009.10.064
Return to citation in text: [1] -
Patel, S. B.; Patel, B. D.; Pannecouque, C.; Bhatt, H. G. Eur. J. Med. Chem. 2016, 117, 230–240. doi:10.1016/j.ejmech.2016.04.019
Return to citation in text: [1] -
Chen, Q.; Bryant, V. C.; Lopez, H.; Kelly, D. L.; Luo, X.; Natarajan, A. Bioorg. Med. Chem. Lett. 2011, 21, 1929–1932. doi:10.1016/j.bmcl.2011.02.055
Return to citation in text: [1] -
Yuan, J.; Ouyang, J.; Cimrová, V.; Leclerc, M.; Najari, A.; Zou, Y. J. Mater. Chem. C 2017, 5, 1858–1879. doi:10.1039/c6tc05381e
Return to citation in text: [1] -
Vasilopoulou, M.; Mohd Yusoff, A. R. b.; Daboczi, M.; Conforto, J.; Gavim, A. E. X.; da Silva, W. J.; Macedo, A. G.; Soultati, A.; Pistolis, G.; Schneider, F. K.; Dong, Y.; Jacoutot, P.; Rotas, G.; Jang, J.; Vougioukalakis, G. C.; Chochos, C. L.; Kim, J.-S.; Gasparini, N. Nat. Commun. 2021, 12, 4868. doi:10.1038/s41467-021-25135-z
Return to citation in text: [1] -
Vishwakarma, V. K.; Nath, S.; Gupta, M.; Dubey, D. K.; Swayamprabha, S. S.; Jou, J.-H.; Pal, S. K.; Sudhakar, A. A. ACS Appl. Electron. Mater. 2019, 1, 1959–1969. doi:10.1021/acsaelm.9b00477
Return to citation in text: [1] -
Huang, T.; Liu, D.; Jiang, J.; Jiang, W. Chem. – Eur. J. 2019, 25, 10926–10937. doi:10.1002/chem.201902116
Return to citation in text: [1] -
Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] -
Ellanki, A. R.; Islam, A.; Rama, V. S.; Pulipati, R. P.; Rambabu, D.; Rama Krishna, G.; Malla Reddy, C.; Mukkanti, K.; Vanaja, G. R.; Kalle, A. M.; Shiva Kumar, K.; Pal, M. Bioorg. Med. Chem. Lett. 2012, 22, 3455–3459. doi:10.1016/j.bmcl.2012.03.091
Return to citation in text: [1] -
Głowacka, I. E.; Grzonkowski, P.; Lisiecki, P.; Kalinowski, Ł.; Piotrowska, D. G. Arch. Pharm. (Weinheim, Ger.) 2019, 352, 1800302. doi:10.1002/ardp.201800302
Return to citation in text: [1] -
Klein, M.; Dinér, P.; Dorin-Semblat, D.; Doerig, C.; Grøtli, M. Org. Biomol. Chem. 2009, 7, 3421–3429. doi:10.1039/b906482f
Return to citation in text: [1] -
Elliott, P. I. P. Organometallic complexes with 1,2,3-triazole-derived ligands. In Organometallic chemistry; Fairlamb, I.; Lynam, J.; Dane, S. B. J., Eds.; Specialist Periodical Reports, 0301-0074, Vol. 39; The Royal Society of Chemistry: Cambridge, UK, 2014; pp 1–25. doi:10.1039/9781849737692-00001
Return to citation in text: [1] -
Crowley, J. D.; McMorran, D. A. “Click-Triazole” Coordination Chemistry: Exploiting 1,4-Disubstituted-1,2,3-Triazoles as Ligands. In Click triazoles; Košmrlj, J.; Buckley, B. R., Eds.; Topics in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2012; pp 31–83. doi:10.1007/7081_2011_67
Return to citation in text: [1] -
Aromí, G.; Barrios, L. A.; Roubeau, O.; Gamez, P. Coord. Chem. Rev. 2011, 255, 485–546. doi:10.1016/j.ccr.2010.10.038
Return to citation in text: [1] -
Jindabot, S.; Teerachanan, K.; Thongkam, P.; Kiatisevi, S.; Khamnaen, T.; Phiriyawirut, P.; Charoenchaidet, S.; Sooksimuang, T.; Kongsaeree, P.; Sangtrirutnugul, P. J. Organomet. Chem. 2014, 750, 35–40. doi:10.1016/j.jorganchem.2013.10.046
Return to citation in text: [1] -
Yang, Y.; Hu, W.; Ye, X.; Wang, D.; Shi, X. Adv. Synth. Catal. 2016, 358, 2583–2588. doi:10.1002/adsc.201600243
Return to citation in text: [1] -
Connell, T. U.; James, J. L.; White, A. R.; Donnelly, P. S. Chem. – Eur. J. 2015, 21, 14146–14155. doi:10.1002/chem.201501630
Return to citation in text: [1] -
Preston, D.; Sutton, J. J.; Gordon, K. C.; Crowley, J. D. Angew. Chem., Int. Ed. 2018, 57, 8659–8663. doi:10.1002/anie.201804745
Return to citation in text: [1] -
Lo, W. K. C.; Huff, G. S.; Cubanski, J. R.; Kennedy, A. D. W.; McAdam, C. J.; McMorran, D. A.; Gordon, K. C.; Crowley, J. D. Inorg. Chem. 2015, 54, 1572–1587. doi:10.1021/ic502557w
Return to citation in text: [1] -
Lakshman, M. K.; Singh, M. K.; Parrish, D.; Balachandran, R.; Day, B. W. J. Org. Chem. 2010, 75, 2461–2473. doi:10.1021/jo902342z
Return to citation in text: [1] -
Bruschi, C.; Gui, X.; Salaeh‐arae, N.; Barchi, T.; Fuhr, O.; Lebedkin, S.; Klopper, W.; Bizzarri, C. Eur. J. Inorg. Chem. 2021, 4074–4084. doi:10.1002/ejic.202100653
Return to citation in text: [1] -
Maiti, S.; Roy, N.; Babu, L. T.; Moharana, P.; Athira, C. C.; Darsana Sreedhar, E.; De, S.; Ashok Kumar, S. K.; Paira, P. New J. Chem. 2020, 44, 920–931. doi:10.1039/c9nj03131f
Return to citation in text: [1] -
Kumar, M.; Joshi, G.; Arora, S.; Singh, T.; Biswas, S.; Sharma, N.; Bhat, Z. R.; Tikoo, K.; Singh, S.; Kumar, R. Molecules 2021, 26, 1490. doi:10.3390/molecules26051490
Return to citation in text: [1] -
Deleuze-Masquefa, C.; Moarbess, G.; Bonnet, P.-A.; Pinguet, F.; Bazarbachi, A.; Bressolle, F. Synthesis of imidazo[l,2-a]quinoxalines for treating cancers. WO Patent WO2009043934, April 9, 2009.
Return to citation in text: [1] -
Liu, C.-H.; Wang, B.; Li, W.-Z.; Yun, L.-H.; Liu, Y.; Su, R.-B.; Li, J.; Liu, H. Bioorg. Med. Chem. 2004, 12, 4701–4707. doi:10.1016/j.bmc.2004.06.026
Return to citation in text: [1] -
Deleuze-Masquéfa, C.; Gerebtzoff, G.; Subra, G.; Fabreguettes, J.-R.; Ovens, A.; Carraz, M.; Strub, M.-P.; Bompart, J.; George, P.; Bonnet, P.-A. Bioorg. Med. Chem. 2004, 12, 1129–1139. doi:10.1016/j.bmc.2003.11.034
Return to citation in text: [1] -
Sasmal, P. K.; Streu, C. N.; Meggers, E. Chem. Commun. 2013, 49, 1581–1587. doi:10.1039/c2cc37832a
Return to citation in text: [1] -
Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r
Return to citation in text: [1] -
Kinzel, N. W.; Werlé, C.; Leitner, W. Angew. Chem., Int. Ed. 2021, 60, 11628–11686. doi:10.1002/anie.202006988
Return to citation in text: [1] -
Bizzarri, C.; Spuling, E.; Knoll, D. M.; Volz, D.; Bräse, S. Coord. Chem. Rev. 2018, 373, 49–82. doi:10.1016/j.ccr.2017.09.011
Return to citation in text: [1] -
Zhao, G.-W.; Zhao, J.-H.; Hu, Y.-X.; Zhang, D.-Y.; Li, X. Synth. Met. 2016, 212, 131–141. doi:10.1016/j.synthmet.2015.12.014
Return to citation in text: [1] [2] -
Haas, K. L.; Franz, K. J. Chem. Rev. 2009, 109, 4921–4960. doi:10.1021/cr900134a
Return to citation in text: [1] -
Zhang, R.; Yuan, J. Acc. Chem. Res. 2020, 53, 1316–1329. doi:10.1021/acs.accounts.0c00172
Return to citation in text: [1] -
Ching, H. Y. V.; Wang, X.; He, M.; Perujo Holland, N.; Guillot, R.; Slim, C.; Griveau, S.; Bertrand, H. C.; Policar, C.; Bedioui, F.; Fontecave, M. Inorg. Chem. 2017, 56, 2966–2976. doi:10.1021/acs.inorgchem.6b03078
Return to citation in text: [1] [2] [3] -
Merillas, B.; Cuéllar, E.; Diez-Varga, A.; Torroba, T.; García-Herbosa, G.; Fernández, S.; Lloret-Fillol, J.; Martín-Alvarez, J. M.; Miguel, D.; Villafañe, F. Inorg. Chem. 2020, 59, 11152–11165. doi:10.1021/acs.inorgchem.0c01654
Return to citation in text: [1] -
Mukherjee, J.; Siewert, I. Eur. J. Inorg. Chem. 2020, 4319–4333. doi:10.1002/ejic.202000738
Return to citation in text: [1] -
Raszeja, L. J.; Siegmund, D.; Cordes, A. L.; Güldenhaupt, J.; Gerwert, K.; Hahn, S.; Metzler-Nolte, N. Chem. Commun. 2017, 53, 905–908. doi:10.1039/c6cc07553c
Return to citation in text: [1] -
Hernández Mejías, Á. D.; Poirot, A.; Rmili, M.; Leygue, N.; Wolff, M.; Saffon-Merceron, N.; Benoist, E.; Fery-Forgues, S. Dalton Trans. 2021, 50, 1313–1323. doi:10.1039/d0dt03577g
Return to citation in text: [1] -
Hostachy, S.; Policar, C.; Delsuc, N. Coord. Chem. Rev. 2017, 351, 172–188. doi:10.1016/j.ccr.2017.05.004
Return to citation in text: [1] -
Swinehart, D. F. J. Chem. Educ. 1962, 39, 333. doi:10.1021/ed039p333
Return to citation in text: [1] -
Kim, T. Y.; Elliott, A. B. S.; Shaffer, K. J.; John McAdam, C.; Gordon, K. C.; Crowley, J. D. Polyhedron 2013, 52, 1391–1398. doi:10.1016/j.poly.2012.05.003
Return to citation in text: [1] [2]
| 11. | Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702 |
| 31. | Sasmal, P. K.; Streu, C. N.; Meggers, E. Chem. Commun. 2013, 49, 1581–1587. doi:10.1039/c2cc37832a |
| 32. | Prier, C. K.; Rankic, D. A.; MacMillan, D. W. C. Chem. Rev. 2013, 113, 5322–5363. doi:10.1021/cr300503r |
| 33. | Kinzel, N. W.; Werlé, C.; Leitner, W. Angew. Chem., Int. Ed. 2021, 60, 11628–11686. doi:10.1002/anie.202006988 |
| 34. | Bizzarri, C.; Spuling, E.; Knoll, D. M.; Volz, D.; Bräse, S. Coord. Chem. Rev. 2018, 373, 49–82. doi:10.1016/j.ccr.2017.09.011 |
| 35. | Zhao, G.-W.; Zhao, J.-H.; Hu, Y.-X.; Zhang, D.-Y.; Li, X. Synth. Met. 2016, 212, 131–141. doi:10.1016/j.synthmet.2015.12.014 |
| 1. | Gedefaw, D.; Prosa, M.; Bolognesi, M.; Seri, M.; Andersson, M. R. Adv. Energy Mater. 2017, 7, 1700575. doi:10.1002/aenm.201700575 |
| 5. | Chen, Q.; Bryant, V. C.; Lopez, H.; Kelly, D. L.; Luo, X.; Natarajan, A. Bioorg. Med. Chem. Lett. 2011, 21, 1929–1932. doi:10.1016/j.bmcl.2011.02.055 |
| 22. | Preston, D.; Sutton, J. J.; Gordon, K. C.; Crowley, J. D. Angew. Chem., Int. Ed. 2018, 57, 8659–8663. doi:10.1002/anie.201804745 |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 4. | Patel, S. B.; Patel, B. D.; Pannecouque, C.; Bhatt, H. G. Eur. J. Med. Chem. 2016, 117, 230–240. doi:10.1016/j.ejmech.2016.04.019 |
| 23. | Lo, W. K. C.; Huff, G. S.; Cubanski, J. R.; Kennedy, A. D. W.; McAdam, C. J.; McMorran, D. A.; Gordon, K. C.; Crowley, J. D. Inorg. Chem. 2015, 54, 1572–1587. doi:10.1021/ic502557w |
| 24. | Lakshman, M. K.; Singh, M. K.; Parrish, D.; Balachandran, R.; Day, B. W. J. Org. Chem. 2010, 75, 2461–2473. doi:10.1021/jo902342z |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 3. | Ajani, O. O.; Obafemi, C. A.; Nwinyi, O. C.; Akinpelu, D. A. Bioorg. Med. Chem. 2010, 18, 214–221. doi:10.1016/j.bmc.2009.10.064 |
| 19. | Jindabot, S.; Teerachanan, K.; Thongkam, P.; Kiatisevi, S.; Khamnaen, T.; Phiriyawirut, P.; Charoenchaidet, S.; Sooksimuang, T.; Kongsaeree, P.; Sangtrirutnugul, P. J. Organomet. Chem. 2014, 750, 35–40. doi:10.1016/j.jorganchem.2013.10.046 |
| 20. | Yang, Y.; Hu, W.; Ye, X.; Wang, D.; Shi, X. Adv. Synth. Catal. 2016, 358, 2583–2588. doi:10.1002/adsc.201600243 |
| 42. | Hernández Mejías, Á. D.; Poirot, A.; Rmili, M.; Leygue, N.; Wolff, M.; Saffon-Merceron, N.; Benoist, E.; Fery-Forgues, S. Dalton Trans. 2021, 50, 1313–1323. doi:10.1039/d0dt03577g |
| 43. | Hostachy, S.; Policar, C.; Delsuc, N. Coord. Chem. Rev. 2017, 351, 172–188. doi:10.1016/j.ccr.2017.05.004 |
| 2. | Xia, R.; Guo, T.; He, J.; Chen, M.; Su, S.; Jiang, S.; Tang, X.; Chen, Y.; Xue, W. Monatsh. Chem. 2019, 150, 1325–1334. doi:10.1007/s00706-019-02449-9 |
| 21. | Connell, T. U.; James, J. L.; White, A. R.; Donnelly, P. S. Chem. – Eur. J. 2015, 21, 14146–14155. doi:10.1002/chem.201501630 |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 11. | Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702 |
| 10. | Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d |
| 13. | Ellanki, A. R.; Islam, A.; Rama, V. S.; Pulipati, R. P.; Rambabu, D.; Rama Krishna, G.; Malla Reddy, C.; Mukkanti, K.; Vanaja, G. R.; Kalle, A. M.; Shiva Kumar, K.; Pal, M. Bioorg. Med. Chem. Lett. 2012, 22, 3455–3459. doi:10.1016/j.bmcl.2012.03.091 |
| 14. | Głowacka, I. E.; Grzonkowski, P.; Lisiecki, P.; Kalinowski, Ł.; Piotrowska, D. G. Arch. Pharm. (Weinheim, Ger.) 2019, 352, 1800302. doi:10.1002/ardp.201800302 |
| 15. | Klein, M.; Dinér, P.; Dorin-Semblat, D.; Doerig, C.; Grøtli, M. Org. Biomol. Chem. 2009, 7, 3421–3429. doi:10.1039/b906482f |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 41. | Raszeja, L. J.; Siegmund, D.; Cordes, A. L.; Güldenhaupt, J.; Gerwert, K.; Hahn, S.; Metzler-Nolte, N. Chem. Commun. 2017, 53, 905–908. doi:10.1039/c6cc07553c |
| 10. | Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d |
| 16. | Elliott, P. I. P. Organometallic complexes with 1,2,3-triazole-derived ligands. In Organometallic chemistry; Fairlamb, I.; Lynam, J.; Dane, S. B. J., Eds.; Specialist Periodical Reports, 0301-0074, Vol. 39; The Royal Society of Chemistry: Cambridge, UK, 2014; pp 1–25. doi:10.1039/9781849737692-00001 |
| 17. | Crowley, J. D.; McMorran, D. A. “Click-Triazole” Coordination Chemistry: Exploiting 1,4-Disubstituted-1,2,3-Triazoles as Ligands. In Click triazoles; Košmrlj, J.; Buckley, B. R., Eds.; Topics in Heterocyclic Chemistry; Springer: Heidelberg, Germany, 2012; pp 31–83. doi:10.1007/7081_2011_67 |
| 18. | Aromí, G.; Barrios, L. A.; Roubeau, O.; Gamez, P. Coord. Chem. Rev. 2011, 255, 485–546. doi:10.1016/j.ccr.2010.10.038 |
| 35. | Zhao, G.-W.; Zhao, J.-H.; Hu, Y.-X.; Zhang, D.-Y.; Li, X. Synth. Met. 2016, 212, 131–141. doi:10.1016/j.synthmet.2015.12.014 |
| 7. | Vasilopoulou, M.; Mohd Yusoff, A. R. b.; Daboczi, M.; Conforto, J.; Gavim, A. E. X.; da Silva, W. J.; Macedo, A. G.; Soultati, A.; Pistolis, G.; Schneider, F. K.; Dong, Y.; Jacoutot, P.; Rotas, G.; Jang, J.; Vougioukalakis, G. C.; Chochos, C. L.; Kim, J.-S.; Gasparini, N. Nat. Commun. 2021, 12, 4868. doi:10.1038/s41467-021-25135-z |
| 8. | Vishwakarma, V. K.; Nath, S.; Gupta, M.; Dubey, D. K.; Swayamprabha, S. S.; Jou, J.-H.; Pal, S. K.; Sudhakar, A. A. ACS Appl. Electron. Mater. 2019, 1, 1959–1969. doi:10.1021/acsaelm.9b00477 |
| 9. | Huang, T.; Liu, D.; Jiang, J.; Jiang, W. Chem. – Eur. J. 2019, 25, 10926–10937. doi:10.1002/chem.201902116 |
| 36. | Haas, K. L.; Franz, K. J. Chem. Rev. 2009, 109, 4921–4960. doi:10.1021/cr900134a |
| 37. | Zhang, R.; Yuan, J. Acc. Chem. Res. 2020, 53, 1316–1329. doi:10.1021/acs.accounts.0c00172 |
| 1. | Gedefaw, D.; Prosa, M.; Bolognesi, M.; Seri, M.; Andersson, M. R. Adv. Energy Mater. 2017, 7, 1700575. doi:10.1002/aenm.201700575 |
| 6. | Yuan, J.; Ouyang, J.; Cimrová, V.; Leclerc, M.; Najari, A.; Zou, Y. J. Mater. Chem. C 2017, 5, 1858–1879. doi:10.1039/c6tc05381e |
| 10. | Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d |
| 11. | Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702 |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 38. | Ching, H. Y. V.; Wang, X.; He, M.; Perujo Holland, N.; Guillot, R.; Slim, C.; Griveau, S.; Bertrand, H. C.; Policar, C.; Bedioui, F.; Fontecave, M. Inorg. Chem. 2017, 56, 2966–2976. doi:10.1021/acs.inorgchem.6b03078 |
| 39. | Merillas, B.; Cuéllar, E.; Diez-Varga, A.; Torroba, T.; García-Herbosa, G.; Fernández, S.; Lloret-Fillol, J.; Martín-Alvarez, J. M.; Miguel, D.; Villafañe, F. Inorg. Chem. 2020, 59, 11152–11165. doi:10.1021/acs.inorgchem.0c01654 |
| 40. | Mukherjee, J.; Siewert, I. Eur. J. Inorg. Chem. 2020, 4319–4333. doi:10.1002/ejic.202000738 |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 25. | Bruschi, C.; Gui, X.; Salaeh‐arae, N.; Barchi, T.; Fuhr, O.; Lebedkin, S.; Klopper, W.; Bizzarri, C. Eur. J. Inorg. Chem. 2021, 4074–4084. doi:10.1002/ejic.202100653 |
| 26. | Maiti, S.; Roy, N.; Babu, L. T.; Moharana, P.; Athira, C. C.; Darsana Sreedhar, E.; De, S.; Ashok Kumar, S. K.; Paira, P. New J. Chem. 2020, 44, 920–931. doi:10.1039/c9nj03131f |
| 10. | Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 38. | Ching, H. Y. V.; Wang, X.; He, M.; Perujo Holland, N.; Guillot, R.; Slim, C.; Griveau, S.; Bertrand, H. C.; Policar, C.; Bedioui, F.; Fontecave, M. Inorg. Chem. 2017, 56, 2966–2976. doi:10.1021/acs.inorgchem.6b03078 |
| 45. | Kim, T. Y.; Elliott, A. B. S.; Shaffer, K. J.; John McAdam, C.; Gordon, K. C.; Crowley, J. D. Polyhedron 2013, 52, 1391–1398. doi:10.1016/j.poly.2012.05.003 |
| 38. | Ching, H. Y. V.; Wang, X.; He, M.; Perujo Holland, N.; Guillot, R.; Slim, C.; Griveau, S.; Bertrand, H. C.; Policar, C.; Bedioui, F.; Fontecave, M. Inorg. Chem. 2017, 56, 2966–2976. doi:10.1021/acs.inorgchem.6b03078 |
| 10. | Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d |
| 11. | Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702 |
| 10. | Chattopadhyay, B.; Vera, C. I. R.; Chuprakov, S.; Gevorgyan, V. Org. Lett. 2010, 12, 2166–2169. doi:10.1021/ol100745d |
| 11. | Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702 |
| 30. | Deleuze-Masquéfa, C.; Gerebtzoff, G.; Subra, G.; Fabreguettes, J.-R.; Ovens, A.; Carraz, M.; Strub, M.-P.; Bompart, J.; George, P.; Bonnet, P.-A. Bioorg. Med. Chem. 2004, 12, 1129–1139. doi:10.1016/j.bmc.2003.11.034 |
| 11. | Roy, S.; Khatua, H.; Das, S. K.; Chattopadhyay, B. Angew. Chem., Int. Ed. 2019, 58, 11439–11443. doi:10.1002/anie.201904702 |
| 27. | Kumar, M.; Joshi, G.; Arora, S.; Singh, T.; Biswas, S.; Sharma, N.; Bhat, Z. R.; Tikoo, K.; Singh, S.; Kumar, R. Molecules 2021, 26, 1490. doi:10.3390/molecules26051490 |
| 28. | Deleuze-Masquefa, C.; Moarbess, G.; Bonnet, P.-A.; Pinguet, F.; Bazarbachi, A.; Bressolle, F. Synthesis of imidazo[l,2-a]quinoxalines for treating cancers. WO Patent WO2009043934, April 9, 2009. |
| 12. | Bertrand, H. C.; Clède, S.; Guillot, R.; Lambert, F.; Policar, C. Inorg. Chem. 2014, 53, 6204–6223. doi:10.1021/ic5007007 |
| 29. | Liu, C.-H.; Wang, B.; Li, W.-Z.; Yun, L.-H.; Liu, Y.; Su, R.-B.; Li, J.; Liu, H. Bioorg. Med. Chem. 2004, 12, 4701–4707. doi:10.1016/j.bmc.2004.06.026 |
| 45. | Kim, T. Y.; Elliott, A. B. S.; Shaffer, K. J.; John McAdam, C.; Gordon, K. C.; Crowley, J. D. Polyhedron 2013, 52, 1391–1398. doi:10.1016/j.poly.2012.05.003 |
© 2022 Holzhauer et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.