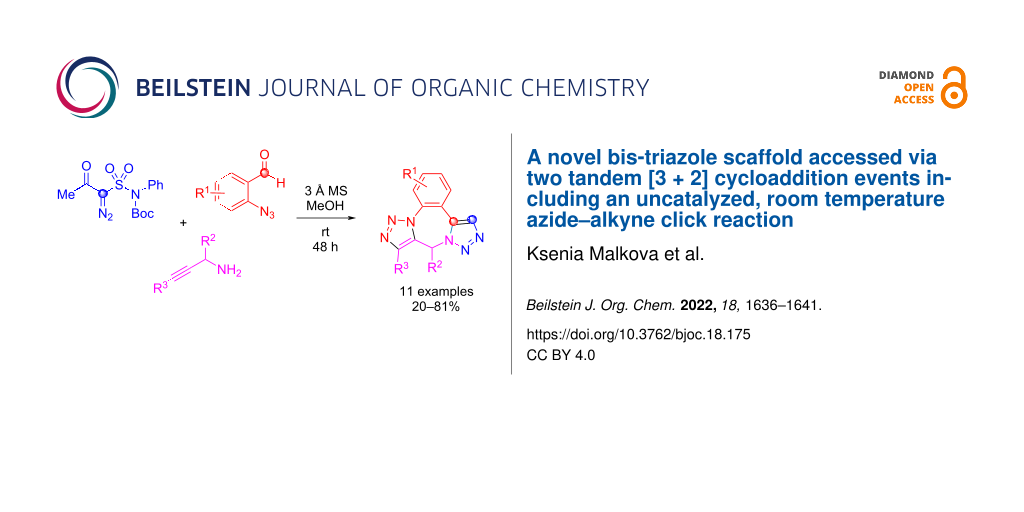
Abstract
The previously described α-acetyl-α-diazomethanesulfonamide was employed in a three-component reaction with azide-containing benzaldehydes and propargylamines. Besides the initial formation of the triazole core, the reaction proceeded further, in uncatalyzed fashion at room temperature and yielded, after intramolecular azide–alkyne click reaction novel, structurally intriguing bistriazoles.

Graphical Abstract
Introduction
1,2,3-Triazoles are well-established heterocycles in drug discovery [1] and are even considered pharmacophores (i.e., structural motifs defining the compound’s biological activity profile) on their own [2]. Therefore, synthetic methods allowing to construct a 1,2,3-triazole heterocycle are a valuable part of the drug discovery chemistry toolbox. For the same reason, development of new methods [3] to either build 1,2,3-triazoles de novo and/or incorporate them into polycyclic scaffolds is a worthy undertaking which can help discover biological activity associated with hitherto unattainable scaffolds.
Recently, we reported a novel, metal-free synthesis of 1,5-disubstituted 1,2,3-triazoles via a three-component reaction of α-acetyl-α-diazomethanesulfonamide (1) with aldehydes and amines [4]. The reaction proceeded, presumably, through the formation of the initial 1,2,3-triazoline adduct 2 [5] which underwent aromatization with the loss of sulfur dioxide and N-Boc-aniline. The multicomponent character and the fairly large scope of this reaction allows to place pairwise reactive groups in the aldehyde and the amine components, which would set a scene for further elaboration of the product’s molecular scaffold. Pondering various opportunities for post-condensational modifications of the 1,5-disubstituted 1,2,3-triazole core according to this strategy, we turned our attention to such powerful transformation as the azide–alkyne [3 + 2] cycloaddition (also known as the azide–alkyne click reaction) [6]. Indeed, if an alkyne and an azido group were strategically positioned within the structure of the amine and the aldehyde components for the reaction with 1, subsequent intramolecular azide–alkyne cycloaddition would be a feasible event which would create a polycyclic bis-1,2,3-triazole framework (Figure 1). Herein, we report on a successful realization of this strategy.
![[1860-5397-18-175-1]](/bjoc/content/figures/1860-5397-18-175-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: (a) Previously developed three-component approach to 1,5-disubstituted 1,2,3-triazoles; (b) double cycloaddition strategy investigated in this work.
Figure 1: (a) Previously developed three-component approach to 1,5-disubstituted 1,2,3-triazoles; (b) double ...
Results and Discussion
To test the possibility of a tandem double cycloaddition reaction between 1, an alkyne-containing amine and an azide-containing aldehyde, we set up a reaction of 1 with o-azidobenzaldehyde (3a) and propargylamine. The reaction was allowed to go to completion in 48 h at room temperature whereupon the reaction mixture was absorbed on silica and subjected to column chromatography for isolation of the product. To our sheer amazement, the product turned out to be not the initial adduct 4a but rather 9H-benzo[f]bis([1,2,3]triazolo)[1,5-a:1',5'-d][1,4]diazepine (5a), i.e., the product of the tandem three-component 1,2,3-triazole synthesis followed by intramolecular azide–alkyne click reaction which, apparently, proceeded at room temperature. Product 5a was isolated in respectable 78% yield; therefore, the reaction conditions were not further optimized (Scheme 1). The structure of tetracyclic product 5a was unequivocally confirmed by 1H and 13C NMR as well as single-crystal X-ray analysis.
![[1860-5397-18-175-i1]](/bjoc/content/inline/1860-5397-18-175-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Results of a trial reaction between 1, o-azidobenzaldehyde (3a) and propargylamine.
Scheme 1: Results of a trial reaction between 1, o-azidobenzaldehyde (3a) and propargylamine.
Compound 5a is representative of the hitherto undescribed bistriazole benzodiazepine scaffold. However, 5,6,7,8-tetrahydro-4H-[1,2,3]triazolo[1,5-d][1,4]diazepine (A) and 5,6,7,8-tetrahydro-4H-[1,2,3]triazolo[1,5-a][1,4]diazepine (B) scaffolds are of high medicinal importance, as evident from the literature. The range of biologically active compounds based on these two closely related scaffolds (both incorporated as fragments in the structure of compound 5a) include compound 6 for the treatment of cognitive impairment [7], BET bromodomain inhibitors 7 [8] and 8 [9] for cancer treatment, σ1 receptor modulator 9 for diverse disorders [10] and bacterial regulatory RNA binder 10 [11] (scaffold A) as well as antidiuretic 11 [12], glycogen phosphorylase inhibitor 12 [13], MK2 kinase inhibitor 13 [14], ENL YEATS domain inhibitor 14 for leukemia treatment [15] and hepatitis C NS5B polymerase inhibitor 15 [16] (scaffold B, Figure 2).
![[1860-5397-18-175-2]](/bjoc/content/figures/1860-5397-18-175-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Diversely bioactive compounds based on scaffolds A and B.
Figure 2: Diversely bioactive compounds based on scaffolds A and B.
The very fact that it was impossible to isolate intermediate 4a from the reaction depicted in Scheme 1 speaks for the unusual facility with which the intramolecular azide–alkyne click reaction took place. Normally, intermolecular click reactions are copper-catalyzed [17-20]. Intramolecular positioning of the click reaction partners may eliminate the need for the metal-based catalyst but the reaction still requires thermal activation [21]. Thus, to our knowledge, the room temperature intramolecular azide–alkyne cycloaddition is unprecedented (plasmon-assisted click reaction at low temperature has been recently reported [22]). Excited by our initial finding, we tested various azide-containing aromatic aldehydes 3a–i in the reaction with 1 and propargylamine (Scheme 2).
![[1860-5397-18-175-i2]](/bjoc/content/inline/1860-5397-18-175-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Three-component reaction of 1, propargylamine and various o-azidobenzaldehydes.
Scheme 2: Three-component reaction of 1, propargylamine and various o-azidobenzaldehydes.
The yields of products 5 were generally fair to good for all the aldehydes tested, even for doubly azide-substituted aldehyde 3c. However, the yield diminished somewhat for chloro-substituted aldehydes 5g–h. Stronger electron-withdrawing cyano group (3i) lowered the product yields to 20% due to low conversion of 3i.
Introducing even a stronger electron acceptor, a nitro group (aldehyde 3j), led to only a trace amount of the respective product detected by 1H NMR analysis of the crude reaction mixture. Likewise, heterocyclic azido aldehydes 3k,l failed to react with 1 and propargylamine (Figure 3).
![[1860-5397-18-175-3]](/bjoc/content/figures/1860-5397-18-175-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Aromatic azido aldehydes 3j–l that failed to react with 1 and propargylamine.
Figure 3: Aromatic azido aldehydes 3j–l that failed to react with 1 and propargylamine.
Having established the scope and limitations of the 9H-benzo[f]bis([1,2,3]triazolo)[1,5-a:1',5'-d][1,4]diazepine (5) synthesis, we proceeded to look at the variations in the alkyne-containing amine component for this transformation. Introduction of an α-methyl substitution in propargylamine (compound 16) was tolerated and the respective reaction gave product 17 in a yield comparable to (and somewhat better than) that of unsubstituted compound 5a. Homologation of propargylamine made a significant impact on the course of the reaction. The use of homopropargylamine (18) in the reaction of 1 with aldehyde 3a abolished the facility of the intramolecular azide–alkyne click reaction which now required heating at 120 °C for 2 hours for the eight-membered (1,5-diazocane) ring to form (notably, the presence of the azide–alkyne intermediate before the click reaction was established by 1H NMR analysis of the reaction mixture). However, the product of this two-step, one-pot reaction (19) was isolated in respectable 61% yield. The structure of compound 19 was confirmed by the single-crystal X-ray analysis which demonstrated that the compound crystalized in two distinct conformers. Finally, we were curious to see if terminally substituted propargylamine 20 would react with 1 and aldehyde 3a under the same reaction conditions. To our delight, the additional phenyl substituent did not dramatically influence the course of the reaction although the yield of product 21 was diminished compared to that of unsubstituted compound 5a. The structure of compound 21 was also confirmed by the single-crystal X-ray analysis (Scheme 3).
![[1860-5397-18-175-i3]](/bjoc/content/inline/1860-5397-18-175-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Variations of the amine component in the reactions with 1 and 3a.
Scheme 3: Variations of the amine component in the reactions with 1 and 3a.
All compounds were tested against lung cancer cell lines A549 and NCI-H460 and did not show any appreciable effect on cell proliferation in concentrations up to 250 μM. This validates these novel compounds as non-cytotoxic probes for interrogation of various biological targets.
Conclusion
The previously described α-acetyl-α-diazomethanesulfonamide was employed in a three-component reaction with azide-containing benzaldehydes and propargylamines. Besides the initial formation of the triazole core, the reaction proceeded further, in uncatalyzed fashion at room temperature and yielded structurally intriguing bistriazoles whose structure was unequivocally confirmed by single-crystal X-ray analysis. Compounds are non-cytotoxic which makes them suitable for interrogation of various biological targets.
Supporting Information
Deposition numbers 2183765 (for 5a), 2183766 (for 19) and 2183767 (for 21) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service http://www.ccdc.cam.ac.uk/structures.
| Supporting Information File 1: General experimental information, X-ray crystallographic data, synthetic procedures, analytical data and NMR spectra for the reported compounds. | ||
| Format: PDF | Size: 3.2 MB | Download |
References
-
Serafini, M.; Pirali, T.; Tron, G. C. Adv. Heterocycl. Chem. 2021, 134, 101–148. doi:10.1016/bs.aihch.2020.10.001
Return to citation in text: [1] -
Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem. – Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432
Return to citation in text: [1] -
Nemallapudi, B. R.; Guda, D. R.; Ummadi, N.; Avula, B.; Zyryanov, G. V.; Reddy, C. S.; Gundala, S. Polycyclic Aromat. Compd. 2022, 42, 3874–3892. doi:10.1080/10406638.2020.1866038
Return to citation in text: [1] -
Bubyrev, A.; Malkova, K.; Kantin, G.; Dar’in, D.; Krasavin, M. J. Org. Chem. 2021, 86, 17516–17522. doi:10.1021/acs.joc.1c02309
Return to citation in text: [1] -
Bubyrev, A.; Adamchik, M.; Dar’in, D.; Kantin, G.; Krasavin, M. J. Org. Chem. 2021, 86, 13454–13464. doi:10.1021/acs.joc.1c01552
Return to citation in text: [1] -
Devaraj, N. K.; Finn, M. G. Chem. Rev. 2021, 121, 6697–6698. doi:10.1021/acs.chemrev.1c00469
Return to citation in text: [1] -
Mekonnen, B.; Butera, J. A.; Huang, J.; Patel, H.; Jiang, Q.; Herr, R. J.; Mayhew, N. J.; Freeman, E. E. Benzodiazepine Derivatives, Compositions, And Methods for Treating Cognitive Impairment. PCT Int. Appl. WO2019/246300A1, Dec 26, 2019.
Return to citation in text: [1] -
Sheppard, G. S.; Wang, L.; Fidanze, S. D.; Hasvold, L. A.; Liu, D.; Pratt, J. K.; Park, C. H.; Longenecker, K.; Qiu, W.; Torrent, M.; Kovar, P. J.; Bui, M.; Faivre, E.; Huang, X.; Lin, X.; Wilcox, D.; Zhang, L.; Shen, Y.; Albert, D. H.; Magoc, T. J.; Rajaraman, G.; Kati, W. M.; McDaniel, K. F. J. Med. Chem. 2020, 63, 5585–5623. doi:10.1021/acs.jmedchem.0c00628
Return to citation in text: [1] -
Sharp, P. P.; Garnier, J.-M.; Hatfaludi, T.; Xu, Z.; Segal, D.; Jarman, K. E.; Jousset, H.; Garnham, A.; Feutrill, J. T.; Cuzzupe, A.; Hall, P.; Taylor, S.; Walkley, C. R.; Tyler, D.; Dawson, M. A.; Czabotar, P.; Wilks, A. F.; Glaser, S.; Huang, D. C. S.; Burns, C. J. ACS Med. Chem. Lett. 2017, 8, 1298–1303. doi:10.1021/acsmedchemlett.7b00389
Return to citation in text: [1] -
Cuevas-Cordobés, F.; Pericás-Brondo, M. A. Tricyclic Triazolic Compounds. Eur. Pat. Appl. EP3164403A1, May 10, 2017.
Return to citation in text: [1] -
Armstrong, I.; Aldhumani, A. H.; Schopis, J. L.; Fang, F.; Parsons, E.; Zeng, C.; Hossain, M. I.; Bergmeier, S. C.; Hines, J. V. Bioorg. Med. Chem. 2020, 28, 115696. doi:10.1016/j.bmc.2020.115696
Return to citation in text: [1] -
Ferring, B. V.; Ashworth, D. M.; Pitt, G. R. W.; Hudson, P.; Yea, C. M.; Franklin, R. J.; Semple, G. Fused Azepine Derivatives And Their Use As Antidiuretic Agents. PCT Int. Appl. WO0200626A1, Jan 3, 2002.
Return to citation in text: [1] -
Yan, Z.; Ma, C.; Wang, Y.; Shuai, L.; Guo, Y.; Zhang, L. Russ. J. Bioorg. Chem. 2022, 48, 589–595. doi:10.1134/s1068162022030232
Return to citation in text: [1] -
McComas, C. C.; Serrano-Wu, M. H.; Vacca, J. P. Fused Quadracyclic Compounds, Compositions and Uses Thereof. Eur. Pat. Appl. EP3400228A1, Nov 14, 2018.
Return to citation in text: [1] -
Ma, X. R.; Xu, L.; Xu, S.; Klein, B. J.; Wang, H.; Das, S.; Li, K.; Yang, K. S.; Sohail, S.; Chapman, A.; Kutateladze, T. G.; Shi, X.; Liu, W. R.; Wen, H. J. Med. Chem. 2021, 64, 10997–11013. doi:10.1021/acs.jmedchem.1c00367
Return to citation in text: [1] -
Zhong, M.; Peng, E.; Huang, N.; Huang, Q.; Huq, A.; Lau, M.; Colonno, R.; Li, L. Bioorg. Med. Chem. Lett. 2018, 28, 963–968. doi:10.1016/j.bmcl.2018.01.029
Return to citation in text: [1] -
Shao, C.; Wang, X.; Xu, J.; Zhao, J.; Zhang, Q.; Hu, Y. J. Org. Chem. 2010, 75, 7002–7005. doi:10.1021/jo101495k
Return to citation in text: [1] -
Özçubukçu, S.; Ozkal, E.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2009, 11, 4680–4683. doi:10.1021/ol9018776
Return to citation in text: [1] -
Rodionov, V. O.; Presolski, S. I.; Gardinier, S.; Lim, Y.-H.; Finn, M. G. J. Am. Chem. Soc. 2007, 129, 12696–12704. doi:10.1021/ja072678l
Return to citation in text: [1] -
Lipshutz, B. H.; Taft, B. R. Angew. Chem., Int. Ed. 2006, 45, 8235–8238. doi:10.1002/anie.200603726
Return to citation in text: [1] -
Oliva, A. I.; Christmann, U.; Font, D.; Cuevas, F.; Ballester, P.; Buschmann, H.; Torrens, A.; Yenes, S.; Pericàs, M. A. Org. Lett. 2008, 10, 1617–1619. doi:10.1021/ol800291t
Return to citation in text: [1] -
Guselnikova, O.; Váňa, J.; Phuong, L. T.; Panov, I.; Rulíšek, L.; Trelin, A.; Postnikov, P.; Švorčík, V.; Andris, E.; Lyutakov, O. Chem. Sci. 2021, 12, 5591–5598. doi:10.1039/d0sc05898j
Return to citation in text: [1]
| 22. | Guselnikova, O.; Váňa, J.; Phuong, L. T.; Panov, I.; Rulíšek, L.; Trelin, A.; Postnikov, P.; Švorčík, V.; Andris, E.; Lyutakov, O. Chem. Sci. 2021, 12, 5591–5598. doi:10.1039/d0sc05898j |
| 17. | Shao, C.; Wang, X.; Xu, J.; Zhao, J.; Zhang, Q.; Hu, Y. J. Org. Chem. 2010, 75, 7002–7005. doi:10.1021/jo101495k |
| 18. | Özçubukçu, S.; Ozkal, E.; Jimeno, C.; Pericàs, M. A. Org. Lett. 2009, 11, 4680–4683. doi:10.1021/ol9018776 |
| 19. | Rodionov, V. O.; Presolski, S. I.; Gardinier, S.; Lim, Y.-H.; Finn, M. G. J. Am. Chem. Soc. 2007, 129, 12696–12704. doi:10.1021/ja072678l |
| 20. | Lipshutz, B. H.; Taft, B. R. Angew. Chem., Int. Ed. 2006, 45, 8235–8238. doi:10.1002/anie.200603726 |
| 21. | Oliva, A. I.; Christmann, U.; Font, D.; Cuevas, F.; Ballester, P.; Buschmann, H.; Torrens, A.; Yenes, S.; Pericàs, M. A. Org. Lett. 2008, 10, 1617–1619. doi:10.1021/ol800291t |
| 1. | Serafini, M.; Pirali, T.; Tron, G. C. Adv. Heterocycl. Chem. 2021, 134, 101–148. doi:10.1016/bs.aihch.2020.10.001 |
| 5. | Bubyrev, A.; Adamchik, M.; Dar’in, D.; Kantin, G.; Krasavin, M. J. Org. Chem. 2021, 86, 13454–13464. doi:10.1021/acs.joc.1c01552 |
| 15. | Ma, X. R.; Xu, L.; Xu, S.; Klein, B. J.; Wang, H.; Das, S.; Li, K.; Yang, K. S.; Sohail, S.; Chapman, A.; Kutateladze, T. G.; Shi, X.; Liu, W. R.; Wen, H. J. Med. Chem. 2021, 64, 10997–11013. doi:10.1021/acs.jmedchem.1c00367 |
| 4. | Bubyrev, A.; Malkova, K.; Kantin, G.; Dar’in, D.; Krasavin, M. J. Org. Chem. 2021, 86, 17516–17522. doi:10.1021/acs.joc.1c02309 |
| 16. | Zhong, M.; Peng, E.; Huang, N.; Huang, Q.; Huq, A.; Lau, M.; Colonno, R.; Li, L. Bioorg. Med. Chem. Lett. 2018, 28, 963–968. doi:10.1016/j.bmcl.2018.01.029 |
| 3. | Nemallapudi, B. R.; Guda, D. R.; Ummadi, N.; Avula, B.; Zyryanov, G. V.; Reddy, C. S.; Gundala, S. Polycyclic Aromat. Compd. 2022, 42, 3874–3892. doi:10.1080/10406638.2020.1866038 |
| 13. | Yan, Z.; Ma, C.; Wang, Y.; Shuai, L.; Guo, Y.; Zhang, L. Russ. J. Bioorg. Chem. 2022, 48, 589–595. doi:10.1134/s1068162022030232 |
| 2. | Agalave, S. G.; Maujan, S. R.; Pore, V. S. Chem. – Asian J. 2011, 6, 2696–2718. doi:10.1002/asia.201100432 |
| 14. | McComas, C. C.; Serrano-Wu, M. H.; Vacca, J. P. Fused Quadracyclic Compounds, Compositions and Uses Thereof. Eur. Pat. Appl. EP3400228A1, Nov 14, 2018. |
| 9. | Sharp, P. P.; Garnier, J.-M.; Hatfaludi, T.; Xu, Z.; Segal, D.; Jarman, K. E.; Jousset, H.; Garnham, A.; Feutrill, J. T.; Cuzzupe, A.; Hall, P.; Taylor, S.; Walkley, C. R.; Tyler, D.; Dawson, M. A.; Czabotar, P.; Wilks, A. F.; Glaser, S.; Huang, D. C. S.; Burns, C. J. ACS Med. Chem. Lett. 2017, 8, 1298–1303. doi:10.1021/acsmedchemlett.7b00389 |
| 11. | Armstrong, I.; Aldhumani, A. H.; Schopis, J. L.; Fang, F.; Parsons, E.; Zeng, C.; Hossain, M. I.; Bergmeier, S. C.; Hines, J. V. Bioorg. Med. Chem. 2020, 28, 115696. doi:10.1016/j.bmc.2020.115696 |
| 8. | Sheppard, G. S.; Wang, L.; Fidanze, S. D.; Hasvold, L. A.; Liu, D.; Pratt, J. K.; Park, C. H.; Longenecker, K.; Qiu, W.; Torrent, M.; Kovar, P. J.; Bui, M.; Faivre, E.; Huang, X.; Lin, X.; Wilcox, D.; Zhang, L.; Shen, Y.; Albert, D. H.; Magoc, T. J.; Rajaraman, G.; Kati, W. M.; McDaniel, K. F. J. Med. Chem. 2020, 63, 5585–5623. doi:10.1021/acs.jmedchem.0c00628 |
| 12. | Ferring, B. V.; Ashworth, D. M.; Pitt, G. R. W.; Hudson, P.; Yea, C. M.; Franklin, R. J.; Semple, G. Fused Azepine Derivatives And Their Use As Antidiuretic Agents. PCT Int. Appl. WO0200626A1, Jan 3, 2002. |
| 7. | Mekonnen, B.; Butera, J. A.; Huang, J.; Patel, H.; Jiang, Q.; Herr, R. J.; Mayhew, N. J.; Freeman, E. E. Benzodiazepine Derivatives, Compositions, And Methods for Treating Cognitive Impairment. PCT Int. Appl. WO2019/246300A1, Dec 26, 2019. |
| 6. | Devaraj, N. K.; Finn, M. G. Chem. Rev. 2021, 121, 6697–6698. doi:10.1021/acs.chemrev.1c00469 |
| 10. | Cuevas-Cordobés, F.; Pericás-Brondo, M. A. Tricyclic Triazolic Compounds. Eur. Pat. Appl. EP3164403A1, May 10, 2017. |
© 2022 Malkova et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.