Abstract
The Pauson–Khand reaction (PKR) is one of the key methods for the construction of cyclopentenone derivatives, which can in turn undergo diverse chemical transformations to yield more complex biologically active molecules. Despite the increasing availability of fluorinated building blocks and methodologies to incorporate fluorine in compounds with biological interest, there have been few significant advances focused on the fluoro-Pauson–Khand reaction, both in the inter- and intramolecular versions. Furthermore, the use of vinyl fluorides as olefinic counterparts had been completely overlooked. In this review, we collect the advances both on the stoichiometric and catalytic intermolecular and intramolecular fluoro-Pauson–Khand reaction, with special attention to the PKR of enynes containing a fluoride moiety.
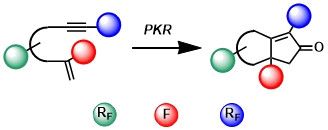
Graphical Abstract
Introduction
The prevalence of fluorine-containing molecules in drug-discovery programs is nowadays unquestionable [1-3]. The presence of fluorine atoms or fluorine-containing units at strategic positions in a drug candidate may result in not only an increase in its potency, but also, perhaps more importantly, bring about an enhanced pharmacokinetic profile resulting in a more “drug-like” molecule [4]. Subtle changes in physicochemical properties such as acidity/basicity, lipophilicity, preferred conformation or hydrogen bond forming ability, among others, may result in a dramatic effect in the therapeutic potential of a drug candidate [5,6]. All these properties may be fine-tuned by the selective incorporation of fluorine into the structure of the molecule [7]. In addition to therapeutic use, fluorine-containing molecules have also found key applications in the field of medical diagnosis. Two of the most powerful imaging techniques used nowadays, positron-emission tomography (PET) and magnetic resonance imaging (MRI), are routinely carried out using fluorine-containing organic compounds [8,9]. The former takes advantage of the superb properties of the 18F-radioisotope as positron emitter (t1/2 = 109 min, 97% β+ emission), while the latter benefits from the excellent performance of the 19F isotope in NMR (100% natural abundance, high sensitivity, lack of endogenous background signal). In addition, the use of fluorinated organic compounds in other key industrial fields is also of paramount importance. The agrochemical and materials industries, together with the aforementioned pharmaceutical industry, are perhaps the fields where organofluorinated compounds have exerted the most profound influence [10-12].
Regarding the development of new synthetic methodologies for the preparation of such molecules, these can be divided in two main categories: fluorination reactions [13-16] and the use of fluorinated building blocks [17]. The difference between them is that while in the former a fluorine atom or a fluorinated unit (e.g., a CF3 group) is introduced into a non-fluorinated molecule, the latter takes advantage of a substrate that already contains fluorine in order to achieve more complex fluorinated structures. According to these definitions, the chemistry that will be discussed in this review belongs to the fluorinated building blocks category. More specifically, the participation of fluorine-containing olefins and alkynes in the Pauson–Khand cyclization, with a special focus on the intramolecular version using fluorinated enynes, which will be discussed in detail.
The focus of this review is to highlight the efforts made in the field of the Pauson–Khand reaction with fluorinated compounds for the preparation of bicyclic derivatives.
Review
The Pauson–Khand reaction
The Pauson–Khand reaction (PKR) formally consists of a [2 + 2 + 1]-cycloaddition between an alkyne, an olefin and carbon monoxide, resulting in the regioselective formation of a cyclopentenone derivative (Scheme 1) [18-22]. This cobalt-mediated reaction was initially discovered by Pauson and Khand in the early 70s [23-25] and has since become a powerful transformation widely used in the synthesis of polycyclic complex molecules. The intermolecular variant shows a wide alkyne scope, but in terms of the olefin counterpart is limited to the use of ethylene or strained alkenes, such as norbornene and norbornadiene. The high prevalence of five-membered ring systems in natural products, pharmaceuticals and other added-value compounds accounts for the great applicability that this reaction has found [26-32]. Despite the increasing demand of fluorinated compounds and the impressive development of the PKR, the combination of these two fields has been understudied, making it an exciting field of research.
![[1860-5397-16-138-i1]](/bjoc/content/inline/1860-5397-16-138-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Schematic representation of the Pauson–Khand reaction.
Scheme 1: Schematic representation of the Pauson–Khand reaction.
In order to study the influence on the reaction outcome, a fluorine atom or fluorine-containing group can be installed at either unsaturated counterpart, bound to either the olefin and/or the alkyne (vide infra) (Scheme 2). Of course, in the intramolecular version, the fluorine atom or fluorinated group can also form a part of the linker. The reaction yields are dependent on the degree of substitution, bulkiness, and electronic effects of the substituents of both the alkyne and alkene moieties. In general, electron-deficient alkynes are poor substrates for the PKR as they are deactivated in the cobalt-complexation step, and the highest yields are usually obtained with terminal alkynes. The scenario is similar in the case of fluorinated substrates, with the intramolecular version being much more developed than the intermolecular one.
![[1860-5397-16-138-i2]](/bjoc/content/inline/1860-5397-16-138-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Substrates included in this review.
Scheme 2: Substrates included in this review.
The regio- and stereochemistry of this transformation is predictable, in most cases. Since this is an important issue throughout the review, a concise remark about the mechanism of this transformation is outlined below (Scheme 3). This mechanism was proposed by Magnus over 30 years ago and is still valid nowadays [33], although only the first intermediate (I) has been isolated and characterized [34]. However, contributions by Nakamura, Pericàs, and others support the initial proposal both experimentally and theoretically [35,36]. Firstly, two coordination vacancies are freed after the extrusion of two carbon monoxide ligands from the starting cobalt species, allowing the alkyne group to bind to the cobalt metal centers. The subsequent coordination of the olefin counterpart requires the extrusion of a third carbon monoxide ligand, leading to pentacarbonyl complex II. This highly endotermic process is the rate-limiting step and long reaction times are generally associated to this. However, the reaction can be accelerated in conditions that facilitate the dissociation of CO ligands such as heating, microwave irradiation [37,38], visible light, or ultrasonication [39]. Alternatively, mild oxidizing additives such as amine oxides, aminophosphines, phosphine oxides, and sulfoxides may be used as promoters to facilitate the dissociation step, by oxidatively removing one of the CO ligands in form of CO2 [40]. The most common oxidants are N-morpholine N-oxide (NMO), trimethylamine N-oxide (TMANO), and dimethyl sulfoxide (DMSO). Once a new coordination vacancy has been opened on one of the cobalt centers, coordination of the olefin sets the stage for the subsequent C–C bond forming steps. The olefin is inserted into the less hindered Co–C bond, determining both the regio- and stereochemical outcome of the overall process. A carbon monoxide ligand then undergoes migratory insertion into one of the Co–C bonds in cobaltacycle V, followed by reductive elimination to release the final product (Scheme 3).
![[1860-5397-16-138-i3]](/bjoc/content/inline/1860-5397-16-138-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Commonly accepted mechanism for the Pauson–Khand reaction.
Scheme 3: Commonly accepted mechanism for the Pauson–Khand reaction.
As mentioned above, the regiochemistry of this transformation is, in most cases, predictable for unsymmetrical alkynes (things are more complex regarding the olefin). Generally, the most sterically encumbered substituent of the alkyne occupies the proximal position in the enone system. This is dictated by the migratory insertion of the olefin into the most accessible Co–C bond (Scheme 3). This trend is strictly followed by terminal alkynes, for which exclusive formation of the α-substituted enone is observed, while mixtures are usually obtained with internal dissymmetric alkynes, although the major product follows the aforementioned trend [41]. On the other hand, for alkynes bearing groups of comparable steric demand at both ends of the alkyne, electronic effects come into play. Here, the more electron-rich substituent occupies the α position, and the more electron-poor the β position (Scheme 4). This selectivity could be rationalized by the insertion of the olefin into the weakest Co–C bond. These electronic effects have been shown to be less important than steric ones, and are often overcome by the latter. Regarding the stereochemistry, exo-products are almost exclusively obtained for norbornene and norbornadiene.
![[1860-5397-16-138-i4]](/bjoc/content/inline/1860-5397-16-138-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Many deviations from the classic reaction conditions have been described, including the use of metals other than cobalt (such as rhodium, iridium, titanium, ruthenium, nickel, and palladium), or the use of CO surrogates such as aldehydes, alcohols and formates. Recently, its utility in flow chemistry has also been described [42].
Intramolecular Pauson–Khand reactions of fluorine-containing compounds
The utility of the Pauson–Khand reaction in the preparation of polycyclic compounds bearing both nitrogenated and cyclopentenone rings, two ubiquitous domains in drugs and natural products, has been reported in various contributions using 1,n-enynes, particularly 4-aza-1,7-enynes as starting materials [43-45]. However, the synthesis of fluorinated 1,n-enynes as well as the corresponding Pauson–Khand adducts has, until recently, scarcely been described in the literature. The intramolecular version of this reaction has recently gained recognition since it facilitates the synthesis of cyclopentenone-fused ring systems, which tend to be difficult to construct. The Pauson–Khand reaction has also been used as a key step in the synthesis of a number of biologically-relevant compounds, including fluorine-containing piperidine-fused cycles. Of course, where the fluorinated group is positioned in the final compound depends on whether it is attached to the alkene or alkyne counterpart of the substrate (Scheme 5).
![[1860-5397-16-138-i5]](/bjoc/content/inline/1860-5397-16-138-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Variability at the acetylenic and olefinic counterpart.
Scheme 5: Variability at the acetylenic and olefinic counterpart.
Regarding the intramolecular PKR of fluorinated enynes, only a few examples have been described. The first example was reported in 2001 by Ishizaki and co-workers [46]. In this study, a wide variety of 1,6-enynes bearing fluorine atoms or fluorine-containing groups at the alkenyl or alkynyl positions were synthesized and evaluated as substrates in the intramolecular PKR with dicobalt octacarbonyl [Co2(CO)8] in CH2Cl2 and promoted by NMO. In general, the presence of fluorinated groups on the alkenyl moiety of the 1,6-enyne resulted in low yields (lower than 35%) of the corresponding cyclized products, due to the poor reactivity of the fluorinated olefin (Scheme 6). For example, difluoroalkene-containing compound 1 decomposed and no cyclized product was formed. In this NMO-promoted PKR, monofluoroolefinic enyne 2 afforded the defluorinated cyclopentenone 7 in 37% yield. Similarly, trifluoromethyl-substituted olefin 3 also lost the chlorine atom upon cyclization to give 8 as a single diastereoisomer, albeit in a low 14% yield. The reaction of trifluoromethyl-substituted allylic alcohols 4 and 5 afforded the corresponding cyclized products 9 and 10 (31% and 34% yield, respectively) as inseparable mixtures of diastereoisomers. Finally, 1,6-enyne 6, bearing a 4-fluorophenyl group on the olefin, stereoselectively produced trans-oriented arylcyclopentenone 11 in 23% yield.
![[1860-5397-16-138-i6]](/bjoc/content/inline/1860-5397-16-138-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Pauson–Khand reaction of fluoroolefinic enynes reported by the group of Ishizaki [46].
Scheme 6: Pauson–Khand reaction of fluoroolefinic enynes reported by the group of Ishizaki [46].
When investigating the intramolecular PKR of enynes bearing fluorine groups on alkynyl moiety (12, 14, 16), several trends could be observed (Scheme 7). Firstly, fluoroaromatic enynes 12a–c afforded the corresponding cyclized products 13a–c in low yields (14–42%). However, no cyclized product was observed when using the trifluoromethyl ketone derivative 12d. Secondly, PKR with enynes 14 containing fluorinated propargyl alcohol groups yielded diastereoisomeric mixtures of pyrrolidine ring-fused cyclopentenones 15 in good yields (67–85%) but low diastereoselectivities. Finally, the reaction of dimethyl malonate-derived fluoroaromatic enynes 16 afforded the corresponding cyclopentenone products 17 in higher yields (85–92%). Thus, the reaction of enynes bearing fluorinated groups attached to the alkyne moiety was found to afford the corresponding cyclized products in moderate to high yields, except for those bearing marked electron withdrawing groups (Scheme 7).
![[1860-5397-16-138-i7]](/bjoc/content/inline/1860-5397-16-138-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: PKR of enynes bearing fluorinated groups on the alkynyl moiety, reported by the group of Ishizaki [46]. Reaction conditions: i) Co(CO)8 (1.1 equiv), toluene, rt, 1 h; ii) NMO (6–12 equiv), rt, 1 h.
Scheme 7: PKR of enynes bearing fluorinated groups on the alkynyl moiety, reported by the group of Ishizaki [46]....
In 2005, Billard and co-workers reported the PKR of α-trifluoromethylated homoallylamine derivatives (Scheme 8) [47]. Both 1,7-enynes 18a and 18b (n = 1) underwent PKR in the presence of 1 equiv of Co2(CO)8 and 10 equiv of NMO, yielding bicyclic derivatives 19 in moderate yields and high diastereoselectivity (de > 95%). The observed diastereoselectivity was rationalized considering two transition states of the PKR, and assuming the CF3 group occupies an axial position due to the steric and electrostatic repulsions that occur in the equatorial position. Consequently, in the most favorable transition state there is no steric hindrance between the CF3 group and the ethylenic hydrogen, leading to the observed diastereoisomer. On the other hand, the use of 1,9-enyne 18c (n = 3) did not afford the corresponding bicyclic compound since the double and triple bonds are too distant.
![[1860-5397-16-138-i8]](/bjoc/content/inline/1860-5397-16-138-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Intramolecular PKR of 1,7-enynes reported by the group of Billard [47].
Scheme 8: Intramolecular PKR of 1,7-enynes reported by the group of Billard [47].
In a following paper by Billard and co-workers, the PKR of oxygen-containing 1,7-enynes was assayed, affording trifluoromethylated oxygenated bicyclic enones (Scheme 9) [48]. Under classical stoichiometric conditions (reaction with Co2(CO)8 followed by the addition of NMO), and starting from the pure anti diastereoisomer of 1,7-enyne 20, the expected bicyclic enone was obtained in good yield and high diastereoselectivity (de > 95%). An attempt to extend the PKR to the formation of a fused tricyclic structure, starting from 1,7-enyne 21, was unsuccessful and no tricyclic product was formed.
![[1860-5397-16-138-i9]](/bjoc/content/inline/1860-5397-16-138-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Intramolecular PKR of 1,7-enynes reported by the group of Billard [48].
Scheme 9: Intramolecular PKR of 1,7-enynes reported by the group of Billard [48].
Bonnet-Delpon and co-workers reported the one-pot synthesis of several CF3-containing N-tethered amines in good yields (54–86% over 2 steps) [49]. These products were subjected to metathesis reactions in the presence of Grubbs catalyst affording the corresponding CF3-containing dehydropiperidine derivatives in excellent yields. Additionally, enynes 22 and 23 were evaluated as substrates in the intramolecular PKR, yielding the corresponding CF3-containing heterobicyclic derivatives in 68% (85:15 ratio of trans/cis stereoisomers) and 80% yield (18:82 ratio of trans/cis stereoisomers), respectively (Scheme 10).
![[1860-5397-16-138-i10]](/bjoc/content/inline/1860-5397-16-138-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Intramolecular PKR of 1,7-enynes by the group of Bonnet-Delpon [49]. Reaction conditions: i) Co(CO)8 (1.2 equiv), CH2Cl2, 30 min; ii) NMO (9 equiv), 0 °C then 3 h at rt.
Scheme 10: Intramolecular PKR of 1,7-enynes by the group of Bonnet-Delpon [49]. Reaction conditions: i) Co(CO)8 (1...
Ichikawa and co-workers described an attractive route to synthesize pyrrolidine ring-fused fluorinated cyclopentenone analogs via intramolecular PKR starting from 2-bromo-3,3,3-trifluoroprop-1-ene [50,51]. To this end, N-propargyl-N-[2-(trifluoromethyl)allyl]amides 24 were treated with dicobalt octacarbonyl to afford the cobalt alkyne complex, which was then heated in CH3CN. Under these conditions, trifluoromethylated cyclopentenone 25a was obtained in high yield (81%) and diastereoselectivity (anti/syn = 94:6) (Scheme 11). The cyclization of internal alkyne substrate 24b yielded pyrrolidine ring-fused cyclopentenone 25b in similar yield but lower diastereoselectivity. Finally, N-propargyl-N-[2-(trifluoromethyl)allyl] ether 24d, containing an ether linkage instead of the aforementioned sulfonamide linkage, gave furan ring-fused cyclopentenone 25d in both lower yield (53%) and lower diastereoselectivity.
![[1860-5397-16-138-i11]](/bjoc/content/inline/1860-5397-16-138-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Intramolecular PKR of 1,6-enynes reported by the group of Ichikawa [50].
Scheme 11: Intramolecular PKR of 1,6-enynes reported by the group of Ichikawa [50].
A catalytic PKR of fluorinated 1,7-enyne amides 26 using catalytic amounts of [Rh(COD)Cl]2 was reported in 2008 by Hammond and co-workers [52]. The authors concluded that the reaction was highly sensitive to experimental parameters such as solvent, concentration, temperature, catalyst and silver salt. Under standard reaction conditions, no reaction was observed in the absence of a silver salt, and the best results were obtained in the presence of AgOTf (20 mol %), giving the corresponding gem-difluorinated bicyclic lactam 27 in 43% yield (Scheme 12). This reaction was limited to unsubstituted alkynes, as the PKR did not occur with a phenyl-substituted alkyne. Unfortunately, no asymmetric induction was observed.
![[1860-5397-16-138-i12]](/bjoc/content/inline/1860-5397-16-138-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Intramolecular Rh(I)-catalyzed PKR reported by the group of Hammond [52].
Scheme 12: Intramolecular Rh(I)-catalyzed PKR reported by the group of Hammond [52].
In 2011, Osipov and co-workers investigated the cobalt-mediated PKR of allenynes 28 in order to synthesize trifluoromethylated nitrogen- and sulfur-based bicyclic compounds (Scheme 13) [53]. Using this methodology, the corresponding cyclopentenones 29 were isolated in generally good yields, except for sulfur-containing derivative 29c, due to the oxidation of the sulfide under the reaction conditions. In contrast, the phosphorus analog 29d was obtained in 45% yield, which could be explained by favorable electronic and steric effects of the phosphonate group.
![[1860-5397-16-138-i13]](/bjoc/content/inline/1860-5397-16-138-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Intramolecular PKR of allenynes reported by the group of Osipov [53].
Scheme 13: Intramolecular PKR of allenynes reported by the group of Osipov [53].
In the same work, the authors also evaluated the PKR of CF3-substituted enynes 30. In this case, bicyclic products 31 were formed as mixtures of separable diastereoisomers, which could be isolated in higher yields than the products of the corresponding reaction with allenynes (Scheme 14).
![[1860-5397-16-138-i14]](/bjoc/content/inline/1860-5397-16-138-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Intramolecular PKR of 1,7-enynes reported by the group of Osipov [53].
Scheme 14: Intramolecular PKR of 1,7-enynes reported by the group of Osipov [53].
In 2012, Konno and co-workers studied the intramolecular PKR using fluorine-containing 1,6-enynes 32 (Scheme 15) [54]. The PKR of fluorinated propargyl allyl ether 32a afforded the corresponding cis bicyclic product 33a in moderate yield and high diastereoselectivity (dr > 20:1). Other fluorinated allyl propargyl ethers 32b–d afforded the corresponding bicyclic PK-adducts 33 in moderate chemical yields but with high cis-selectivity. On the other hand, spirocyclic derivative 33e was formed in a significantly lower yield. Surprisingly, a subtle change of the fluoroalkyl group from a CF3 group to a CHF2 group completely inhibited the reaction.
![[1860-5397-16-138-i15]](/bjoc/content/inline/1860-5397-16-138-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Intramolecular PKR of fluorine-containing 1,6-enynes reported by the Konno group [54].
Scheme 15: Intramolecular PKR of fluorine-containing 1,6-enynes reported by the Konno group [54].
Within the frame of a broader study, our group reported a single example of an intramolecular PKR using an Ellman’s imine-derived CF3-containing enyne, bearing the trifluoromethylethynyl group at the ortho position (Scheme 16) [55]. One of the key steps in the preparation of the starting 1,n-enynes was a highly diastereoselective allylation reaction of chiral Ellman’s sulfinylimines. Based on this strategy, chiral 1,7-enynes 34 were prepared in three steps from sulfinylimines derived of o-iodobenzaldehydes. A variety of fluorinated compounds bearing fluorine or fluoroalkyl groups attached to the aryl moieties were efficiently prepared (Scheme 15). In this report, the suitability of enynes bearing CF3-substituted alkyne moieties (R = CF3) to participate in intramolecular PK reactions was also demonstrated. Furthermore, several other substrates bearing fluorine at different positions were included (Scheme 16). The process took place with moderate to high chemical yields and diastereoselectivities. This transformation can be performed on a multigram scale, and is characterized by a broad substrate scope, functional group compatibility, and high chemo- and diastereoselectivity.
![[1860-5397-16-138-i16]](/bjoc/content/inline/1860-5397-16-138-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: Diastereoselective PKR with enantioenriched fluorinated enynes 34 [55].
Scheme 16: Diastereoselective PKR with enantioenriched fluorinated enynes 34 [55].
Martínez-Solorio and co-workers reported an intramolecular PKR of Si−O tethered 1,7-enynes 35, affording cyclopentaoxasilinones 36 with high diastereoselectivity [56]. In contrast to previous silicon-based tethers, which reacted in low yields and resulted in unexpected byproducts, this transformation could be performed on a multigram scale and showed a wide substrate scope and functional group compatibility, as well as high diastereoselectivity [57,58]. In this work, Si−O tethered 1,7-enynes underwent the PKR after treatment with 1.05 equiv of Co2(CO)8 using 4-fluorobenzyl(methyl)sulfide (4-FBnSMe) as an additive, which is commercially available and can be easily recovered by flash chromatography. Under the aforementioned conditions, cyclopentaoxasilinone 36a was isolated in 81% yield. A systematic study of the scope showed that unsubstituted enyne 35b only afforded the desired product in 25% yield. In contrast, isopropyl and phenyl substituted enynes yielded cyclopentaoxasilinone 36c,d in 74 and 79% yield, respectively. Furthermore, electron-withdrawing para-substituted arenes were obtained in good yields and demonstrate excellent functional group compatibility; MeO– (36e, 65%), −CN (36f, 72%), −CO2Me (36g, 76%) and fluorinated groups such as p-CF3C6H4 (36h, 77%) (Scheme 17).
![[1860-5397-16-138-i17]](/bjoc/content/inline/1860-5397-16-138-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Intramolecular PKR reported by the group of Martinez-Solorio [56].
Scheme 17: Intramolecular PKR reported by the group of Martinez-Solorio [56].
Concerning the intramolecular PKR with fluorine atoms or fluorinated groups at the vinylic position, very few examples have been described to date. In this context, our group recently explored the reactivity of 1,n-enynes bearing a vinyl fluoride moiety as the olefin counterpart in the intramolecular PKR [59] (Scheme 18).
![[1860-5397-16-138-i18]](/bjoc/content/inline/1860-5397-16-138-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Fluorine substitution at the olefinic counterpart.
Scheme 18: Fluorine substitution at the olefinic counterpart.
The study of the behavior of this kind of compounds in the PK reaction started with fluorinated enynes derived from malonates and those containing heteroatoms as linkers. The synthesis of the starting enynes 37 was accomplished following various approaches (Scheme 19).
![[1860-5397-16-138-i19]](/bjoc/content/inline/1860-5397-16-138-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: Synthesis of fluorinated enynes 37 [59].
Scheme 19: Synthesis of fluorinated enynes 37 [59].
The first of these (via a) was based on a report by Hammond and co-workers, in which they detailed the Markovnikov hydrofluorination of alkynes using HF.DMPU coupled with a gold catalyst [60]. Accordingly, the appropriate propargylmalonate derivatives were fluorinated to give fluoroalkene intermediates, which were then converted into malonate-based enynes 37 (Z = CO2R) through a simple propargylation procedure. In addition, the fluoroallyl alcohol 38a was employed as a starting material to obtain the corresponding propargyl ethers 37 (Z = O) by Williamson’s synthesis using propargyl bromides in moderate yields (via b). Activation of the fluoroallyl alcohol by conversion into the corresponding mesylate 38b proved sufficient for the synthesis of N-tethered substrates 37 (Z = NTs), through simple nucleophilic substitution (via c).
It is noteworthy that under standard PK reaction conditions, fluoroenyne 39 evolves to diene 40. The formation of compound 40 is possible by elimination of HF from the PK product 41, meaning that the fluoro-PKR does initially take place and that the desired product 41 could be isolated by avoiding the subsequent elimination reaction. This hypothesis was confirmed when the less basic DMSO was used as the promoter instead of NMO, allowing the successful isolation of 41 (Scheme 20) [59].
![[1860-5397-16-138-i20]](/bjoc/content/inline/1860-5397-16-138-i20.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 20: Fluorine-containing substrates in PKR [59].
Scheme 20: Fluorine-containing substrates in PKR [59].
Under the optimized conditions, using stoichiometric Co2(CO)8 and DMSO as the promoter, the process worked well and moderate to good yields were obtained for derivatives 42 bearing aryl substituents at the propargyl moiety, regardless of their electronic nature (Scheme 21). Alkyl-substituted derivatives proved to be similarly successful substrates; however, the TMS-substituted derivative was obtained in a significantly lower yield. In terms of linkers, heteroatoms were well-tolerated, although the use of malononitrile resulted exclusively in the elimination product despite testing a variety of reaction conditions. More complex biorelevant examples such as isatin derivatives were also suitable substrates, affording the corresponding spirocyclic derivatives, albeit in low yields (Scheme 21) [59]. Unfortunately, thioether and sulfone-based linkers were unsuitable in this reaction, and the starting materials were recovered in all cases.
![[1860-5397-16-138-i21]](/bjoc/content/inline/1860-5397-16-138-i21.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 21: Pauson Khand reaction for fluorinated enynes by the Fustero group: scope and limitations [59].
Scheme 21: Pauson Khand reaction for fluorinated enynes by the Fustero group: scope and limitations [59].
As a comparison, the same authors also explored the reactivity of the corresponding chloro- and bromoenynes 43 as olefinic counterparts for the intramolecular PKR (Scheme 22) [59].
![[1860-5397-16-138-i22]](/bjoc/content/inline/1860-5397-16-138-i22.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 22: Synthesis of chloro and bromo analogues [59].
Scheme 22: Synthesis of chloro and bromo analogues [59].
In these cases, the corresponding halogenated PKR adducts 44 could not be detected in the crude reaction mixtures. Instead, the major isolated species was dimer 45. However, the fact that 45 bears the cyclopentenone core suggests that the desired PKR does indeed take place, albeit as an intermediate before a secondary transformation to form the final dimer (Scheme 23) [59]. The authors rationalized the formation of 45 by considering that the inherent weakening of the C—X bond going down the halogen series may favor the generation of radical A with chloride and bromide derivatives, especially given the tertiary position of the halide and the stoichiometric quantities of cobalt present in the reaction mixture.
![[1860-5397-16-138-i23]](/bjoc/content/inline/1860-5397-16-138-i23.svg?scale=2.0&max-width=1024&background=FFFFFF)
The same group also studied the intramolecular PKR of chiral fluorine-containing N-tethered 1,7-enynes 48 for the stereoselective construction of enantioenriched bicyclic alkaloid analogues 49, containing a fluorine atom in the bridge position [61]. For this purpose, 1,7-enynes 48 were prepared from Ellman’s tert-butane sulfinylimines, followed by a diastereoselective addition of propargylmagnesium bromide to obtain a variety of sulfinyl amide intermediates 46 in good yields and high diastereoselectivities. Subsequent oxidation to the corresponding sulfonamides 47, followed by the introduction of the fluoroallyl group via N-alkylation with previously described mesylate 38 provided fluorinated enynes 48 (Scheme 24).
![[1860-5397-16-138-i24]](/bjoc/content/inline/1860-5397-16-138-i24.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 24: Synthesis of fluorine-containing N-tethered 1,7-enynes [61].
Scheme 24: Synthesis of fluorine-containing N-tethered 1,7-enynes [61].
The enantioenriched starting materials 48 were then evaluated in the Co-mediated PKR to yield the bicyclic products 49 in moderate to good yields and excellent diastereoselectivities (dr > 20:1) (Scheme 25). The substrate scope revealed that the reaction was tolerant of a wide range of substituents at the stereogenic center, such as heteroaromatic, aromatic, and aliphatic substituents (both linear and cyclic). Regarding aromatic substituents, electron-neutral and electron-rich rings with several substitution patterns performed well. However, pyridine-based 48j resulted in a low yield. Noteworthy, the PKR of chiral enynes 48 led to a bridgehead quaternary stereocenter containing a C–F bond in a single step. Besides the intrinsic difficulty in generating quaternary stereocenters, the goal achieved is even more significant given the attention that the asymmetric introduction of fluorine at sp3 carbon centers has received in recent years [62,63]. A gram-scale synthesis was also successfully performed in five steps starting from the corresponding aldehyde in a 41% global yield.
![[1860-5397-16-138-i25]](/bjoc/content/inline/1860-5397-16-138-i25.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 25: Intramolecular PKR of chiral N-tethered fluorinated 1,7-enynes [61].
Scheme 25: Intramolecular PKR of chiral N-tethered fluorinated 1,7-enynes [61].
Bicyclic product 49a underwent diastereoselective (dr > 20:1) hydrogenation using palladium over activated charcoal under an atmosphere of hydrogen to afford saturated derivative 50 (Scheme 26). On the other hand, the tert-butanesulfonyl group could be removed through treatment of 49a with trifluoromethanesulfonic acid in the presence of anisole to form 51.
![[1860-5397-16-138-i26]](/bjoc/content/inline/1860-5397-16-138-i26.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 26: Examples of further modifications to the Pauson−Khand adducts [61].
Scheme 26: Examples of further modifications to the Pauson−Khand adducts [61].
In a recent report, Fustero and co-workers synthesized a series of N-tethered 1,7-enynes 53 bearing fluorinated substituents starting from fluorinated tert-butanesulfinyl imines 52 (Scheme 27), which were later evaluated in the cobalt-mediated PKR [64].
![[1860-5397-16-138-i27]](/bjoc/content/inline/1860-5397-16-138-i27.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 27: Asymmetric synthesis the fluorinated enynes 53.
Scheme 27: Asymmetric synthesis the fluorinated enynes 53.
The chiral enynes were treated with a stoichiometric amount of Co2(CO)8 in CH2Cl2 affording the corresponding cobalt complexes that, upon addition of an excess of NMO, underwent an efficient intramolecular PKR to afford the corresponding bicyclic cyclopentenones 54 as single diastereoisomers (Scheme 28). In general, yields were moderate to good, and high diastereoselectivities were observed in almost all cases.
![[1860-5397-16-138-i28]](/bjoc/content/inline/1860-5397-16-138-i28.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 28: Intramolecular PKR of chiral N-tethered 1,7-enynes 53 [64].
Scheme 28: Intramolecular PKR of chiral N-tethered 1,7-enynes 53 [64].
The PKR tolerated both mono- and disubstituted olefins well, but when a trisubstituted olefin was assayed in the PKR, the initial enyne was recovered, in agreement with previous precedents describing that trisubstituted alkenes are very unreactive substrates in this kind of process (Scheme 28) [65]. The influence of the introduction a vinyl fluoride moiety on the PKR was also investigated (Scheme 29). The resulting cyclopentenones 57 were obtained in a lower yield (52%) but high diastereoselectivity, similar to those previously reported [59,61].
![[1860-5397-16-138-i29]](/bjoc/content/inline/1860-5397-16-138-i29.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 29: Intramolecular PKR of chiral N-tethered 1,7-enyne bearing a vinyl fluoride [64].
Scheme 29: Intramolecular PKR of chiral N-tethered 1,7-enyne bearing a vinyl fluoride [64].
The authors also explored the PKR in a catalytic version based on a biphasic system of ethylene glycol/toluene, which generally enhanced both yields and stereoselectivities, as well as simplifying purification of the products [66]. This methodology (7 mol % catalyst, atmospheric CO pressure, and 15% v/v of ethylene glycol in toluene) was shown to be suitable for substrates bearing differing fluorinated groups, as well as one example with a phenyl-substituted alkyne, giving rise to products 54 in good yields and diastereoselectivities (Scheme 30).
![[1860-5397-16-138-i30]](/bjoc/content/inline/1860-5397-16-138-i30.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 30: Catalytic intramolecular PKR of chiral N-tethered 1,7-enynes [64].
Scheme 30: Catalytic intramolecular PKR of chiral N-tethered 1,7-enynes [64].
Intermolecular Pauson–Khand reactions of fluorine-containing compounds
In contrast to the previously discussed intramolecular PKR of fluorinated enynes, the study of intermolecular PKR reactions is far scarcer, and is limited by the poor reactivity and selectivity of simple alkenes. In this regard, most examples have been restricted to the use of ethylene or strained alkenes such as cyclopropene, norbornene, norbornadiene, (E)-cyclooctene, or bicyclo[3.2.0]hept-6-ene [67-69]. The first example of an intermolecular version of fluorinated compounds was reported by Riera, Fustero and co-workers in 2010 [70]. In this seminal study, four model fluorinated alkyne precursors 58 were synthesized (Scheme 31).
![[1860-5397-16-138-i31]](/bjoc/content/inline/1860-5397-16-138-i31.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 31: Model fluorinated alkynes used by Riera and Fustero [70].
Scheme 31: Model fluorinated alkynes used by Riera and Fustero [70].
With this small family of fluorinated alkynes, the authors studied the PKR using norbonadiene as the olefin partner under thermal conditions using a stoichiometric amount of Co2(CO)8. Products 59 were obtained in moderate to excellent yields, as single regioisomers, and 59c as a 1:1 mixture of diastereoisomers. The most striking feature was the unexpected regiochemical outcome of this study; the fluorinated moiety occupied the α-position in the final cyclopentenone ring in all cases (Scheme 32). This was expected for terminal alkyne 58a, since this is the substitution pattern always found (see Scheme 4). On the other hand, for alkynes bearing substituents of similar steric bulk, the electron withdrawing group is expected to occupy the β-position. However, for alkynes 58b,c the opposite regiochemistry was found. Finally, the use of alkyne 58d with two electron-withdrawing groups resulted in the regioselective formation of product 59d in excellent yield, indicating an inherent trend of the fluoroalkyl group to occupy the α-position regardless of the steric or electronic nature of the other substituent. These results contrast with those obtained for non-fluorinated analogue (ethyl 2-butynoate) for which the expected regioisomer 60 was formed, bearing the methyl group at the α-position and the electron-withdrawing ester group in the β-position. These results may suggest that fluoroalkyl groups behave as bulky substituents rather than as electron-withdrawing ones, perhaps due to the purely inductive nature of the latter [71].
![[1860-5397-16-138-i32]](/bjoc/content/inline/1860-5397-16-138-i32.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 32: PKR with norbornadiene and fluorinated alkynes 58 [71].
Scheme 32: PKR with norbornadiene and fluorinated alkynes 58 [71].
In the same work, the authors described the conjugated addition of several nucleophiles to model substrate 59d (Scheme 33). In this sense, while the addition of hard organometallic nucleophiles, such as lithium dialkylcuprates or Grignard reagents, failed, softer nucleophiles such as nitroalkanes cleanly added to the β-position, providing the Michael adduct 61. Unexpectedly, the conjugate addition reaction resulted in concomitant detrifluoromethylation. Furthermore, the Lewis acid-mediated retro Diels–Alder reaction was carried out uneventfully on product 61, affording the corresponding cyclopentenone 62 in moderate yield (Scheme 33).
![[1860-5397-16-138-i33]](/bjoc/content/inline/1860-5397-16-138-i33.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 33: Nucleophilic addition/detrifluoromethylation and retro Diels-Alder reactions [70].
Scheme 33: Nucleophilic addition/detrifluoromethylation and retro Diels-Alder reactions [70].
In order to rationalize the unexpected loss of the trifluoromethyl group upon nucleophilic addition, the authors suggested the tentative mechanism depicted in Scheme 34. As shown, γ-fluoride loss from enolate I, formed after the conjugate Michael addition, would lead to the formation of difluoroenone II. This could in turn undergo a nucleophilic addition of water, followed by a retro-aldol reaction affording the final product (Scheme 34). This mechanism was experimentally supported by the beneficial effect of water and the observation of HF-loss by 19F NMR.
![[1860-5397-16-138-i34]](/bjoc/content/inline/1860-5397-16-138-i34.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 34: Tentative mechanism for the nucleophilic addition/retro-aldol reaction sequence.
Scheme 34: Tentative mechanism for the nucleophilic addition/retro-aldol reaction sequence.
The catalytic version of this process was also studied. The best results in terms of efficiency and stereoselectivity were obtained using the less reactive triphenylphosphine dicobaltpentacarbonyl complex 63 as the catalyst (Scheme 35).
![[1860-5397-16-138-i35]](/bjoc/content/inline/1860-5397-16-138-i35.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 35: Catalytic PKR with norbornadiene [70].
Scheme 35: Catalytic PKR with norbornadiene [70].
In a later study [72], Riera and Fustero generalized the use of trifluoromethylalkynes as substrates for the PKR. The copper-catalyzed trifluoromethylation of terminal alkynes described by Qing and co-workers [73] allowed the efficient preparation of a small library of substrates bearing aryl, alkyl, and alkenyl substitutents. These were isolated after complexation to Co2(CO)8 as the corresponding adducts 64, due to difficulties in their isolation. Subsequent heating with norbornadiene afforded the corresponding products 59 in good to excellent yields (Scheme 36).
![[1860-5397-16-138-i36]](/bjoc/content/inline/1860-5397-16-138-i36.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 36: Scope of the PKR of trifluoromethylalkynes with norbornadiene [72].
Scheme 36: Scope of the PKR of trifluoromethylalkynes with norbornadiene [72].
The authors then studied the elimination of the trifluoromethyl group from this library of PK adducts, building upon their own experience in the field (vide supra, Scheme 34). Thus, by subjecting enones 59 to treatment with DBU in wet nitromethane under reflux, clean conjugate addition/detrifluoromethylation was observed, in this case followed by retro-Michael reaction of nitromethane achieving enones 65 in moderate to good yields (Scheme 37). Interestingly, the overall reaction sequence results in the formal inversion of the regiochemistry for terminal alkynes, affording the products substituted at the β-position. This regiochemistry switch might be regarded as the Holy Grail in Pauson–Khand chemistry.
![[1860-5397-16-138-i37]](/bjoc/content/inline/1860-5397-16-138-i37.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 37: DBU-mediated detrifluoromethylation [72].
Scheme 37: DBU-mediated detrifluoromethylation [72].
The synthetic potential of this methodology was demonstrated by the formal total synthesis of α-cuparenone [74,75], a bicyclic sesquiterpene that belongs to the cuparene family isolated from Thuja orientalis [76]. More specifically, the authors designed a simple route to enone 67, a key intermediate in several previous total syntheses [77,78]. Its synthesis was achieved in two steps, namely a nickel-catalyzed conjugated addition of trimethylaluminum to form 66, followed by Lewis acid-mediated retro Diels–Alder reaction (Scheme 38).
![[1860-5397-16-138-i38]](/bjoc/content/inline/1860-5397-16-138-i38.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 38: A simple route to enone 67, a common intermediate in the total synthesis of α-cuparenone.
Scheme 38: A simple route to enone 67, a common intermediate in the total synthesis of α-cuparenone.
In another study by Riera and co-workers, they studied the influence of the olefin counterpart on the regioselectivity of the reaction [79]. Two olefins other than norbornadiene were used in this study, namely norbornene and ethylene (Scheme 39). Contrary to the results observed with norbonadiene and ethylene, which both furnished a single regioisomer, the use of norbornene afforded mixtures of regioisomers, although the same α-CF3 isomer was favored in all cases. The aforementioned DBU-mediated detrifluoromethylation (Scheme 37) was achieved for substrates 68 in most cases with low to moderate yields, while the corresponding products could not be isolated starting from substrates 69.
![[1860-5397-16-138-i39]](/bjoc/content/inline/1860-5397-16-138-i39.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 39: Effect of the olefin partner in the regioselectivity of the PKR with trifluoromethyl alkynes [79].
Scheme 39: Effect of the olefin partner in the regioselectivity of the PKR with trifluoromethyl alkynes [79].
In 2012, Konno and co-workers reported the synthesis of 2-fluoralkyl-2-cyclopentenones through an intermolecular Co-mediated PKR of various trifluoromethyl alkynes with 2-norbornene (Scheme 40) [54]. In this process, the authors did not use NMO as a promoter due to its negative effect on the reaction yield. Under the described conditions, the cyclized products 70 were obtained as regioisomeric mixtures in moderate to high yields. The influence of alkyne substitution was studied, and alkynes bearing an electron-withdrawing or electron-donating group on the benzene ring afforded the corresponding cyclopentenones in high yields but as regioisomeric mixtures (ratio A/B ca. 70:30). Interestingly, a notable improvement of the regioselectivity was observed for trifluoromethyl alkynes bearing an alkyl substituent (R = Alk) or an ethoxycarbonyl group (R= CO2Et). When a difluoromethylated alkyne (RF = CF2H) was used, the reaction took place very smoothly to preferentially afford the cyclopentenone with opposite regioselectivity (ratio A/B 31:69) to that observed for the other examples. This result contrasts with the lack of reactivity observed for CF2H-substituted enynes in the intramolecular version (see Scheme 15). Although the PKR was also investigated for other alkenes such as cyclohexene, maleic anhydride, ethylene carbonate, and 1-octene, the desired products were not formed under the reaction conditions.
![[1860-5397-16-138-i40]](/bjoc/content/inline/1860-5397-16-138-i40.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 40: Intermolecular PKR of trifluoromethylalkynes with 2-norbornene reported by the group of Konno [54].
Scheme 40: Intermolecular PKR of trifluoromethylalkynes with 2-norbornene reported by the group of Konno [54].
In the same year, Helaja and co-workers examined the electronic effects of the alkyne substituent on the regioselectivity of the microwave-assisted PKR with norbornene [80]. The electronic effects were evaluated by altering one functional group in the para-position of the starting diarylalkynes (Scheme 41). In this regard, electron-donating substituents such as, methoxy, dimethylamine, and methyl favored the α-position in the final cycloadduct (71A), whereas electron-withdrawing substituents such as dimethylaminium, trifluoromethyl, and acetyl favored the β-regioisomer (71B). The 4-fluorine substituted diarylalkynes had a very weak EWG effect yielding an equimolar mixture of both regioisomers. The experimental results were confirmed by a DFT study of the NBO charges of the α-alkyne carbons, which also showed a correlation with the regioselectivity.
![[1860-5397-16-138-i41]](/bjoc/content/inline/1860-5397-16-138-i41.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 41: Intermolecular PKR of diarylalkynes with 2-norbornene reported by the group of Helaja [80].
Scheme 41: Intermolecular PKR of diarylalkynes with 2-norbornene reported by the group of Helaja [80].
In 2016, León and Fernández reported the first intermolecular PKR of internal alkynylboronic esters with norbornadiene [81]. For this purpose, terminal alkynes were converted into their corresponding alkynylboronic pinacol esters, and evaluated in the intermolecular PKR with norbornadiene. Under the optimal reaction conditions, Co2(CO)8 (1 equiv), norbornadiene (3 equiv) and NMO (6 equiv) in dichloromethane, the internal alkynes afforded the corresponding α,β-substituted cyclopentenones with total stereo- and regioselectivity; the exo-stereoisomer with the B(pin) substituent in the β-position was obtained exclusively. The PKR was compatible with aromatic groups containing substituents with differing electronic properties, heteroaryl groups, benzopinacol, 1,8-diaminonaphthalene, olefinic, and aliphatic groups. However, the PKR failed when hindered alkynes were used. The authors reported two examples of fluorine-containing alkynylboronic pinacol esters, which afforded the corresponding β-substituted cyclopentenones 72 in good yields (Scheme 42).
![[1860-5397-16-138-i42]](/bjoc/content/inline/1860-5397-16-138-i42.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 42: Intermolecular PKR reported by León and Fernández [81].
Scheme 42: Intermolecular PKR reported by León and Fernández [81].
In a recent report, Marek, Zhang, Ma and co-workers described a RhII-catalyzed cyclopropenation reaction of internal alkynes with a difluorodiazoethane reagent, offering efficient access to a broad range of enantioenriched difluoromethylated cyclopropenes with almost quantitative yields and up to 97% ee [82]. This asymmetric carbene transfer reaction was performed using chiral RhII complexes, more specifically the Hashimoto catalyst ([Rh2(S-TCPTTL)4]) was found to be the most efficient in terms of yield and enantioselectivity. In this study, the authors described an example of an intermolecular PKR using the strained difluoromethylated cyclopropene 73 which, upon reaction with the hexacarbonyldicobalt complex prepared by treatment of the 1-butyne with Co2(CO)8, afforded the corresponding bicyclic cyclopropane-fused cyclopentenone 74 in high yield and high enantioselectivity (Scheme 43).
![[1860-5397-16-138-i43]](/bjoc/content/inline/1860-5397-16-138-i43.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 43: PKR reported with cyclopropene 73 [82].
Scheme 43: PKR reported with cyclopropene 73 [82].
Conclusion
In conclusion, as highlighted in this review, the PKR is still a hot area of chemical research as it provides access to cyclopentenones from accessible starting materials under relatively mild conditions. The increasing demand of fine chemicals bearing fluorine atoms at strategic positions, together with the ubiquitous presence of the cyclopentenone ring in added-value compounds, has attracted researchers’ interest throughout the past decades. Since its discovery, numerous research groups have devoted their efforts to develop alternative ways to expand the scope of the PKR and the combination of fluorinated building blocks with this [2 + 2 + 1] cycloaddition has been efficiently applied in the preparation of fluorinated compounds of biological interest. The addition of fluorine-containing olefins and alkynes to the arsenal of substrates for the PKR has resulted in a major contribution in the field. Not only has the scope of accessible products been significantly expanded, but a better understanding of the factors governing the regioselectivity has been achieved, including the possibility to formally reverse the regiochemistry in some cases. Despite the important advances highlighted in this review, there remains room for improvement in the scope of this useful reaction with fluorinated compounds. Nowadays, most described protocols for the fluorinated PKR are based on the cobalt-catalyzed version, and only a few examples have used other transition metal complexes. In this regard, significant advances can come from the careful selection of the metal complex and the CO source. The encouraging results described in this review will definitely pave the way for future applications in the fluoro-PKR, and put this emerging reaction at the forefront of drug design.
References
-
Mei, H.; Han, J.; Fustero, S.; Medio‐Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840
Return to citation in text: [1] -
Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392
Return to citation in text: [1] -
Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879
Return to citation in text: [1] -
Shultz, M. D. J. Med. Chem. 2019, 62, 1701–1714. doi:10.1021/acs.jmedchem.8b00686
Return to citation in text: [1] -
O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a
Return to citation in text: [1] -
Hiyama, T. In Organofluorine Compounds: Chemistry and Applications; Yamamoto, H., Ed.; Springer: New York, NY, USA, 2000. doi:10.1007/978-3-662-04164-2
Return to citation in text: [1] -
Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566
Return to citation in text: [1] -
Tredwell, M.; Gouverneur, V. Angew. Chem., Int. Ed. 2012, 51, 11426–11437. doi:10.1002/anie.201204687
Return to citation in text: [1] -
Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. Chem. Soc. Rev. 2013, 42, 7971–7982. doi:10.1039/c3cs60129c
Return to citation in text: [1] -
Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014
Return to citation in text: [1] -
Vincent, J.-M. Chem. Commun. 2012, 48, 11382–11391. doi:10.1039/c2cc34750d
Return to citation in text: [1] -
Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f
Return to citation in text: [1] -
Wu, J. Tetrahedron Lett. 2014, 55, 4289–4294. doi:10.1016/j.tetlet.2014.06.006
See for nucleophilic fluorination.
Return to citation in text: [1] -
Hollingworth, C.; Gouverneur, V. Chem. Commun. 2012, 48, 2929–2942. doi:10.1039/c2cc16158c
See for nucleophilic fluorination.
Return to citation in text: [1] -
Baudoux, J.; Cahard, D. Org. React. 2008, 69, 1–326. doi:10.1002/0471264180.or069.02
See for nucleophilic fluorination.
Return to citation in text: [1] -
Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583
See for radical fluorination.
Return to citation in text: [1] -
Kuehnel, M. F.; Lentz, D.; Braun, T. Angew. Chem., Int. Ed. 2013, 52, 3328–3348. doi:10.1002/anie.201205260
Return to citation in text: [1] -
Ricker, J. D.; Geary, L. M. Top. Catal. 2017, 60, 609–619. doi:10.1007/s11244-017-0741-0
See for a review on the Pauson−Khand reaction.
Return to citation in text: [1] -
Ríos Torres, R., Ed. The Pauson-Khand Reaction. Scope, Variations and Applications; John Wiley & Sons: Chichester, UK, 2012. doi:10.1002/9781119941934
Return to citation in text: [1] -
Lee, H.-W.; Kwong, F.-Y. Eur. J. Org. Chem. 2010, 789–811. doi:10.1002/ejoc.200900892
See for a review on the Pauson−Khand reaction.
Return to citation in text: [1] -
Park, J. H.; Chang, K.-M.; Chung, Y. K. Coord. Chem. Rev. 2009, 253, 2461–2480. doi:10.1016/j.ccr.2009.08.005
See for a review on the Pauson−Khand reaction.
Return to citation in text: [1] -
Blanco-Urgoiti, J.; Añorbe, L.; Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2004, 33, 32–42. doi:10.1039/b300976a
See for a review on the Pauson−Khand reaction.
Return to citation in text: [1] -
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. J. Chem. Soc. D 1971, 36a. doi:10.1039/c2971000036a
Return to citation in text: [1] -
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. J. Chem. Soc., Perkin Trans. 1 1973, 975–977. doi:10.1039/p19730000975
Return to citation in text: [1] -
Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E.; Foreman, M. I. J. Chem. Soc., Perkin Trans. 1 1973, 977–981. doi:10.1039/p19730000977
Return to citation in text: [1] -
García-Lacuna, J.; Domínguez, G.; Blanco-Urgoiti, J.; Pérez-Castells, J. Org. Biomol. Chem. 2019, 17, 9489–9501. doi:10.1039/c9ob02124h
Return to citation in text: [1] -
Hugelshofer, C. L.; Palani, V.; Sarpong, R. J. Am. Chem. Soc. 2019, 141, 8431–8435. doi:10.1021/jacs.9b03576
Return to citation in text: [1] -
Zhao, N.; Xie, S.; Tian, P.; Tong, R.; Ning, C.; Xu, J. Org. Chem. Front. 2019, 6, 2014–2022. doi:10.1039/c9qo00384c
Return to citation in text: [1] -
Hu, N.; Dong, C.; Zhang, C.; Liang, G. Angew. Chem., Int. Ed. 2019, 58, 6659–6662. doi:10.1002/anie.201902043
Return to citation in text: [1] -
Kaneko, H.; Takahashi, S.; Kogure, N.; Kitajima, M.; Takayama, H. J. Org. Chem. 2019, 84, 5645–5654. doi:10.1021/acs.joc.9b00586
Return to citation in text: [1] -
Cabré, A.; Khaizourane, H.; Garçon, M.; Verdaguer, X.; Riera, A. Org. Lett. 2018, 20, 3953–3957. doi:10.1021/acs.orglett.8b01525
Return to citation in text: [1] -
Huang, Z.; Huang, J.; Qu, Y.; Zhang, W.; Gong, J.; Yang, Z. Angew. Chem., Int. Ed. 2018, 57, 8744–8748. doi:10.1002/anie.201805143
Return to citation in text: [1] -
Magnus, P.; Principe, L. M. Tetrahedron Lett. 1985, 26, 4851–4854. doi:10.1016/s0040-4039(00)94968-2
Return to citation in text: [1] -
Banide, E. V.; Müller-Bunz, H.; Manning, A. R.; Evans, P.; McGlinchey, M. J. Angew. Chem., Int. Ed. 2007, 46, 2907–2910. doi:10.1002/anie.200605171
Return to citation in text: [1] -
Yamanaka, M.; Nakamura, E. J. Am. Chem. Soc. 2001, 123, 1703–1708. doi:10.1021/ja005565+
Return to citation in text: [1] -
Pericàs, M. A.; Balsells, J.; Castro, J.; Marchueta, I.; Moyano, A.; Riera, A.; Vázquez, J.; Verdaguer, X. Pure Appl. Chem. 2002, 74, 167–174. doi:10.1351/pac200274010167
Return to citation in text: [1] -
Iqbal, M.; Vyse, N.; Dauvergne, J.; Evans, P. Tetrahedron Lett. 2002, 43, 7859–7862. doi:10.1016/s0040-4039(02)01912-3
Return to citation in text: [1] -
Rodríguez, A. M.; Prieto, P. Tetrahedron 2016, 72, 7443–7448. doi:10.1016/j.tet.2016.09.048
Return to citation in text: [1] -
Son, S. U.; Lee, S. I.; Chung, Y. K.; Kim, S.-W.; Hyeon, T. Org. Lett. 2002, 4, 277–279. doi:10.1021/ol017043k
Return to citation in text: [1] -
Jeong, N.; Chung, Y. K.; Lee, B. Y.; Lee, S. H.; Yoo, S.-E. Synlett 1991, 204–206. doi:10.1055/s-1991-20681
Return to citation in text: [1] -
Fager-Jokela, E.; Muuronen, M.; Khaizourane, H.; Vázquez-Romero, A.; Verdaguer, X.; Riera, A.; Helaja, J. J. Org. Chem. 2014, 79, 10999–11010. doi:10.1021/jo502035t
Return to citation in text: [1] -
Lam, F. L.; Lee, H. W.; Wang, J.; Kwong, F. Y. Recent Advancement of Catalytic Pauson–Khand-type Reactions. In Pauson Khand Reaction. Scope, Variations and Applications; Ríos Torres, R., Ed.; John Wiley & Sons: Chichester, UK, 2012; pp 181–210. doi:10.1002/9781119941934.ch7
Return to citation in text: [1] -
Ockey, D. A.; Lewis, M. A.; Schore, N. E. Tetrahedron 2003, 59, 5377–5381. doi:10.1016/s0040-4020(03)00779-8
Return to citation in text: [1] -
Mukai, C.; Yoshida, T.; Sorimachi, M.; Odani, A. Org. Lett. 2006, 8, 83–86. doi:10.1021/ol052562z
Return to citation in text: [1] -
Miller, K. A.; Shanahan, C. S.; Martin, S. F. Tetrahedron 2008, 64, 6884–6900. doi:10.1016/j.tet.2008.02.066
Return to citation in text: [1] -
Ishizaki, M.; Suzuki, D.; Hoshino, O. J. Fluorine Chem. 2001, 111, 81–90. doi:10.1016/s0022-1139(01)00436-5
Return to citation in text: [1] [2] [3] -
Ferry, A.; Billard, T.; Langlois, B. R. Synlett 2005, 1027–1029. doi:10.1055/s-2005-864812
Return to citation in text: [1] [2] -
Harthong, S.; Billard, T.; Langlois, B. R. Synthesis 2005, 2253–2263. doi:10.1055/s-2005-872076
Return to citation in text: [1] [2] -
Magueur, G.; Legros, J.; Meyer, F.; Ourévitch, M.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2005, 1258–1265. doi:10.1002/ejoc.200400719
Return to citation in text: [1] [2] -
Nadano, R.; Ichikawa, J. Chem. Lett. 2007, 36, 22–23. doi:10.1246/cl.2007.22
Return to citation in text: [1] [2] -
Nadano, R.; Fuchibe, K.; Ikeda, M.; Takahashi, H.; Ichikawa, J. Chem. – Asian J. 2010, 5, 1875–1883. doi:10.1002/asia.201000139
Return to citation in text: [1] -
Arimitsu, S.; Bottom, R. L.; Hammond, G. B. J. Fluorine Chem. 2008, 129, 1047–1051. doi:10.1016/j.jfluchem.2008.05.010
Return to citation in text: [1] [2] -
Vorobyeva, D. V.; Mailyan, A. K.; Peregudov, A. S.; Karimova, N. M.; Vasilyeva, T. P.; Bushmarinov, I. S.; Bruneau, C.; Dixneuf, P. H.; Osipov, S. N. Tetrahedron 2011, 67, 3524–3532. doi:10.1016/j.tet.2011.03.031
Return to citation in text: [1] [2] [3] -
Konno, T.; Kida, T.; Tani, A.; Ishihara, T. J. Fluorine Chem. 2012, 144, 147–156. doi:10.1016/j.jfluchem.2012.08.006
Return to citation in text: [1] [2] [3] [4] -
Fustero, S.; Lázaro, R.; Aiguabella, N.; Riera, A.; Simón-Fuentes, A.; Barrio, P. Org. Lett. 2014, 16, 1224–1227. doi:10.1021/ol500142c
Return to citation in text: [1] [2] -
Gallagher, A. G.; Tian, H.; Torres-Herrera, O. A.; Yin, S.; Xie, A.; Lange, D. M.; Wilson, J. K.; Mueller, L. G.; Gau, M. R.; Carroll, P. J.; Martinez-Solorio, D. Org. Lett. 2019, 21, 8646–8651. doi:10.1021/acs.orglett.9b03255
Return to citation in text: [1] [2] -
Dobbs, A. P.; Miller, I. J.; Martinović, S. Beilstein J. Org. Chem. 2007, 3, No. 21. doi:10.1186/1860-5397-3-21
Return to citation in text: [1] -
Ishaq, S.; Porter, M. J. Synth. Commun. 2006, 36, 547–557. doi:10.1080/00397910500406120
Return to citation in text: [1] -
Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509
Return to citation in text: [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] -
Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. doi:10.1021/ja508369z
Return to citation in text: [1] -
Llobat, A.; Román, R.; Mateu, N.; Sedgwick, D. M.; Barrio, P.; Medio-Simón, M.; Fustero, S. Org. Lett. 2019, 21, 7294–7297. doi:10.1021/acs.orglett.9b02557
Return to citation in text: [1] [2] [3] [4] [5] -
Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a
Return to citation in text: [1] -
Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778
Return to citation in text: [1] -
Llobat, A.; Sedgwick, D. M.; Cabré, A.; Román, R.; Mateu, N.; Escorihuela, J.; Medio‐Simón, M.; Soloshonok, V.; Han, J.; Riera, A.; Fustero, S. Adv. Synth. Catal. 2020, 362, 1378–1384. doi:10.1002/adsc.201901504
Return to citation in text: [1] [2] [3] [4] -
Pérez-Serrano, L.; Blanco-Urgoiti, J.; Casarrubios, L.; Domínguez, G.; Pérez-Castells, J. J. Org. Chem. 2000, 65, 3513–3519. doi:10.1021/jo0001232
Return to citation in text: [1] -
Cabré, A.; Verdaguer, X.; Riera, A. Synthesis 2018, 50, 3891–3896. doi:10.1055/s-0037-1610441
Return to citation in text: [1] -
Gibson, S. E.; Mainolfi, N. Angew. Chem., Int. Ed. 2005, 44, 3022–3037. doi:10.1002/anie.200462235
Return to citation in text: [1] -
Lledó, A.; Fuster, A.; Revés, M.; Verdaguer, X.; Riera, A. Chem. Commun. 2013, 49, 3055–3057. doi:10.1039/c3cc41005f
Return to citation in text: [1] -
Marchueta, I.; Verdaguer, X.; Moyano, A.; Pericàs, M. A.; Riera, A. Org. Lett. 2001, 3, 3193–3196. doi:10.1021/ol016505r
Return to citation in text: [1] -
Kizirian, J.-C.; Aiguabella, N.; Pesquer, A.; Fustero, S.; Bello, P.; Verdaguer, X.; Riera, A. Org. Lett. 2010, 12, 5620–5623. doi:10.1021/ol102283c
Return to citation in text: [1] [2] [3] [4] -
For a systematic study on the effect of electron-withdrawing groups of different nature (inductive vs mesomeric effect) on the regioseectivity of the PKR see reference [41].
Return to citation in text: [1] [2] -
Aiguabella, N.; del Pozo, C.; Verdaguer, X.; Fustero, S.; Riera, A. Angew. Chem., Int. Ed. 2013, 52, 5355–5359. doi:10.1002/anie.201300907
Return to citation in text: [1] [2] [3] -
Chu, L.; Qing, F.-L. J. Am. Chem. Soc. 2010, 132, 7262–7263. doi:10.1021/ja102175w
Return to citation in text: [1] -
Matsuda, T.; Tsuboi, T.; Murakami, M. J. Am. Chem. Soc. 2007, 129, 12596–12597. doi:10.1021/ja0732779
Return to citation in text: [1] -
Hodgson, D. M.; Chung, Y. K.; Nuzzo, I.; Freixas, G.; Kulikiewicz, K. K.; Cleator, E.; Paris, J.-M. J. Am. Chem. Soc. 2007, 129, 4456–4462. doi:10.1021/ja0672932
Return to citation in text: [1] -
Chetty, G. L.; Dev, S. Tetrahedron Lett. 1964, 5, 73–77. doi:10.1016/s0040-4039(00)90332-0
Return to citation in text: [1] -
Zhang, P.; Le, H.; Kyne, R. E.; Morken, J. P. J. Am. Chem. Soc. 2011, 133, 9716–9719. doi:10.1021/ja2039248
Return to citation in text: [1] -
Nakashima, H.; Sato, M.; Taniguchi, T.; Ogasawara, K. Tetrahedron Lett. 2000, 41, 2639–2642. doi:10.1016/s0040-4039(00)00235-5
Return to citation in text: [1] -
Aiguabella, N.; Arce, E. M.; del Pozo, C.; Verdaguer, X.; Riera, A. Molecules 2014, 19, 1763–1774. doi:10.3390/molecules19021763
Return to citation in text: [1] [2] -
Fager-Jokela, E.; Muuronen, M.; Patzschke, M.; Helaja, J. J. Org. Chem. 2012, 77, 9134–9147. doi:10.1021/jo3016902
Return to citation in text: [1] [2] -
León, T.; Fernández, E. Chem. Commun. 2016, 52, 9363–9366. doi:10.1039/c6cc04717c
Return to citation in text: [1] [2] -
Zhang, Z.-Q.; Zheng, M.-M.; Xue, X.-S.; Marek, I.; Zhang, F.-G.; Ma, J.-A. Angew. Chem., Int. Ed. 2019, 58, 18191–18196. doi:10.1002/anie.201911701
Return to citation in text: [1] [2]
| 49. | Magueur, G.; Legros, J.; Meyer, F.; Ourévitch, M.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2005, 1258–1265. doi:10.1002/ejoc.200400719 |
| 49. | Magueur, G.; Legros, J.; Meyer, F.; Ourévitch, M.; Crousse, B.; Bonnet-Delpon, D. Eur. J. Org. Chem. 2005, 1258–1265. doi:10.1002/ejoc.200400719 |
| 70. | Kizirian, J.-C.; Aiguabella, N.; Pesquer, A.; Fustero, S.; Bello, P.; Verdaguer, X.; Riera, A. Org. Lett. 2010, 12, 5620–5623. doi:10.1021/ol102283c |
| 50. | Nadano, R.; Ichikawa, J. Chem. Lett. 2007, 36, 22–23. doi:10.1246/cl.2007.22 |
| 51. | Nadano, R.; Fuchibe, K.; Ikeda, M.; Takahashi, H.; Ichikawa, J. Chem. – Asian J. 2010, 5, 1875–1883. doi:10.1002/asia.201000139 |
| 72. | Aiguabella, N.; del Pozo, C.; Verdaguer, X.; Fustero, S.; Riera, A. Angew. Chem., Int. Ed. 2013, 52, 5355–5359. doi:10.1002/anie.201300907 |
| 71. | For a systematic study on the effect of electron-withdrawing groups of different nature (inductive vs mesomeric effect) on the regioseectivity of the PKR see reference [41]. |
| 70. | Kizirian, J.-C.; Aiguabella, N.; Pesquer, A.; Fustero, S.; Bello, P.; Verdaguer, X.; Riera, A. Org. Lett. 2010, 12, 5620–5623. doi:10.1021/ol102283c |
| 71. | For a systematic study on the effect of electron-withdrawing groups of different nature (inductive vs mesomeric effect) on the regioseectivity of the PKR see reference [41]. |
| 54. | Konno, T.; Kida, T.; Tani, A.; Ishihara, T. J. Fluorine Chem. 2012, 144, 147–156. doi:10.1016/j.jfluchem.2012.08.006 |
| 54. | Konno, T.; Kida, T.; Tani, A.; Ishihara, T. J. Fluorine Chem. 2012, 144, 147–156. doi:10.1016/j.jfluchem.2012.08.006 |
| 53. | Vorobyeva, D. V.; Mailyan, A. K.; Peregudov, A. S.; Karimova, N. M.; Vasilyeva, T. P.; Bushmarinov, I. S.; Bruneau, C.; Dixneuf, P. H.; Osipov, S. N. Tetrahedron 2011, 67, 3524–3532. doi:10.1016/j.tet.2011.03.031 |
| 76. | Chetty, G. L.; Dev, S. Tetrahedron Lett. 1964, 5, 73–77. doi:10.1016/s0040-4039(00)90332-0 |
| 53. | Vorobyeva, D. V.; Mailyan, A. K.; Peregudov, A. S.; Karimova, N. M.; Vasilyeva, T. P.; Bushmarinov, I. S.; Bruneau, C.; Dixneuf, P. H.; Osipov, S. N. Tetrahedron 2011, 67, 3524–3532. doi:10.1016/j.tet.2011.03.031 |
| 52. | Arimitsu, S.; Bottom, R. L.; Hammond, G. B. J. Fluorine Chem. 2008, 129, 1047–1051. doi:10.1016/j.jfluchem.2008.05.010 |
| 72. | Aiguabella, N.; del Pozo, C.; Verdaguer, X.; Fustero, S.; Riera, A. Angew. Chem., Int. Ed. 2013, 52, 5355–5359. doi:10.1002/anie.201300907 |
| 53. | Vorobyeva, D. V.; Mailyan, A. K.; Peregudov, A. S.; Karimova, N. M.; Vasilyeva, T. P.; Bushmarinov, I. S.; Bruneau, C.; Dixneuf, P. H.; Osipov, S. N. Tetrahedron 2011, 67, 3524–3532. doi:10.1016/j.tet.2011.03.031 |
| 74. | Matsuda, T.; Tsuboi, T.; Murakami, M. J. Am. Chem. Soc. 2007, 129, 12596–12597. doi:10.1021/ja0732779 |
| 75. | Hodgson, D. M.; Chung, Y. K.; Nuzzo, I.; Freixas, G.; Kulikiewicz, K. K.; Cleator, E.; Paris, J.-M. J. Am. Chem. Soc. 2007, 129, 4456–4462. doi:10.1021/ja0672932 |
| 73. | Chu, L.; Qing, F.-L. J. Am. Chem. Soc. 2010, 132, 7262–7263. doi:10.1021/ja102175w |
| 52. | Arimitsu, S.; Bottom, R. L.; Hammond, G. B. J. Fluorine Chem. 2008, 129, 1047–1051. doi:10.1016/j.jfluchem.2008.05.010 |
| 72. | Aiguabella, N.; del Pozo, C.; Verdaguer, X.; Fustero, S.; Riera, A. Angew. Chem., Int. Ed. 2013, 52, 5355–5359. doi:10.1002/anie.201300907 |
| 55. | Fustero, S.; Lázaro, R.; Aiguabella, N.; Riera, A.; Simón-Fuentes, A.; Barrio, P. Org. Lett. 2014, 16, 1224–1227. doi:10.1021/ol500142c |
| 55. | Fustero, S.; Lázaro, R.; Aiguabella, N.; Riera, A.; Simón-Fuentes, A.; Barrio, P. Org. Lett. 2014, 16, 1224–1227. doi:10.1021/ol500142c |
| 54. | Konno, T.; Kida, T.; Tani, A.; Ishihara, T. J. Fluorine Chem. 2012, 144, 147–156. doi:10.1016/j.jfluchem.2012.08.006 |
| 56. | Gallagher, A. G.; Tian, H.; Torres-Herrera, O. A.; Yin, S.; Xie, A.; Lange, D. M.; Wilson, J. K.; Mueller, L. G.; Gau, M. R.; Carroll, P. J.; Martinez-Solorio, D. Org. Lett. 2019, 21, 8646–8651. doi:10.1021/acs.orglett.9b03255 |
| 80. | Fager-Jokela, E.; Muuronen, M.; Patzschke, M.; Helaja, J. J. Org. Chem. 2012, 77, 9134–9147. doi:10.1021/jo3016902 |
| 79. | Aiguabella, N.; Arce, E. M.; del Pozo, C.; Verdaguer, X.; Riera, A. Molecules 2014, 19, 1763–1774. doi:10.3390/molecules19021763 |
| 54. | Konno, T.; Kida, T.; Tani, A.; Ishihara, T. J. Fluorine Chem. 2012, 144, 147–156. doi:10.1016/j.jfluchem.2012.08.006 |
| 77. | Zhang, P.; Le, H.; Kyne, R. E.; Morken, J. P. J. Am. Chem. Soc. 2011, 133, 9716–9719. doi:10.1021/ja2039248 |
| 78. | Nakashima, H.; Sato, M.; Taniguchi, T.; Ogasawara, K. Tetrahedron Lett. 2000, 41, 2639–2642. doi:10.1016/s0040-4039(00)00235-5 |
| 79. | Aiguabella, N.; Arce, E. M.; del Pozo, C.; Verdaguer, X.; Riera, A. Molecules 2014, 19, 1763–1774. doi:10.3390/molecules19021763 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 60. | Okoromoba, O. E.; Han, J.; Hammond, G. B.; Xu, B. J. Am. Chem. Soc. 2014, 136, 14381–14384. doi:10.1021/ja508369z |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 81. | León, T.; Fernández, E. Chem. Commun. 2016, 52, 9363–9366. doi:10.1039/c6cc04717c |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 82. | Zhang, Z.-Q.; Zheng, M.-M.; Xue, X.-S.; Marek, I.; Zhang, F.-G.; Ma, J.-A. Angew. Chem., Int. Ed. 2019, 58, 18191–18196. doi:10.1002/anie.201911701 |
| 57. | Dobbs, A. P.; Miller, I. J.; Martinović, S. Beilstein J. Org. Chem. 2007, 3, No. 21. doi:10.1186/1860-5397-3-21 |
| 58. | Ishaq, S.; Porter, M. J. Synth. Commun. 2006, 36, 547–557. doi:10.1080/00397910500406120 |
| 80. | Fager-Jokela, E.; Muuronen, M.; Patzschke, M.; Helaja, J. J. Org. Chem. 2012, 77, 9134–9147. doi:10.1021/jo3016902 |
| 56. | Gallagher, A. G.; Tian, H.; Torres-Herrera, O. A.; Yin, S.; Xie, A.; Lange, D. M.; Wilson, J. K.; Mueller, L. G.; Gau, M. R.; Carroll, P. J.; Martinez-Solorio, D. Org. Lett. 2019, 21, 8646–8651. doi:10.1021/acs.orglett.9b03255 |
| 81. | León, T.; Fernández, E. Chem. Commun. 2016, 52, 9363–9366. doi:10.1039/c6cc04717c |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 1. | Mei, H.; Han, J.; Fustero, S.; Medio‐Simon, M.; Sedgwick, D. M.; Santi, C.; Ruzziconi, R.; Soloshonok, V. A. Chem. – Eur. J. 2019, 25, 11797–11819. doi:10.1002/chem.201901840 |
| 2. | Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 422–518. doi:10.1021/acs.chemrev.5b00392 |
| 3. | Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432–2506. doi:10.1021/cr4002879 |
| 41. | Fager-Jokela, E.; Muuronen, M.; Khaizourane, H.; Vázquez-Romero, A.; Verdaguer, X.; Riera, A.; Helaja, J. J. Org. Chem. 2014, 79, 10999–11010. doi:10.1021/jo502035t |
| 82. | Zhang, Z.-Q.; Zheng, M.-M.; Xue, X.-S.; Marek, I.; Zhang, F.-G.; Ma, J.-A. Angew. Chem., Int. Ed. 2019, 58, 18191–18196. doi:10.1002/anie.201911701 |
| 8. | Tredwell, M.; Gouverneur, V. Angew. Chem., Int. Ed. 2012, 51, 11426–11437. doi:10.1002/anie.201204687 |
| 9. | Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. Chem. Soc. Rev. 2013, 42, 7971–7982. doi:10.1039/c3cs60129c |
| 37. | Iqbal, M.; Vyse, N.; Dauvergne, J.; Evans, P. Tetrahedron Lett. 2002, 43, 7859–7862. doi:10.1016/s0040-4039(02)01912-3 |
| 38. | Rodríguez, A. M.; Prieto, P. Tetrahedron 2016, 72, 7443–7448. doi:10.1016/j.tet.2016.09.048 |
| 61. | Llobat, A.; Román, R.; Mateu, N.; Sedgwick, D. M.; Barrio, P.; Medio-Simón, M.; Fustero, S. Org. Lett. 2019, 21, 7294–7297. doi:10.1021/acs.orglett.9b02557 |
| 7. | Liang, T.; Neumann, C. N.; Ritter, T. Angew. Chem., Int. Ed. 2013, 52, 8214–8264. doi:10.1002/anie.201206566 |
| 39. | Son, S. U.; Lee, S. I.; Chung, Y. K.; Kim, S.-W.; Hyeon, T. Org. Lett. 2002, 4, 277–279. doi:10.1021/ol017043k |
| 5. | O'Hagan, D. Chem. Soc. Rev. 2008, 37, 308–319. doi:10.1039/b711844a |
| 6. | Hiyama, T. In Organofluorine Compounds: Chemistry and Applications; Yamamoto, H., Ed.; Springer: New York, NY, USA, 2000. doi:10.1007/978-3-662-04164-2 |
| 34. | Banide, E. V.; Müller-Bunz, H.; Manning, A. R.; Evans, P.; McGlinchey, M. J. Angew. Chem., Int. Ed. 2007, 46, 2907–2910. doi:10.1002/anie.200605171 |
| 62. | Champagne, P. A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Chem. Rev. 2015, 115, 9073–9174. doi:10.1021/cr500706a |
| 63. | Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V. A.; Coelho, J. A. S.; Toste, F. D. Chem. Rev. 2018, 118, 3887–3964. doi:10.1021/acs.chemrev.7b00778 |
| 4. | Shultz, M. D. J. Med. Chem. 2019, 62, 1701–1714. doi:10.1021/acs.jmedchem.8b00686 |
| 35. | Yamanaka, M.; Nakamura, E. J. Am. Chem. Soc. 2001, 123, 1703–1708. doi:10.1021/ja005565+ |
| 36. | Pericàs, M. A.; Balsells, J.; Castro, J.; Marchueta, I.; Moyano, A.; Riera, A.; Vázquez, J.; Verdaguer, X. Pure Appl. Chem. 2002, 74, 167–174. doi:10.1351/pac200274010167 |
| 61. | Llobat, A.; Román, R.; Mateu, N.; Sedgwick, D. M.; Barrio, P.; Medio-Simón, M.; Fustero, S. Org. Lett. 2019, 21, 7294–7297. doi:10.1021/acs.orglett.9b02557 |
| 18. |
Ricker, J. D.; Geary, L. M. Top. Catal. 2017, 60, 609–619. doi:10.1007/s11244-017-0741-0
See for a review on the Pauson−Khand reaction. |
| 19. | Ríos Torres, R., Ed. The Pauson-Khand Reaction. Scope, Variations and Applications; John Wiley & Sons: Chichester, UK, 2012. doi:10.1002/9781119941934 |
| 20. |
Lee, H.-W.; Kwong, F.-Y. Eur. J. Org. Chem. 2010, 789–811. doi:10.1002/ejoc.200900892
See for a review on the Pauson−Khand reaction. |
| 21. |
Park, J. H.; Chang, K.-M.; Chung, Y. K. Coord. Chem. Rev. 2009, 253, 2461–2480. doi:10.1016/j.ccr.2009.08.005
See for a review on the Pauson−Khand reaction. |
| 22. |
Blanco-Urgoiti, J.; Añorbe, L.; Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. Chem. Soc. Rev. 2004, 33, 32–42. doi:10.1039/b300976a
See for a review on the Pauson−Khand reaction. |
| 26. | García-Lacuna, J.; Domínguez, G.; Blanco-Urgoiti, J.; Pérez-Castells, J. Org. Biomol. Chem. 2019, 17, 9489–9501. doi:10.1039/c9ob02124h |
| 27. | Hugelshofer, C. L.; Palani, V.; Sarpong, R. J. Am. Chem. Soc. 2019, 141, 8431–8435. doi:10.1021/jacs.9b03576 |
| 28. | Zhao, N.; Xie, S.; Tian, P.; Tong, R.; Ning, C.; Xu, J. Org. Chem. Front. 2019, 6, 2014–2022. doi:10.1039/c9qo00384c |
| 29. | Hu, N.; Dong, C.; Zhang, C.; Liang, G. Angew. Chem., Int. Ed. 2019, 58, 6659–6662. doi:10.1002/anie.201902043 |
| 30. | Kaneko, H.; Takahashi, S.; Kogure, N.; Kitajima, M.; Takayama, H. J. Org. Chem. 2019, 84, 5645–5654. doi:10.1021/acs.joc.9b00586 |
| 31. | Cabré, A.; Khaizourane, H.; Garçon, M.; Verdaguer, X.; Riera, A. Org. Lett. 2018, 20, 3953–3957. doi:10.1021/acs.orglett.8b01525 |
| 32. | Huang, Z.; Huang, J.; Qu, Y.; Zhang, W.; Gong, J.; Yang, Z. Angew. Chem., Int. Ed. 2018, 57, 8744–8748. doi:10.1002/anie.201805143 |
| 61. | Llobat, A.; Román, R.; Mateu, N.; Sedgwick, D. M.; Barrio, P.; Medio-Simón, M.; Fustero, S. Org. Lett. 2019, 21, 7294–7297. doi:10.1021/acs.orglett.9b02557 |
| 17. | Kuehnel, M. F.; Lentz, D.; Braun, T. Angew. Chem., Int. Ed. 2013, 52, 3328–3348. doi:10.1002/anie.201205260 |
| 33. | Magnus, P.; Principe, L. M. Tetrahedron Lett. 1985, 26, 4851–4854. doi:10.1016/s0040-4039(00)94968-2 |
| 61. | Llobat, A.; Román, R.; Mateu, N.; Sedgwick, D. M.; Barrio, P.; Medio-Simón, M.; Fustero, S. Org. Lett. 2019, 21, 7294–7297. doi:10.1021/acs.orglett.9b02557 |
| 13. |
Wu, J. Tetrahedron Lett. 2014, 55, 4289–4294. doi:10.1016/j.tetlet.2014.06.006
See for nucleophilic fluorination. |
| 14. |
Hollingworth, C.; Gouverneur, V. Chem. Commun. 2012, 48, 2929–2942. doi:10.1039/c2cc16158c
See for nucleophilic fluorination. |
| 15. |
Baudoux, J.; Cahard, D. Org. React. 2008, 69, 1–326. doi:10.1002/0471264180.or069.02
See for nucleophilic fluorination. |
| 16. |
Sibi, M. P.; Landais, Y. Angew. Chem., Int. Ed. 2013, 52, 3570–3572. doi:10.1002/anie.201209583
See for radical fluorination. |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 10. | Fujiwara, T.; O’Hagan, D. J. Fluorine Chem. 2014, 167, 16–29. doi:10.1016/j.jfluchem.2014.06.014 |
| 11. | Vincent, J.-M. Chem. Commun. 2012, 48, 11382–11391. doi:10.1039/c2cc34750d |
| 12. | Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Chem. Soc. Rev. 2011, 40, 3496–3508. doi:10.1039/c0cs00221f |
| 23. | Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. J. Chem. Soc. D 1971, 36a. doi:10.1039/c2971000036a |
| 24. | Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E. J. Chem. Soc., Perkin Trans. 1 1973, 975–977. doi:10.1039/p19730000975 |
| 25. | Khand, I. U.; Knox, G. R.; Pauson, P. L.; Watts, W. E.; Foreman, M. I. J. Chem. Soc., Perkin Trans. 1 1973, 977–981. doi:10.1039/p19730000977 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 42. | Lam, F. L.; Lee, H. W.; Wang, J.; Kwong, F. Y. Recent Advancement of Catalytic Pauson–Khand-type Reactions. In Pauson Khand Reaction. Scope, Variations and Applications; Ríos Torres, R., Ed.; John Wiley & Sons: Chichester, UK, 2012; pp 181–210. doi:10.1002/9781119941934.ch7 |
| 40. | Jeong, N.; Chung, Y. K.; Lee, B. Y.; Lee, S. H.; Yoo, S.-E. Synlett 1991, 204–206. doi:10.1055/s-1991-20681 |
| 41. | Fager-Jokela, E.; Muuronen, M.; Khaizourane, H.; Vázquez-Romero, A.; Verdaguer, X.; Riera, A.; Helaja, J. J. Org. Chem. 2014, 79, 10999–11010. doi:10.1021/jo502035t |
| 65. | Pérez-Serrano, L.; Blanco-Urgoiti, J.; Casarrubios, L.; Domínguez, G.; Pérez-Castells, J. J. Org. Chem. 2000, 65, 3513–3519. doi:10.1021/jo0001232 |
| 59. | Román, R.; Mateu, N.; López, I.; Medio-Simón, M.; Fustero, S.; Barrio, P. Org. Lett. 2019, 21, 2569–2573. doi:10.1021/acs.orglett.9b00509 |
| 61. | Llobat, A.; Román, R.; Mateu, N.; Sedgwick, D. M.; Barrio, P.; Medio-Simón, M.; Fustero, S. Org. Lett. 2019, 21, 7294–7297. doi:10.1021/acs.orglett.9b02557 |
| 64. | Llobat, A.; Sedgwick, D. M.; Cabré, A.; Román, R.; Mateu, N.; Escorihuela, J.; Medio‐Simón, M.; Soloshonok, V.; Han, J.; Riera, A.; Fustero, S. Adv. Synth. Catal. 2020, 362, 1378–1384. doi:10.1002/adsc.201901504 |
| 64. | Llobat, A.; Sedgwick, D. M.; Cabré, A.; Román, R.; Mateu, N.; Escorihuela, J.; Medio‐Simón, M.; Soloshonok, V.; Han, J.; Riera, A.; Fustero, S. Adv. Synth. Catal. 2020, 362, 1378–1384. doi:10.1002/adsc.201901504 |
| 48. | Harthong, S.; Billard, T.; Langlois, B. R. Synthesis 2005, 2253–2263. doi:10.1055/s-2005-872076 |
| 48. | Harthong, S.; Billard, T.; Langlois, B. R. Synthesis 2005, 2253–2263. doi:10.1055/s-2005-872076 |
| 47. | Ferry, A.; Billard, T.; Langlois, B. R. Synlett 2005, 1027–1029. doi:10.1055/s-2005-864812 |
| 70. | Kizirian, J.-C.; Aiguabella, N.; Pesquer, A.; Fustero, S.; Bello, P.; Verdaguer, X.; Riera, A. Org. Lett. 2010, 12, 5620–5623. doi:10.1021/ol102283c |
| 47. | Ferry, A.; Billard, T.; Langlois, B. R. Synlett 2005, 1027–1029. doi:10.1055/s-2005-864812 |
| 70. | Kizirian, J.-C.; Aiguabella, N.; Pesquer, A.; Fustero, S.; Bello, P.; Verdaguer, X.; Riera, A. Org. Lett. 2010, 12, 5620–5623. doi:10.1021/ol102283c |
| 46. | Ishizaki, M.; Suzuki, D.; Hoshino, O. J. Fluorine Chem. 2001, 111, 81–90. doi:10.1016/s0022-1139(01)00436-5 |
| 64. | Llobat, A.; Sedgwick, D. M.; Cabré, A.; Román, R.; Mateu, N.; Escorihuela, J.; Medio‐Simón, M.; Soloshonok, V.; Han, J.; Riera, A.; Fustero, S. Adv. Synth. Catal. 2020, 362, 1378–1384. doi:10.1002/adsc.201901504 |
| 46. | Ishizaki, M.; Suzuki, D.; Hoshino, O. J. Fluorine Chem. 2001, 111, 81–90. doi:10.1016/s0022-1139(01)00436-5 |
| 67. | Gibson, S. E.; Mainolfi, N. Angew. Chem., Int. Ed. 2005, 44, 3022–3037. doi:10.1002/anie.200462235 |
| 68. | Lledó, A.; Fuster, A.; Revés, M.; Verdaguer, X.; Riera, A. Chem. Commun. 2013, 49, 3055–3057. doi:10.1039/c3cc41005f |
| 69. | Marchueta, I.; Verdaguer, X.; Moyano, A.; Pericàs, M. A.; Riera, A. Org. Lett. 2001, 3, 3193–3196. doi:10.1021/ol016505r |
| 43. | Ockey, D. A.; Lewis, M. A.; Schore, N. E. Tetrahedron 2003, 59, 5377–5381. doi:10.1016/s0040-4020(03)00779-8 |
| 44. | Mukai, C.; Yoshida, T.; Sorimachi, M.; Odani, A. Org. Lett. 2006, 8, 83–86. doi:10.1021/ol052562z |
| 45. | Miller, K. A.; Shanahan, C. S.; Martin, S. F. Tetrahedron 2008, 64, 6884–6900. doi:10.1016/j.tet.2008.02.066 |
| 64. | Llobat, A.; Sedgwick, D. M.; Cabré, A.; Román, R.; Mateu, N.; Escorihuela, J.; Medio‐Simón, M.; Soloshonok, V.; Han, J.; Riera, A.; Fustero, S. Adv. Synth. Catal. 2020, 362, 1378–1384. doi:10.1002/adsc.201901504 |
| 46. | Ishizaki, M.; Suzuki, D.; Hoshino, O. J. Fluorine Chem. 2001, 111, 81–90. doi:10.1016/s0022-1139(01)00436-5 |
| 66. | Cabré, A.; Verdaguer, X.; Riera, A. Synthesis 2018, 50, 3891–3896. doi:10.1055/s-0037-1610441 |
© 2020 Escorihuela et al.; licensee Beilstein-Institut.
This is an Open Access article under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/4.0). Please note that the reuse, redistribution and reproduction in particular requires that the authors and source are credited.
The license is subject to the Beilstein Journal of Organic Chemistry terms and conditions: (https://www.beilstein-journals.org/bjoc)