Abstract
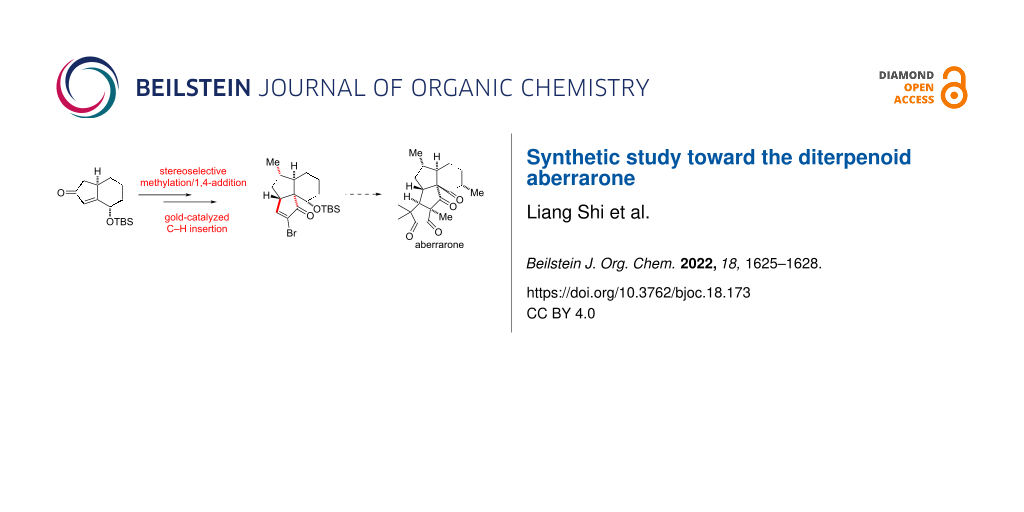
An approach to aberrarone, an antimalarial diterpenoid natural product with tetracyclic skeleton is reported. Key to the stereoselective preparation of the 6-5-5 tricyclic skeleton includes the mediation of Nagata reagent for constructing the C1 all-carbon quaternary centers and gold-catalyzed cyclopentenone synthesis through C–H insertion.

Graphical Abstract
Introduction
Marine natural products have found myriad use in new drug development, exemplified by ET-743 and eribulin [1]. Back in 1990s, Rodriguez and co-workers isolated a rich array of terpenoid natural products from the Caribbean sea whip, Pseudopterogorgia elisabethae with unprecedented carbon skeleton, most of which showed antitumor, antituberculosis and antimalarial activities [2-6]. Among these structurally intriguing natural products, aberrarone (1) shows antimalarial activity against the chloroquine-resistant strain of Plasmodium falciparum (IC50 = 10 μg/mL) [7]. Structurally, aberrarone possesses an unusual tetracyclic carbon skeleton yet-to-be found in Pseudopterogorgia elisabethae species, although related cyclohexane-angularly-fused triquinane systems have been found in waihoensene (3), conidiogenone (4), lycopodium alkaloids magellamine (5) and lycojaponicumin C (6) (Figure 1). Its seven stereogenic centers, including two all-carbon quaternary centers, together with the non-enolizable cyclic α-diketone moiety collectively render aberrarone as an attractive but challenging synthetic target. Its congener elisabanolide (2) with a lactone in the D ring shows their potential biosynthetic relationship [2]. These natural products have been popular synthetic targets mainly due to their intriguing structural features. For example, several total syntheses of 3–6 have been reported [8-29]. Previously, two synthetic studies of aberrarone were reported [30,31] and more recently, Carreira and co-workers reported [32] the first total synthesis of aberrarone through an impressive cascade reaction including a gold-catalyzed Nazarov cyclization, a cyclopropanation followed by intramolecular aldol reaction to forge the A, B and D rings. Impressed by the structural features and biological profiles, our group embarked a project on the total synthesis of this natural product. Herein, we report our stereoselective synthesis of its 6-5-5 tricyclic skeleton.
![[1860-5397-18-173-1]](/bjoc/content/figures/1860-5397-18-173-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Selected representative natural products with 6-5-5 tricyclic skeleton.
Figure 1: Selected representative natural products with 6-5-5 tricyclic skeleton.
Our retrosynthetic analysis is shown in Scheme 1. For the formation of the D ring with one quaternary carbon stereocenter and 1,2-dikeone moiety, Nazarov cyclization [33] of 7 was proposed for synthesizing this challenging moiety. The corresponding precursor cyclopentenone 8 may be prepared from alkynone 9 through a gold-catalyzed C–H insertion [34]. Alkynone 9 could be achieved through functional transformation from 10, which itself would be prepared through methylation and conjugate addition from Pauson–Khand adduct 11. This cyclopentenone could be readily accessed from 1,7-enyne 12 which could be obtained through the reported procedure [35] from the commercially available 5-hexenoic acid.
![[1860-5397-18-173-i1]](/bjoc/content/inline/1860-5397-18-173-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Retrosynthetic analysis of aberrarone (1).
Scheme 1: Retrosynthetic analysis of aberrarone (1).
Results and Discussion
Our synthetic route commenced from known compound 12 which is readily accessed from 5-hexenoic acid through a reported procedure [35]. In the mediation of Co2(CO)8, the 6-5 bicyclic skeleton [36] was constructed with the right configuration at C6, and the explanation of this stereoselectivity is possible through the conformation of 14 where the OTBS group is in pseudoequatorial position (Scheme 2). Therefore, the Pauson–Khand reaction proceeded to afford 11 containing an α-H at C6. From this intermediate, to our delight, the stereoselective attachment of the requisite methyl group through the corresponding lithium enolate occurred from the convex face of the bicyclic ring system [37]. After these two continuous stereocenters were successfully installed, the expected challenging all-carbon quaternary center at C1 was constructed utilizing the Nagata reagent (Et2AlCN). By using this strategy, the stereogenic center at C1 was synthesized, along with a smooth attachment of the cyanate group served for further functional group transformation to construct the C ring through C–H insertion. The stereochemistry finding of this conjugate addition from the convex face of the 6-5 ring system was further confirmed through X-ray crystallographic analysis.
![[1860-5397-18-173-i2]](/bjoc/content/inline/1860-5397-18-173-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthetic study toward aberrarone (1).
Scheme 2: Synthetic study toward aberrarone (1).
With the key intermediate 10 in hand, we were in a position to test the planned two-step transformation including the palladium-catalyzed reductive cross coupling with HCO2H followed by Pd/C-catalyzed hydrogenation. To our surprise, the hydrogenation turned out to be a difficult transformation due to the steric hindered environment of the trisubstituted double bond, mainly caused by the bulky OTBS group. However, direct subjection of compound 16 to hydrogenation [38] afforded reduction of both triflate and double bond. The plausible pathway for this facile transformation might proceed with first hydrogenation followed by the substitution of the labile triflate ester (for details, see Supporting Information File 1). Moving forward, compound 17 was further converted into alkynone 9 through DIBAL-H reduction, nucleophilic addition and Dess–Martin oxidation. At this stage, the pivotal C–H insertion step was tried under the reported conditions [34], and cyclopentenone 8 was successfully obtained. Further study with cross coupling or halogen–magnesium exchange shows this moiety is inert for functional group transformation. The attempt for constructing the D ring is currently undergoing.
Conclusion
In summary, we have developed an approach to assemble the tricyclic skeleton of aberrarone through stereoselective methylation, conjugate addition and gold-catalyzed C–H insertion from the readily accessed cyclopentenone. Further work to access natural product aberrarone from the key intermediate cyclopentenone 8 is currently underway, and will be reported in due course.
Supporting Information
The crystallographic data of compound 10 (CCDC 2204711) has been deposited at the Cambridge Crystallographic Database Center (http://www.ccdc.cam.ac.uk).
| Supporting Information File 1: Characterization data and 1H NMR, 13C NMR, and HRMS spectra of the compounds. | ||
| Format: PDF | Size: 1.3 MB | Download |
References
-
Altmann, K.-H. Chimia 2017, 71, 646–652. doi:10.2533/chimia.2017.646
Return to citation in text: [1] -
Rodríguez, A. D.; González, E.; Huang, S. D. J. Org. Chem. 1998, 63, 7083–7091. doi:10.1021/jo981385v
Return to citation in text: [1] [2] -
Rodríguez, A. D.; Ramírez, C. Org. Lett. 2000, 2, 507–510. doi:10.1021/ol991362i
Return to citation in text: [1] -
Rodríguez, A. D.; Ramírez, C.; Rodríguez, I. I.; Barnes, C. L. J. Org. Chem. 2000, 65, 1390–1398. doi:10.1021/jo9914869
Return to citation in text: [1] -
Rodríguez, A. D.; Ramírez, C.; Shi, Y.-P. J. Org. Chem. 2000, 65, 6682–6687. doi:10.1021/jo000875w
Return to citation in text: [1] -
Wei, X.; Rodríguez, I. I.; Rodríguez, A. D.; Barnes, C. L. J. Org. Chem. 2007, 72, 7386–7389. doi:10.1021/jo070649n
Return to citation in text: [1] -
Rodríguez, I. I.; Rodríguez, A. D.; Zhao, H. J. Org. Chem. 2009, 74, 7581–7584. doi:10.1021/jo901578r
Return to citation in text: [1] -
Jeon, H.; Winkler, J. D. Synthesis 2021, 53, 475–488. doi:10.1055/s-0040-1705953
Return to citation in text: [1] -
Lee, H.; Kang, T.; Lee, H.-Y. Angew. Chem., Int. Ed. 2017, 56, 8254–8257. doi:10.1002/anie.201704492
Return to citation in text: [1] -
Qu, Y.; Wang, Z.; Zhang, Z.; Zhang, W.; Huang, J.; Yang, Z. J. Am. Chem. Soc. 2020, 142, 6511–6515. doi:10.1021/jacs.0c02143
Return to citation in text: [1] -
Peng, C.; Arya, P.; Zhou, Z.; Snyder, S. A. Angew. Chem., Int. Ed. 2020, 59, 13521–13525. doi:10.1002/anie.202004177
Return to citation in text: [1] -
Rosenbaum, L.-C.; Häfner, M.; Gaich, T. Angew. Chem., Int. Ed. 2021, 60, 2939–2942. doi:10.1002/anie.202011298
Return to citation in text: [1] -
Wang, Y.-P.; Fang, K.; Tu, Y.-Q.; Yin, J.-J.; Zhao, Q.; Ke, T. Nat. Commun. 2022, 13, 2335. doi:10.1038/s41467-022-29947-5
Return to citation in text: [1] -
Hou, S.-H.; Tu, Y.-Q.; Wang, S.-H.; Xi, C.-C.; Zhang, F.-M.; Wang, S.-H.; Li, Y.-T.; Liu, L. Angew. Chem., Int. Ed. 2016, 55, 4456–4460. doi:10.1002/anie.201600529
Return to citation in text: [1] -
Hu, P.; Chi, H. M.; DeBacker, K. C.; Gong, X.; Keim, J. H.; Hsu, I. T.; Snyder, S. A. Nature 2019, 569, 703–707. doi:10.1038/s41586-019-1179-2
Return to citation in text: [1] -
Xu, B.; Xun, W.; Su, S.; Zhai, H. Angew. Chem., Int. Ed. 2020, 59, 16475–16479. doi:10.1002/anie.202007247
Return to citation in text: [1] -
Hirst, G. C.; Johnson, T. O., Jr.; Overman, L. E. J. Am. Chem. Soc. 1993, 115, 2992–2993. doi:10.1021/ja00060a064
Return to citation in text: [1] -
Paquette, L. A.; Friedrich, D.; Pinard, E.; Williams, J. P.; St. Laurent, D.; Roden, B. A. J. Am. Chem. Soc. 1993, 115, 4377–4378. doi:10.1021/ja00063a072
Return to citation in text: [1] -
Sha, C.-K.; Lee, F.-K.; Chang, C.-J. J. Am. Chem. Soc. 1999, 121, 9875–9876. doi:10.1021/ja992315o
Return to citation in text: [1] -
Yen, C.-F.; Liao, C.-C. Angew. Chem., Int. Ed. 2002, 41, 4090–4093. doi:10.1002/1521-3773(20021104)41:21<4090::aid-anie4090>3.0.co;2-%23
Return to citation in text: [1] -
Ishizaki, M.; Niimi, Y.; Hoshino, O.; Hara, H.; Takahashi, T. Tetrahedron 2005, 61, 4053–4065. doi:10.1016/j.tet.2005.02.044
Return to citation in text: [1] -
Kozaka, T.; Miyakoshi, N.; Mukai, C. J. Org. Chem. 2007, 72, 10147–10154. doi:10.1021/jo702136b
Return to citation in text: [1] -
Jiang, S.-Z.; Lei, T.; Wei, K.; Yang, Y.-R. Org. Lett. 2014, 16, 5612–5615. doi:10.1021/ol502679v
Return to citation in text: [1] -
Lin, K.-W.; Ananthan, B.; Tseng, S.-F.; Yan, T.-H. Org. Lett. 2015, 17, 3938–3940. doi:10.1021/acs.orglett.5b01975
Return to citation in text: [1] -
McGee, P.; Bétournay, G.; Barabé, F.; Barriault, L. Angew. Chem., Int. Ed. 2017, 56, 6280–6283. doi:10.1002/anie.201611606
Return to citation in text: [1] -
Liu, J.; Chen, S.; Li, N.; Qiu, F. G. Adv. Synth. Catal. 2019, 361, 3514–3517. doi:10.1002/adsc.201900376
Return to citation in text: [1] -
Huang, B.-B.; Lei, K.; Zhong, L.-R.; Yang, X.; Yao, Z.-J. J. Org. Chem. 2022, 87, 8685–8696. doi:10.1021/acs.joc.2c00871
Return to citation in text: [1] -
Hou, S.-H.; Tu, Y.-Q.; Liu, L.; Zhang, F.-M.; Wang, S.-H.; Zhang, X.-M. Angew. Chem., Int. Ed. 2013, 52, 11373–11376. doi:10.1002/anie.201306369
Return to citation in text: [1] -
Zheng, N.; Zhang, L.; Gong, J.; Yang, Z. Org. Lett. 2017, 19, 2921–2924. doi:10.1021/acs.orglett.7b01154
Return to citation in text: [1] -
Srikrishna, A.; Neetu, G. Tetrahedron 2011, 67, 7581–7585. doi:10.1016/j.tet.2011.07.054
Return to citation in text: [1] -
Kobayashi, T.; Tokumoto, K.; Tsuchitani, Y.; Abe, H.; Ito, H. Tetrahedron 2015, 71, 5918–5924. doi:10.1016/j.tet.2015.05.089
Return to citation in text: [1] -
Amberg, W. M.; Carreira, E. M. J. Am. Chem. Soc. 2022, 144, 15475–15479. doi:10.1021/jacs.2c07150
Return to citation in text: [1] -
Hoffmann, M.; Weibel, J.-M.; de Frémont, P.; Pale, P.; Blanc, A. Org. Lett. 2014, 16, 908–911. doi:10.1021/ol403663j
Return to citation in text: [1] -
Wang, Y.; Zarca, M.; Gong, L.-Z.; Zhang, L. J. Am. Chem. Soc. 2016, 138, 7516–7519. doi:10.1021/jacs.6b04297
Return to citation in text: [1] [2] -
Cantagrel, G.; Meyer, C.; Cossy, J. Synlett 2007, 2983–2986. doi:10.1055/s-2007-992366
Return to citation in text: [1] [2] -
Mukai, C.; Kozaka, T.; Suzuki, Y.; Kim, I. J. Tetrahedron 2004, 60, 2497–2507. doi:10.1016/j.tet.2004.01.041
Return to citation in text: [1] -
Hog, D. T.; Huber, F. M. E.; Jiménez-Osés, G.; Mayer, P.; Houk, K. N.; Trauner, D. Chem. – Eur. J. 2015, 21, 13646–13665. doi:10.1002/chem.201501423
Return to citation in text: [1] -
Jigajinni, V. B.; Wightman, R. H. Tetrahedron Lett. 1982, 23, 117–120. doi:10.1016/s0040-4039(00)97549-x
Return to citation in text: [1]
| 8. | Jeon, H.; Winkler, J. D. Synthesis 2021, 53, 475–488. doi:10.1055/s-0040-1705953 |
| 9. | Lee, H.; Kang, T.; Lee, H.-Y. Angew. Chem., Int. Ed. 2017, 56, 8254–8257. doi:10.1002/anie.201704492 |
| 10. | Qu, Y.; Wang, Z.; Zhang, Z.; Zhang, W.; Huang, J.; Yang, Z. J. Am. Chem. Soc. 2020, 142, 6511–6515. doi:10.1021/jacs.0c02143 |
| 11. | Peng, C.; Arya, P.; Zhou, Z.; Snyder, S. A. Angew. Chem., Int. Ed. 2020, 59, 13521–13525. doi:10.1002/anie.202004177 |
| 12. | Rosenbaum, L.-C.; Häfner, M.; Gaich, T. Angew. Chem., Int. Ed. 2021, 60, 2939–2942. doi:10.1002/anie.202011298 |
| 13. | Wang, Y.-P.; Fang, K.; Tu, Y.-Q.; Yin, J.-J.; Zhao, Q.; Ke, T. Nat. Commun. 2022, 13, 2335. doi:10.1038/s41467-022-29947-5 |
| 14. | Hou, S.-H.; Tu, Y.-Q.; Wang, S.-H.; Xi, C.-C.; Zhang, F.-M.; Wang, S.-H.; Li, Y.-T.; Liu, L. Angew. Chem., Int. Ed. 2016, 55, 4456–4460. doi:10.1002/anie.201600529 |
| 15. | Hu, P.; Chi, H. M.; DeBacker, K. C.; Gong, X.; Keim, J. H.; Hsu, I. T.; Snyder, S. A. Nature 2019, 569, 703–707. doi:10.1038/s41586-019-1179-2 |
| 16. | Xu, B.; Xun, W.; Su, S.; Zhai, H. Angew. Chem., Int. Ed. 2020, 59, 16475–16479. doi:10.1002/anie.202007247 |
| 17. | Hirst, G. C.; Johnson, T. O., Jr.; Overman, L. E. J. Am. Chem. Soc. 1993, 115, 2992–2993. doi:10.1021/ja00060a064 |
| 18. | Paquette, L. A.; Friedrich, D.; Pinard, E.; Williams, J. P.; St. Laurent, D.; Roden, B. A. J. Am. Chem. Soc. 1993, 115, 4377–4378. doi:10.1021/ja00063a072 |
| 19. | Sha, C.-K.; Lee, F.-K.; Chang, C.-J. J. Am. Chem. Soc. 1999, 121, 9875–9876. doi:10.1021/ja992315o |
| 20. | Yen, C.-F.; Liao, C.-C. Angew. Chem., Int. Ed. 2002, 41, 4090–4093. doi:10.1002/1521-3773(20021104)41:21<4090::aid-anie4090>3.0.co;2-%23 |
| 21. | Ishizaki, M.; Niimi, Y.; Hoshino, O.; Hara, H.; Takahashi, T. Tetrahedron 2005, 61, 4053–4065. doi:10.1016/j.tet.2005.02.044 |
| 22. | Kozaka, T.; Miyakoshi, N.; Mukai, C. J. Org. Chem. 2007, 72, 10147–10154. doi:10.1021/jo702136b |
| 23. | Jiang, S.-Z.; Lei, T.; Wei, K.; Yang, Y.-R. Org. Lett. 2014, 16, 5612–5615. doi:10.1021/ol502679v |
| 24. | Lin, K.-W.; Ananthan, B.; Tseng, S.-F.; Yan, T.-H. Org. Lett. 2015, 17, 3938–3940. doi:10.1021/acs.orglett.5b01975 |
| 25. | McGee, P.; Bétournay, G.; Barabé, F.; Barriault, L. Angew. Chem., Int. Ed. 2017, 56, 6280–6283. doi:10.1002/anie.201611606 |
| 26. | Liu, J.; Chen, S.; Li, N.; Qiu, F. G. Adv. Synth. Catal. 2019, 361, 3514–3517. doi:10.1002/adsc.201900376 |
| 27. | Huang, B.-B.; Lei, K.; Zhong, L.-R.; Yang, X.; Yao, Z.-J. J. Org. Chem. 2022, 87, 8685–8696. doi:10.1021/acs.joc.2c00871 |
| 28. | Hou, S.-H.; Tu, Y.-Q.; Liu, L.; Zhang, F.-M.; Wang, S.-H.; Zhang, X.-M. Angew. Chem., Int. Ed. 2013, 52, 11373–11376. doi:10.1002/anie.201306369 |
| 29. | Zheng, N.; Zhang, L.; Gong, J.; Yang, Z. Org. Lett. 2017, 19, 2921–2924. doi:10.1021/acs.orglett.7b01154 |
| 34. | Wang, Y.; Zarca, M.; Gong, L.-Z.; Zhang, L. J. Am. Chem. Soc. 2016, 138, 7516–7519. doi:10.1021/jacs.6b04297 |
| 2. | Rodríguez, A. D.; González, E.; Huang, S. D. J. Org. Chem. 1998, 63, 7083–7091. doi:10.1021/jo981385v |
| 7. | Rodríguez, I. I.; Rodríguez, A. D.; Zhao, H. J. Org. Chem. 2009, 74, 7581–7584. doi:10.1021/jo901578r |
| 37. | Hog, D. T.; Huber, F. M. E.; Jiménez-Osés, G.; Mayer, P.; Houk, K. N.; Trauner, D. Chem. – Eur. J. 2015, 21, 13646–13665. doi:10.1002/chem.201501423 |
| 2. | Rodríguez, A. D.; González, E.; Huang, S. D. J. Org. Chem. 1998, 63, 7083–7091. doi:10.1021/jo981385v |
| 3. | Rodríguez, A. D.; Ramírez, C. Org. Lett. 2000, 2, 507–510. doi:10.1021/ol991362i |
| 4. | Rodríguez, A. D.; Ramírez, C.; Rodríguez, I. I.; Barnes, C. L. J. Org. Chem. 2000, 65, 1390–1398. doi:10.1021/jo9914869 |
| 5. | Rodríguez, A. D.; Ramírez, C.; Shi, Y.-P. J. Org. Chem. 2000, 65, 6682–6687. doi:10.1021/jo000875w |
| 6. | Wei, X.; Rodríguez, I. I.; Rodríguez, A. D.; Barnes, C. L. J. Org. Chem. 2007, 72, 7386–7389. doi:10.1021/jo070649n |
| 38. | Jigajinni, V. B.; Wightman, R. H. Tetrahedron Lett. 1982, 23, 117–120. doi:10.1016/s0040-4039(00)97549-x |
| 34. | Wang, Y.; Zarca, M.; Gong, L.-Z.; Zhang, L. J. Am. Chem. Soc. 2016, 138, 7516–7519. doi:10.1021/jacs.6b04297 |
| 35. | Cantagrel, G.; Meyer, C.; Cossy, J. Synlett 2007, 2983–2986. doi:10.1055/s-2007-992366 |
| 33. | Hoffmann, M.; Weibel, J.-M.; de Frémont, P.; Pale, P.; Blanc, A. Org. Lett. 2014, 16, 908–911. doi:10.1021/ol403663j |
| 36. | Mukai, C.; Kozaka, T.; Suzuki, Y.; Kim, I. J. Tetrahedron 2004, 60, 2497–2507. doi:10.1016/j.tet.2004.01.041 |
| 32. | Amberg, W. M.; Carreira, E. M. J. Am. Chem. Soc. 2022, 144, 15475–15479. doi:10.1021/jacs.2c07150 |
| 30. | Srikrishna, A.; Neetu, G. Tetrahedron 2011, 67, 7581–7585. doi:10.1016/j.tet.2011.07.054 |
| 31. | Kobayashi, T.; Tokumoto, K.; Tsuchitani, Y.; Abe, H.; Ito, H. Tetrahedron 2015, 71, 5918–5924. doi:10.1016/j.tet.2015.05.089 |
| 35. | Cantagrel, G.; Meyer, C.; Cossy, J. Synlett 2007, 2983–2986. doi:10.1055/s-2007-992366 |
© 2022 Shi et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.