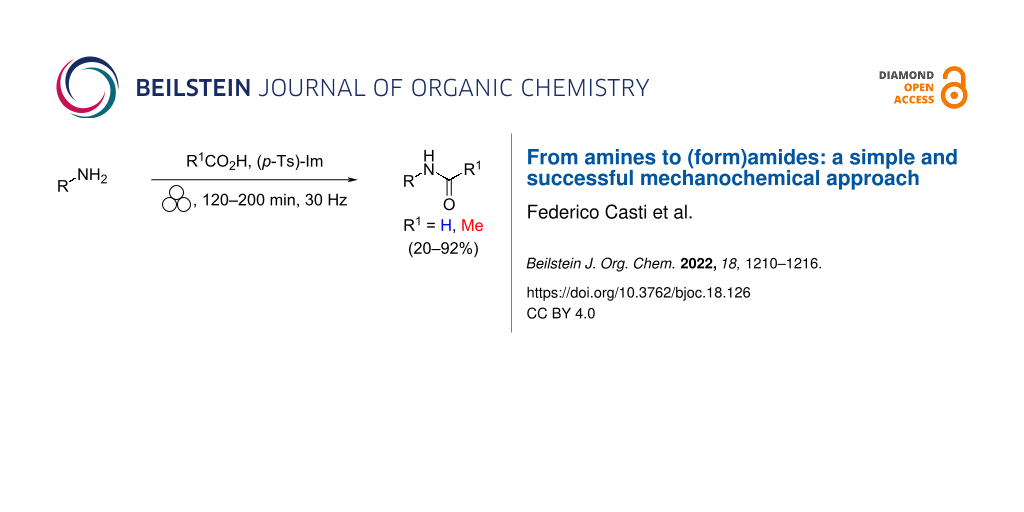
Abstract
Two easily accessible routes for preparing an array of formylated and acetylated amines under mechanochemical conditions are presented. The two methodologies exhibit complementary features as they enable the derivatization of aliphatic and aromatic amines.

Graphical Abstract
Introduction
The preparation of N-formylated and N-acetylated amines plays a crucial role in organic synthesis [1-6]. In one respect, it is relevant to protect the amine group straightforwardly and under mild conditions [7,8]. On the other hand, the formamide and acetamide moieties are found in many active pharmaceutical ingredients (APIs) and natural products [1,9]. N-Formyl derivatives were used as building blocks in Vilsmeier–Haack reactions [10,11] and for preparing molecule drug substances, various heterocycles, formamidines, isocyanates, and isocyanides [12-17]. The large number of procedures reported in the literature witnesses the relevance of this class of compounds [9,18-26]. However, despite the remarkable advancement in the field, many conventional methodologies proceed at high temperatures and require an expensive catalyst and toxic reagents. In addition, these procedures only work in substantial excess of formyl or acyl sources, often used as a solvent.
Mechanochemistry has been established as a powerful tool for the rapid, clean, and environmentally friendly synthesis of organic compounds, avoiding bulk solvent and restrictions of solvent-based chemistry [27-34]. In general, mechanochemistry refers to studying solid-state chemical changes promoted by external mechanical energy, such as grinding or milling. A deeper understanding of its mechanistic aspects laid the basis for further growth in this topic, opening new routes to more efficient mechanochemical reactions [35-42].
In our effort to develop green and sustainable methodologies using mechanochemistry [43-46], we recently studied a protocol for synthesizing isocyanides using p-tosyl chloride (Ts-Cl) in basic conditions, starting from the corresponding formamides [47]. In this work, we aimed to set compatible conditions to access formamides, envisioning the possibility of generating the isocyanide in a one-pot, two-step reaction. However, to the best of our knowledge, despite the notable improvement in the mechanochemical synthesis of the amide moiety [44,48-54], no systematic report of N-formylation and N-acetylation by ball milling has been reported yet. Here, we describe two complementary procedures to prepare formamides and acetamides, applied to primary and secondary aromatic and aliphatic amines. The methodologies directly involve HCO2H derivatives and CH3CO2H and two activating agents for promoting amide coupling.
Results and Discussion
We started our investigation by reacting p-methoxyaniline (1.0 mmol) with ammonium formate (3.0 mmol) (Table 1, entry 1) in a zirconia jar in the presence of one milling ball of the same material (Ø = 8 mm, m = 3.2 g) [22,55] in a horizontal vibratory mill at 30 Hz. Under these conditions, we did not detect the formation of the formamide moiety. At the same time, the desired product 2 was obtained in 16% NMR yield when formic acid was added to the reaction mixture (Table 1, entry 2). Switching to sodium sulfate as a grinding additive significantly enhanced the reaction performance (Table 1, entries 3 and 4). At this stage, we wondered, if using a more efficient dehydrating agent would be mandatory for the reaction to occur. Therefore, we turned our attention toward p-tosylimidazole (p-Ts-Im), a cheap and commercially available reagent directly prepared from p-toluensulfonic acid by reaction with 1,1′-carbonyldiimidazole (CDI). The compound proved very effective for dehydrating oximes under mechanochemical acidic Beckmann conditions [44]. Moreover, it represents a suitable and compatible means in the view of a one-pot methodology for preparing isocyanides directly from amines [56].
Table 1: Optimization of reaction conditions for 2.a,b.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-126-i3.svg?max-width=637&scale=1.0)
|
||||
| Entry | Formic acid (equiv) | Additives (equiv) | Yield of 2 (%)c | Ratio 2:2a |
| 1 | – | HCOONH4 (3.0) | – | – |
| 2 | 1.5 | HCOONH4 (3.0) | 16 | – |
| 3 | 1.5 | Na2SO4 (2.8) | 40 | – |
| 4 | 1.5 | Na2SO4 (2.8)/ MeOH (LAG, η = 0.2) | 45 | – |
| 5 | 1.5 | p-Ts-Im/Et3N 1:1 | 56 | 70:30 |
| 6 | 2 | p-Ts-Im (1.0) | 75 | 90:10 |
| 7d | 2 | p-Ts-Im (1.0) | 85 | 88:12 |
| 8d | 2 | p-Ts-Im (0.1) | 85 | 95:5 |
| 9d | 2 | imidazole (1.0) | 94 | – |
aThe reaction scheme was depicted using the symbolism proposed in [57]. bConditions: compound 1 (1.0 mmol, 123.1 mg), formic acid, and additives in the given ratio were milled in a horizontal vibratory mill in a 15 mL ZrO2 milling jar with one milling ball (Ø = 8 mm, m = 3.2 g) of the same material for 200 minutes at the frequency of 30 Hz. cDetermined by 1H NMR analysis. d20 milling balls (Ø = 3.0 mm, mtot = 6.5 g) were used.
When the amine 1 was reacted in the presence of Et3N, HCOOH, and p-Ts-Im [58] (Table 1, entry 5), the formamide was accompanied by a significant amount of sulfonamide (formamide/sulfonamide ratio: 70:30). Better results were observed when p-Ts-Im was used under acidic conditions (see Table 1, entries 6 and 7). A slight enhancement in yields was observed when 20 balls (Ø = 3 mm, mtot = 6.5 g) were used instead of one ball.
Recent studies on the effect of the size and number of milling balls pointed out that milling balls of larger diameter led to more rapid transformations [59,60]. On the other side, previously reported procedures revealed a beneficial effect on the reaction rate when the volume fraction occupied by balls inside the reactor increases. In addition, it was found that in some certain transformations, small milling balls are beneficial for the reaction rates since the number of stress events is significantly increased [61,62]. The stress energy appears to be less critical in these processes, and we cannot rule out a thermochemical process [63].
Furthermore, we observed that the reaction efficiently took place even in the presence of a catalytic amount of p-Ts-Im without significant differences in the reactivity.
This data led us to question the effective role of p-Ts-Im in promoting the formylation reaction, which could exploit its role as the sole solid auxiliary of grinding. p-Tosylimidazole releases imidazole as a byproduct during the process. Therefore, we wondered whether imidazole could promote the formation of the target formamide 2. Imidazole, compared to p-tosylimidazole, is a cheaper reagent and makes the final purification process easier.
As a matter of fact, by reacting 1.0 mmol of the model substrate with 2.0 mmol of formic acid and 1.0 mmol of imidazole [23], the product was obtained in better yield and with a higher degree of purity. A control experiment performed by reacting p-methoxyaniline (1.0 mmol) and formic acid (2.0 mmol) provided lower conversion into the desired formamide 2 (71% NMR yield, Table S1 in Supporting Information File 1), denoting the input given by imidazole as a promoter of formamide synthesis. Further variation in the ratio of imidazole/formic acid/amine decreases the reaction yield (Table S1 in Supporting Information File 1). These data pointed out the importance of working in the presence of an excess of acid and a stoichiometric amount of imidazole.
The better results recorded in the presence of imidazole may be due to different factors. It is known that imidazole can efficiently promote those processes involving proton transfer under mechanochemical conditions. In the solvent-based procedure, imidazole has already been used in combination with DMF, which works as a solvent and source of a formyl group [23]. In that paper, the authors assumed the formation of formylimidazole as an intermediate in the formylation reaction. However, the intermediate was not detected by NMR or GC–MS analysis, possibly due to its instability [64]. Several studies carried out on our reaction crude at different times did not show the presence of compounds traceable to formylimidazole. The imidazole plays a dual role as promoting reagent and solid grinding additive and can be easily removed by aqueous acid workup. The adoption of imidazole as a grinding additive is required to avoid the slurry obtained by mixing amine and acid while allowing the formation of a waxy solid, which is more suitable for a mechanochemical action. With the optimized conditions in hand, the methodology was successfully applied to the synthesis of several formamides starting from a series of aromatic amines (Scheme 1).
![[1860-5397-18-126-i1]](/bjoc/content/inline/1860-5397-18-126-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Scope of the formylation reaction using imidazole. Reaction conditions: amine (1.0 mmol), formic acid (2.0 mmol), and imidazole (1.0 mmol) were milled in a horizontal vibratory mill in a 15 mL ZrO2 milling jar with 20 milling balls (Ø = 3 mm, mtot = 6.5 g) of the same material for 200 minutes at the frequency of 30 Hz.
Scheme 1: Scope of the formylation reaction using imidazole. Reaction conditions: amine (1.0 mmol), formic ac...
The N-formyl derivative 3 was prepared in good yields as well as formanilides bearing halogen substituents, which were well tolerated (Scheme 1, products 4 and 5). N-(1-Naphthyl)formamide (6) was obtained in satisfying yield, while the methyl and tert-butyl ester moieties affected the reaction outcome (Scheme 1, amides 7 and 8). Secondary amines were also tested under the developed conditions; N-methylaniline provided the desired product 9 in 39% yield, while outstanding results were achieved in the N-formylation of indoline (Scheme 1, formamide 10). The present methodology could also be effectively applied to the synthesis of N-formylmorpholine (Scheme 1, product 11).
Aliphatic primary amines can be more challenging substrates [22]. In fact, when the reaction was tested with phenylethylamine (12), we observed only 10% of conversion into the desired formamide (Table 2, entry 1). We, therefore, chose to resume the use of p-Ts-Im, envisioning it could contribute to improving the reactivity. At this point, we reacted the amine 12 (1.0 mmol) with p-Ts-Im (1.0 mmol), and formic acid (1.0 or 2.0 mmol, Table 2, compare entries 2 and 3) for 200 minutes at the frequency of 30 Hz and observed a significant improvement on the performance of the reaction. To our delight, the complete conversion into the desired formamide 13 was obtained using 1.5 equivalents of p-Ts-Im. Under these experimental conditions, we did not detect the formation of the sulfonamide derivative, preserving complete selectivity towards the target formamide (Table 2, entry 4).
Table 2: Optimization of reaction conditions for product 13.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-126-i4.svg?max-width=637&scale=1.0)
|
|||
| Entry | Formic acid (equiv) | Additives (equiv) | Yield of 13 (%)b |
| 1 | 2 | imidazole (1.0) | 10 |
| 2 | 2 | p-Ts-Im (1.0) | 42 |
| 3 | 1 | p-Ts-Im (1.0) | 42 |
| 4 | 1.5 | p-Ts-Im (1.5) | 99 |
| 5c | 1.5 | p-Ts-Im (1.5) | 95 |
aAmine 12 (1.0 mmol, 121.2 mg), formic acid, and additives in the given ratio were milled in a horizontal vibratory mill in a 15 mL ZrO2 milling jar with 20 milling balls (Ø = 3 mm, mtot = 6.5 g) of the same material for 200 minutes at the frequency of 30 Hz; bdetermined by 1H NMR analysis; cthe mixture was ball-milled for 120 minutes.
Remarkably, the results remain unchanged regarding yields and purity by shortening the reaction time (Table 2, entry 5). Furthermore, these optimized conditions were applied to other amines that provided us with poor results using the methodology previously described (Scheme 2).
![[1860-5397-18-126-i2]](/bjoc/content/inline/1860-5397-18-126-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Scope of the formylation reaction using p-Ts-Im as activating agent. Reaction conditions: amine (1.0 mmol), formic acid (1.5 mmol), and p-Ts-Im (1.5 mmol) were milled in a horizontal vibratory mill in a 15 mL ZrO2 milling jar with 20 milling balls (Ø = 3 mm, mtot = 6.5 g) of the same material for 120 minutes at the frequency of 30 Hz. aIsolated yields. bAcetic acid and p-Ts-Im were ball-milled for 10 minutes, then the amine was added, and the mixture was ball-milled for additional 120 minutes.
Scheme 2: Scope of the formylation reaction using p-Ts-Im as activating agent. Reaction conditions: amine (1....
We obtained the formation of formamides 14 and 15 from cyclohexylamine and benzylamine in high yields (Scheme 2). Excellent results were also achieved using secondary aliphatic amines such as dibenzyl- and methylbenzylamine, which provided amides 16 and 17 in 87% and 74% isolated yields, respectively. Furthermore, the methodology allowed us to synthesize a set of aromatic formanilides (amides 18–20, Scheme 2). Indeed, the deactivated p-nitroaniline was successfully converted into the corresponding formamide 19 as was the poorly nucleophilic diphenylamine (Scheme 2, amide 20).
Lastly, we aimed to apply the procedure to the acylation of a series of amines. To our delight, we successfully extended the methodology to the mechanosynthesis of amides 21–23 (Scheme 2). The best results were obtained when the acetic acid and the p-Ts-Im were ball-milled together before adding the suitable amine. In this way, we could acylate primary aromatic and aliphatic amines under the experimental conditions (Scheme 2, amides 21–23).
Conclusion
In conclusion, we developed two easily accessible ways to obtain a good number of formylated and acylated amines under mechanochemical conditions. The two methodologies exhibit complementary features as they enable the derivatization of different kinds of amines. Imidazole was found a suitable additive for the N-formylation reaction of several aromatic amines. On the other side, p-Ts-Im, activated by the acid reagent, proved an efficient activating agent under mild mechanochemical conditions for the formylation and acylation of those amines with less marked or limited (see protonated aliphatic amines) reactivity.
Supporting Information
| Supporting Information File 1: Experimental section and characterization of synthesized compounds. | ||
| Format: PDF | Size: 2.5 MB | Download |
References
-
Sonawane, R. B.; Rasal, N. K.; Bhange, D. S.; Jagtap, S. V. ChemCatChem 2018, 10, 3907–3913. doi:10.1002/cctc.201800609
Return to citation in text: [1] [2] -
Jackson, A.; Meth-Cohn, O. J. Chem. Soc., Chem. Commun. 1995, 1319. doi:10.1039/c39950001319
Return to citation in text: [1] -
Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606–631. doi:10.1039/b701677h
Return to citation in text: [1] -
Sonawane, R. B.; Rasal, N. K.; Jagtap, S. V. Org. Lett. 2017, 19, 2078–2081. doi:10.1021/acs.orglett.7b00660
Return to citation in text: [1] -
Chikkulapalli, A.; Aavula, S. K.; Mona NP, R.; C., K.; C.H., V. K.; Sulur G., M.; Sumathi, S. Tetrahedron Lett. 2015, 56, 3799–3803. doi:10.1016/j.tetlet.2015.04.077
Return to citation in text: [1] -
Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Chem. Rev. 2009, 109, 2455–2504. doi:10.1021/cr800323s
Return to citation in text: [1] -
Sheehan, J. C.; Yang, D.-D. H. J. Am. Chem. Soc. 1958, 80, 1154–1158. doi:10.1021/ja01538a036
Return to citation in text: [1] -
Avery, M. A. J. Med. Chem. 1999, 42, 5285. doi:10.1021/jm990518h
Return to citation in text: [1] -
Li, C.; Wang, M.; Lu, X.; Zhang, L.; Jiang, J.; Zhang, L. ACS Sustainable Chem. Eng. 2020, 8, 4353–4361. doi:10.1021/acssuschemeng.9b06591
Return to citation in text: [1] [2] -
Vilsmeier, A.; Haack, A. Ber. Dtsch. Chem. Ges. B 1927, 60, 119–122. doi:10.1002/cber.19270600118
Return to citation in text: [1] -
Downie, I. M.; Earle, M. J.; Heaney, H.; Shuhaibar, K. F. Tetrahedron 1993, 49, 4015–4034. doi:10.1016/s0040-4020(01)89915-4
Return to citation in text: [1] -
Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728
Return to citation in text: [1] -
Mayer, J.; Umkehrer, M.; Kalinski, C.; Ross, G.; Kolb, J.; Burdack, C.; Hiller, W. Tetrahedron Lett. 2005, 46, 7393–7396. doi:10.1016/j.tetlet.2005.08.101
Return to citation in text: [1] -
Picci, G.; Mocci, R.; Ciancaleoni, G.; Lippolis, V.; Zielińska‐Błajet, M.; Caltagirone, C. ChemPlusChem 2020, 85, 1389–1395. doi:10.1002/cplu.202000260
Return to citation in text: [1] -
Kakehi, A.; Ito, S.; Hayashi, S.; Fujii, T. Bull. Chem. Soc. Jpn. 1995, 68, 3573–3580. doi:10.1246/bcsj.68.3573
Return to citation in text: [1] -
Han, Y.; Cai, L. Tetrahedron Lett. 1997, 38, 5423–5426. doi:10.1016/s0040-4039(97)01206-9
Return to citation in text: [1] -
Chen, B.-C.; Bednarz, M. S.; Zhao, R.; Sundeen, J. E.; Chen, P.; Shen, Z.; Skoumbourdis, A. P.; Barrish, J. C. Tetrahedron Lett. 2000, 41, 5453–5456. doi:10.1016/s0040-4039(00)00910-2
Return to citation in text: [1] -
Gerack, C.; McElwee-White, L. Molecules 2014, 19, 7689–7713. doi:10.3390/molecules19067689
Return to citation in text: [1] -
Minkin, V. I.; Dorofeenko, G. N. Russ. Chem. Rev. 1960, 29, 599–618. doi:10.1070/rc1960v029n11abeh001257
Return to citation in text: [1] -
Dine, T. M. E.; Evans, D.; Rouden, J.; Blanchet, J. Chem. – Eur. J. 2016, 22, 5894–5898. doi:10.1002/chem.201600234
Return to citation in text: [1] -
Lanigan, R. M.; Sheppard, T. D. Eur. J. Org. Chem. 2013, 7453–7465. doi:10.1002/ejoc.201300573
Return to citation in text: [1] -
Ganapati Reddy, P.; Kishore Kumar, G. D.; Baskaran, S. Tetrahedron Lett. 2000, 41, 9149–9151. doi:10.1016/s0040-4039(00)01636-1
Return to citation in text: [1] [2] [3] -
Suchý, M.; Elmehriki, A. A. H.; Hudson, R. H. E. Org. Lett. 2011, 13, 3952–3955. doi:10.1021/ol201475j
Return to citation in text: [1] [2] [3] -
Kalla, R. M. N.; Lim, J.; Bae, J.; Kim, I. Tetrahedron Lett. 2017, 58, 1595–1599. doi:10.1016/j.tetlet.2017.03.023
Return to citation in text: [1] -
Wan, W.; Hou, J.; Jiang, H.; Yuan, Z.; Ma, G.; Zhao, G.; Hao, J. Eur. J. Org. Chem. 2010, 1778–1786. doi:10.1002/ejoc.200901424
Return to citation in text: [1] -
Chen, Z.; Fu, R.; Chai, W.; Zheng, H.; Sun, L.; Lu, Q.; Yuan, R. Tetrahedron 2014, 70, 2237–2245. doi:10.1016/j.tet.2014.02.042
Return to citation in text: [1] -
Porcheddu, A.; Colacino, E.; De Luca, L.; Delogu, F. ACS Catal. 2020, 10, 8344–8394. doi:10.1021/acscatal.0c00142
Return to citation in text: [1] -
Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478
Return to citation in text: [1] -
Bolt, R. R. A.; Leitch, J. A.; Jones, A. C.; Nicholson, W. I.; Browne, D. L. Chem. Soc. Rev. 2022, 51, 4243–4260. doi:10.1039/d1cs00657f
Return to citation in text: [1] -
James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a
Return to citation in text: [1] -
Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c
Return to citation in text: [1] -
Liu, Y.; Lam, A. H. W.; Fowler, F. W.; Lauher, J. W. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 2002, 389, 39–46. doi:10.1080/713738914
Return to citation in text: [1] -
Tan, D.; Friščić, T. Eur. J. Org. Chem. 2018, 18–33. doi:10.1002/ejoc.201700961
Return to citation in text: [1] -
Cuccu, F.; De Luca, L.; Delogu, F.; Colacino, E.; Solin, N.; Mocci, R.; Porcheddu, A. ChemSusChem 2022, e202200362. doi:10.1002/cssc.202200362
Return to citation in text: [1] -
Boldyreva, E. Chem. Soc. Rev. 2013, 42, 7719–7738. doi:10.1039/c3cs60052a
Return to citation in text: [1] -
Howard, J. L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D. L. Green Chem. 2017, 19, 2798–2802. doi:10.1039/c6gc03139k
Return to citation in text: [1] -
Hwang, S.; Grätz, S.; Borchardt, L. Chem. Commun. 2022, 58, 1661–1671. doi:10.1039/d1cc05697b
Return to citation in text: [1] -
Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a
Return to citation in text: [1] -
Tireli, M.; Juribašić Kulcsár, M.; Cindro, N.; Gracin, D.; Biliškov, N.; Borovina, M.; Ćurić, M.; Halasz, I.; Užarević, K. Chem. Commun. 2015, 51, 8058–8061. doi:10.1039/c5cc01915j
Return to citation in text: [1] -
Puccetti, F.; Lukin, S.; Užarević, K.; Colacino, E.; Halasz, I.; Bolm, C.; Hernández, J. G. Chem. – Eur. J. 2022, 28, e202104409. doi:10.1002/chem.202104409
Return to citation in text: [1] -
Kulla, H.; Fischer, F.; Benemann, S.; Rademann, K.; Emmerling, F. CrystEngComm 2017, 19, 3902–3907. doi:10.1039/c7ce00502d
Return to citation in text: [1] -
Michalchuk, A. A. L.; Emmerling, F. Angew. Chem., Int. Ed. 2022, 61, e202117270. doi:10.1002/anie.202117270
Return to citation in text: [1] -
Porcheddu, A.; Mocci, R.; Brindisi, M.; Cuccu, F.; Fattuoni, C.; Delogu, F.; Colacino, E.; D'Auria, M. V. Green Chem. 2022, 24, 4859–4869. doi:10.1039/d2gc00724j
Return to citation in text: [1] -
Mocci, R.; Colacino, E.; De Luca, L.; Fattuoni, C.; Porcheddu, A.; Delogu, F. ACS Sustainable Chem. Eng. 2021, 9, 2100–2114. doi:10.1021/acssuschemeng.0c07254
Return to citation in text: [1] [2] [3] -
Mocci, R.; De Luca, L.; Delogu, F.; Porcheddu, A. Adv. Synth. Catal. 2016, 358, 3135–3144. doi:10.1002/adsc.201600350
Return to citation in text: [1] -
Mocci, R.; Murgia, S.; De Luca, L.; Colacino, E.; Delogu, F.; Porcheddu, A. Org. Chem. Front. 2018, 5, 531–538. doi:10.1039/c7qo01006k
Return to citation in text: [1] -
Basoccu, F.; Cuccu, F.; Casti, F.; Mocci, R.; Fattuoni, C.; Porcheddu, A. Beilstein J. Org. Chem. 2022, 18, 732–737. doi:10.3762/bjoc.18.73
Return to citation in text: [1] -
Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Angew. Chem., Int. Ed. 2009, 48, 9318–9321. doi:10.1002/anie.200903510
Return to citation in text: [1] -
Gómez-Carpintero, J.; Sánchez, J. D.; González, J. F.; Menéndez, J. C. J. Org. Chem. 2021, 86, 14232–14237. doi:10.1021/acs.joc.1c02350
Return to citation in text: [1] -
Métro, T.-X.; Bonnamour, J.; Reidon, T.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Chem. Commun. 2012, 48, 11781–11783. doi:10.1039/c2cc36352f
Return to citation in text: [1] -
Štrukil, V.; Bartolec, B.; Portada, T.; Đilović, I.; Halasz, I.; Margetić, D. Chem. Commun. 2012, 48, 12100–12102. doi:10.1039/c2cc36613d
Return to citation in text: [1] -
Porte, V.; Thioloy, M.; Pigoux, T.; Métro, T.-X.; Martinez, J.; Lamaty, F. Eur. J. Org. Chem. 2016, 3505–3508. doi:10.1002/ejoc.201600617
Return to citation in text: [1] -
Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Green Chem. 2013, 15, 1116–1120. doi:10.1039/c3gc40302e
Return to citation in text: [1] -
Métro, T.-X.; Martinez, J.; Lamaty, F. ACS Sustainable Chem. Eng. 2017, 5, 9599–9602. doi:10.1021/acssuschemeng.7b03260
Return to citation in text: [1] -
Martina, K.; Baricco, F.; Tagliapietra, S.; Moran, M. J.; Cravotto, G.; Cintas, P. New J. Chem. 2018, 42, 18881–18888. doi:10.1039/c8nj04240c
Return to citation in text: [1] -
Basoccu, F.; Cuccu, F.; Casti, F.; Mocci, R.; Fattuoni, C.; Porcheddu, A. Beilstein J. Org. Chem. 2022, 18, 732–737. doi:10.3762/bjoc.18.73
Return to citation in text: [1] -
Michalchuk, A. A. L.; Boldyreva, E. V.; Belenguer, A. M.; Emmerling, F.; Boldyrev, V. V. Front. Chem. (Lausanne, Switz.) 2021, 9, 685789. doi:10.3389/fchem.2021.685789
Return to citation in text: [1] -
Behrouz, S.; Rad, M. N. S.; Forouhari, E. J. Chem. Res. 2016, 40, 101–106. doi:10.3184/174751916x14531325057887
Return to citation in text: [1] -
Michalchuk, A. A. L.; Tumanov, I. A.; Boldyreva, E. V. CrystEngComm 2019, 21, 2174–2179. doi:10.1039/c8ce02109k
Return to citation in text: [1] -
Michalchuk, A. A. L.; Tumanov, I. A.; Boldyreva, E. V. J. Mater. Sci. 2018, 53, 13380–13389. doi:10.1007/s10853-018-2324-2
Return to citation in text: [1] -
Jicsinszky, L.; Tuza, K.; Cravotto, G.; Porcheddu, A.; Delogu, F.; Colacino, E. Beilstein J. Org. Chem. 2017, 13, 1893–1899. doi:10.3762/bjoc.13.184
Return to citation in text: [1] -
Stolle, A.; Schmidt, R.; Jacob, K. Faraday Discuss. 2014, 170, 267–286. doi:10.1039/c3fd00144j
Return to citation in text: [1] -
The topic is deeply concerning, but the discussion is still a work in progress. More in-depth studies addressing these issues are beyond the scope of this paper.
Return to citation in text: [1] -
Kitagwa, T.; Arita, J.; Nagahata, A. Chem. Pharm. Bull. 1994, 42, 1655–1657. doi:10.1248/cpb.42.1655
Return to citation in text: [1]
| 63. | The topic is deeply concerning, but the discussion is still a work in progress. More in-depth studies addressing these issues are beyond the scope of this paper. |
| 59. | Michalchuk, A. A. L.; Tumanov, I. A.; Boldyreva, E. V. CrystEngComm 2019, 21, 2174–2179. doi:10.1039/c8ce02109k |
| 60. | Michalchuk, A. A. L.; Tumanov, I. A.; Boldyreva, E. V. J. Mater. Sci. 2018, 53, 13380–13389. doi:10.1007/s10853-018-2324-2 |
| 61. | Jicsinszky, L.; Tuza, K.; Cravotto, G.; Porcheddu, A.; Delogu, F.; Colacino, E. Beilstein J. Org. Chem. 2017, 13, 1893–1899. doi:10.3762/bjoc.13.184 |
| 62. | Stolle, A.; Schmidt, R.; Jacob, K. Faraday Discuss. 2014, 170, 267–286. doi:10.1039/c3fd00144j |
| 1. | Sonawane, R. B.; Rasal, N. K.; Bhange, D. S.; Jagtap, S. V. ChemCatChem 2018, 10, 3907–3913. doi:10.1002/cctc.201800609 |
| 2. | Jackson, A.; Meth-Cohn, O. J. Chem. Soc., Chem. Commun. 1995, 1319. doi:10.1039/c39950001319 |
| 3. | Valeur, E.; Bradley, M. Chem. Soc. Rev. 2009, 38, 606–631. doi:10.1039/b701677h |
| 4. | Sonawane, R. B.; Rasal, N. K.; Jagtap, S. V. Org. Lett. 2017, 19, 2078–2081. doi:10.1021/acs.orglett.7b00660 |
| 5. | Chikkulapalli, A.; Aavula, S. K.; Mona NP, R.; C., K.; C.H., V. K.; Sulur G., M.; Sumathi, S. Tetrahedron Lett. 2015, 56, 3799–3803. doi:10.1016/j.tetlet.2015.04.077 |
| 6. | Isidro-Llobet, A.; Álvarez, M.; Albericio, F. Chem. Rev. 2009, 109, 2455–2504. doi:10.1021/cr800323s |
| 12. | Dömling, A. Chem. Rev. 2006, 106, 17–89. doi:10.1021/cr0505728 |
| 13. | Mayer, J.; Umkehrer, M.; Kalinski, C.; Ross, G.; Kolb, J.; Burdack, C.; Hiller, W. Tetrahedron Lett. 2005, 46, 7393–7396. doi:10.1016/j.tetlet.2005.08.101 |
| 14. | Picci, G.; Mocci, R.; Ciancaleoni, G.; Lippolis, V.; Zielińska‐Błajet, M.; Caltagirone, C. ChemPlusChem 2020, 85, 1389–1395. doi:10.1002/cplu.202000260 |
| 15. | Kakehi, A.; Ito, S.; Hayashi, S.; Fujii, T. Bull. Chem. Soc. Jpn. 1995, 68, 3573–3580. doi:10.1246/bcsj.68.3573 |
| 16. | Han, Y.; Cai, L. Tetrahedron Lett. 1997, 38, 5423–5426. doi:10.1016/s0040-4039(97)01206-9 |
| 17. | Chen, B.-C.; Bednarz, M. S.; Zhao, R.; Sundeen, J. E.; Chen, P.; Shen, Z.; Skoumbourdis, A. P.; Barrish, J. C. Tetrahedron Lett. 2000, 41, 5453–5456. doi:10.1016/s0040-4039(00)00910-2 |
| 57. | Michalchuk, A. A. L.; Boldyreva, E. V.; Belenguer, A. M.; Emmerling, F.; Boldyrev, V. V. Front. Chem. (Lausanne, Switz.) 2021, 9, 685789. doi:10.3389/fchem.2021.685789 |
| 10. | Vilsmeier, A.; Haack, A. Ber. Dtsch. Chem. Ges. B 1927, 60, 119–122. doi:10.1002/cber.19270600118 |
| 11. | Downie, I. M.; Earle, M. J.; Heaney, H.; Shuhaibar, K. F. Tetrahedron 1993, 49, 4015–4034. doi:10.1016/s0040-4020(01)89915-4 |
| 58. | Behrouz, S.; Rad, M. N. S.; Forouhari, E. J. Chem. Res. 2016, 40, 101–106. doi:10.3184/174751916x14531325057887 |
| 1. | Sonawane, R. B.; Rasal, N. K.; Bhange, D. S.; Jagtap, S. V. ChemCatChem 2018, 10, 3907–3913. doi:10.1002/cctc.201800609 |
| 9. | Li, C.; Wang, M.; Lu, X.; Zhang, L.; Jiang, J.; Zhang, L. ACS Sustainable Chem. Eng. 2020, 8, 4353–4361. doi:10.1021/acssuschemeng.9b06591 |
| 44. | Mocci, R.; Colacino, E.; De Luca, L.; Fattuoni, C.; Porcheddu, A.; Delogu, F. ACS Sustainable Chem. Eng. 2021, 9, 2100–2114. doi:10.1021/acssuschemeng.0c07254 |
| 7. | Sheehan, J. C.; Yang, D.-D. H. J. Am. Chem. Soc. 1958, 80, 1154–1158. doi:10.1021/ja01538a036 |
| 8. | Avery, M. A. J. Med. Chem. 1999, 42, 5285. doi:10.1021/jm990518h |
| 56. | Basoccu, F.; Cuccu, F.; Casti, F.; Mocci, R.; Fattuoni, C.; Porcheddu, A. Beilstein J. Org. Chem. 2022, 18, 732–737. doi:10.3762/bjoc.18.73 |
| 43. | Porcheddu, A.; Mocci, R.; Brindisi, M.; Cuccu, F.; Fattuoni, C.; Delogu, F.; Colacino, E.; D'Auria, M. V. Green Chem. 2022, 24, 4859–4869. doi:10.1039/d2gc00724j |
| 44. | Mocci, R.; Colacino, E.; De Luca, L.; Fattuoni, C.; Porcheddu, A.; Delogu, F. ACS Sustainable Chem. Eng. 2021, 9, 2100–2114. doi:10.1021/acssuschemeng.0c07254 |
| 45. | Mocci, R.; De Luca, L.; Delogu, F.; Porcheddu, A. Adv. Synth. Catal. 2016, 358, 3135–3144. doi:10.1002/adsc.201600350 |
| 46. | Mocci, R.; Murgia, S.; De Luca, L.; Colacino, E.; Delogu, F.; Porcheddu, A. Org. Chem. Front. 2018, 5, 531–538. doi:10.1039/c7qo01006k |
| 44. | Mocci, R.; Colacino, E.; De Luca, L.; Fattuoni, C.; Porcheddu, A.; Delogu, F. ACS Sustainable Chem. Eng. 2021, 9, 2100–2114. doi:10.1021/acssuschemeng.0c07254 |
| 48. | Declerck, V.; Nun, P.; Martinez, J.; Lamaty, F. Angew. Chem., Int. Ed. 2009, 48, 9318–9321. doi:10.1002/anie.200903510 |
| 49. | Gómez-Carpintero, J.; Sánchez, J. D.; González, J. F.; Menéndez, J. C. J. Org. Chem. 2021, 86, 14232–14237. doi:10.1021/acs.joc.1c02350 |
| 50. | Métro, T.-X.; Bonnamour, J.; Reidon, T.; Sarpoulet, J.; Martinez, J.; Lamaty, F. Chem. Commun. 2012, 48, 11781–11783. doi:10.1039/c2cc36352f |
| 51. | Štrukil, V.; Bartolec, B.; Portada, T.; Đilović, I.; Halasz, I.; Margetić, D. Chem. Commun. 2012, 48, 12100–12102. doi:10.1039/c2cc36613d |
| 52. | Porte, V.; Thioloy, M.; Pigoux, T.; Métro, T.-X.; Martinez, J.; Lamaty, F. Eur. J. Org. Chem. 2016, 3505–3508. doi:10.1002/ejoc.201600617 |
| 53. | Bonnamour, J.; Métro, T.-X.; Martinez, J.; Lamaty, F. Green Chem. 2013, 15, 1116–1120. doi:10.1039/c3gc40302e |
| 54. | Métro, T.-X.; Martinez, J.; Lamaty, F. ACS Sustainable Chem. Eng. 2017, 5, 9599–9602. doi:10.1021/acssuschemeng.7b03260 |
| 64. | Kitagwa, T.; Arita, J.; Nagahata, A. Chem. Pharm. Bull. 1994, 42, 1655–1657. doi:10.1248/cpb.42.1655 |
| 35. | Boldyreva, E. Chem. Soc. Rev. 2013, 42, 7719–7738. doi:10.1039/c3cs60052a |
| 36. | Howard, J. L.; Sagatov, Y.; Repusseau, L.; Schotten, C.; Browne, D. L. Green Chem. 2017, 19, 2798–2802. doi:10.1039/c6gc03139k |
| 37. | Hwang, S.; Grätz, S.; Borchardt, L. Chem. Commun. 2022, 58, 1661–1671. doi:10.1039/d1cc05697b |
| 38. | Howard, J. L.; Cao, Q.; Browne, D. L. Chem. Sci. 2018, 9, 3080–3094. doi:10.1039/c7sc05371a |
| 39. | Tireli, M.; Juribašić Kulcsár, M.; Cindro, N.; Gracin, D.; Biliškov, N.; Borovina, M.; Ćurić, M.; Halasz, I.; Užarević, K. Chem. Commun. 2015, 51, 8058–8061. doi:10.1039/c5cc01915j |
| 40. | Puccetti, F.; Lukin, S.; Užarević, K.; Colacino, E.; Halasz, I.; Bolm, C.; Hernández, J. G. Chem. – Eur. J. 2022, 28, e202104409. doi:10.1002/chem.202104409 |
| 41. | Kulla, H.; Fischer, F.; Benemann, S.; Rademann, K.; Emmerling, F. CrystEngComm 2017, 19, 3902–3907. doi:10.1039/c7ce00502d |
| 42. | Michalchuk, A. A. L.; Emmerling, F. Angew. Chem., Int. Ed. 2022, 61, e202117270. doi:10.1002/anie.202117270 |
| 22. | Ganapati Reddy, P.; Kishore Kumar, G. D.; Baskaran, S. Tetrahedron Lett. 2000, 41, 9149–9151. doi:10.1016/s0040-4039(00)01636-1 |
| 55. | Martina, K.; Baricco, F.; Tagliapietra, S.; Moran, M. J.; Cravotto, G.; Cintas, P. New J. Chem. 2018, 42, 18881–18888. doi:10.1039/c8nj04240c |
| 22. | Ganapati Reddy, P.; Kishore Kumar, G. D.; Baskaran, S. Tetrahedron Lett. 2000, 41, 9149–9151. doi:10.1016/s0040-4039(00)01636-1 |
| 27. | Porcheddu, A.; Colacino, E.; De Luca, L.; Delogu, F. ACS Catal. 2020, 10, 8344–8394. doi:10.1021/acscatal.0c00142 |
| 28. | Ardila‐Fierro, K. J.; Hernández, J. G. ChemSusChem 2021, 14, 2145–2162. doi:10.1002/cssc.202100478 |
| 29. | Bolt, R. R. A.; Leitch, J. A.; Jones, A. C.; Nicholson, W. I.; Browne, D. L. Chem. Soc. Rev. 2022, 51, 4243–4260. doi:10.1039/d1cs00657f |
| 30. | James, S. L.; Adams, C. J.; Bolm, C.; Braga, D.; Collier, P.; Friščić, T.; Grepioni, F.; Harris, K. D. M.; Hyett, G.; Jones, W.; Krebs, A.; Mack, J.; Maini, L.; Orpen, A. G.; Parkin, I. P.; Shearouse, W. C.; Steed, J. W.; Waddell, D. C. Chem. Soc. Rev. 2012, 41, 413–447. doi:10.1039/c1cs15171a |
| 31. | Stolle, A.; Szuppa, T.; Leonhardt, S. E. S.; Ondruschka, B. Chem. Soc. Rev. 2011, 40, 2317–2329. doi:10.1039/c0cs00195c |
| 32. | Liu, Y.; Lam, A. H. W.; Fowler, F. W.; Lauher, J. W. Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A 2002, 389, 39–46. doi:10.1080/713738914 |
| 33. | Tan, D.; Friščić, T. Eur. J. Org. Chem. 2018, 18–33. doi:10.1002/ejoc.201700961 |
| 34. | Cuccu, F.; De Luca, L.; Delogu, F.; Colacino, E.; Solin, N.; Mocci, R.; Porcheddu, A. ChemSusChem 2022, e202200362. doi:10.1002/cssc.202200362 |
| 23. | Suchý, M.; Elmehriki, A. A. H.; Hudson, R. H. E. Org. Lett. 2011, 13, 3952–3955. doi:10.1021/ol201475j |
| 9. | Li, C.; Wang, M.; Lu, X.; Zhang, L.; Jiang, J.; Zhang, L. ACS Sustainable Chem. Eng. 2020, 8, 4353–4361. doi:10.1021/acssuschemeng.9b06591 |
| 18. | Gerack, C.; McElwee-White, L. Molecules 2014, 19, 7689–7713. doi:10.3390/molecules19067689 |
| 19. | Minkin, V. I.; Dorofeenko, G. N. Russ. Chem. Rev. 1960, 29, 599–618. doi:10.1070/rc1960v029n11abeh001257 |
| 20. | Dine, T. M. E.; Evans, D.; Rouden, J.; Blanchet, J. Chem. – Eur. J. 2016, 22, 5894–5898. doi:10.1002/chem.201600234 |
| 21. | Lanigan, R. M.; Sheppard, T. D. Eur. J. Org. Chem. 2013, 7453–7465. doi:10.1002/ejoc.201300573 |
| 22. | Ganapati Reddy, P.; Kishore Kumar, G. D.; Baskaran, S. Tetrahedron Lett. 2000, 41, 9149–9151. doi:10.1016/s0040-4039(00)01636-1 |
| 23. | Suchý, M.; Elmehriki, A. A. H.; Hudson, R. H. E. Org. Lett. 2011, 13, 3952–3955. doi:10.1021/ol201475j |
| 24. | Kalla, R. M. N.; Lim, J.; Bae, J.; Kim, I. Tetrahedron Lett. 2017, 58, 1595–1599. doi:10.1016/j.tetlet.2017.03.023 |
| 25. | Wan, W.; Hou, J.; Jiang, H.; Yuan, Z.; Ma, G.; Zhao, G.; Hao, J. Eur. J. Org. Chem. 2010, 1778–1786. doi:10.1002/ejoc.200901424 |
| 26. | Chen, Z.; Fu, R.; Chai, W.; Zheng, H.; Sun, L.; Lu, Q.; Yuan, R. Tetrahedron 2014, 70, 2237–2245. doi:10.1016/j.tet.2014.02.042 |
| 47. | Basoccu, F.; Cuccu, F.; Casti, F.; Mocci, R.; Fattuoni, C.; Porcheddu, A. Beilstein J. Org. Chem. 2022, 18, 732–737. doi:10.3762/bjoc.18.73 |
| 23. | Suchý, M.; Elmehriki, A. A. H.; Hudson, R. H. E. Org. Lett. 2011, 13, 3952–3955. doi:10.1021/ol201475j |
© 2022 Casti et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.