

Abstract
Continuous flow technology has become the method of choice for many academic and industrial researchers when developing new routes to chemical compounds of interest. With this technology maturing over the last decades, robust and oftentimes automated processes are now commonly exploited to generate fine chemical building blocks. The integration of effective inline analysis and purification tools is thereby frequently exploited to achieve effective and reliable flow processes. This perspective article summarizes recent applications of different inline purification techniques such as chromatography, extractions, and crystallization from academic and industrial laboratories. A discussion of the advantages and drawbacks of these tools is provided as a guide to aid researchers in selecting the most appropriate approach for future applications. It is hoped that this perspective contributes to new developments in this field in the context of process and cost efficiency, sustainability and industrial uptake of new flow chemistry tools developed in academia.
Graphical Abstract
Introduction
Continuous flow chemistry is a mature and widely applied platform technology that exploits intrinsic advantages over batch processing such as better heat and mass transfer, improved safety and scalability as well as the potential of reaction telescoping and automation [1,2]. Consequently, researchers from academia and the fine chemical industry have established flow technology as a complementary and oftentimes superior approach to traditional batch mode synthesis [3-5]. Reactor miniaturization thereby renders many benefits including better control over mixing and heat transfer, whilst offering cost effective small footprint setups with a high degree of modularity. Researchers can choose from creating custom-built flow setups that offer flexibility at low cost or exploit standardized flow reactor modules readily available from various vendors. The growing popularity of flow chemistry over the last two decades has led to many developments to streamline important chemical reactions, overcome limitations due to highly unstable intermediates that would otherwise be prohibitive or achieve readily scalable processes suitable for industrial applications [6-9]. In addition, flow chemistry has become the method of choice in modern research areas including photo- [10-13], electrochemistry [14-16], and biocatalysis [17-19] due to the advantageous dimensioning inherent in flow chemistry setups. With further advances in these fields underway, many applications also achieve more sustainable chemical processing by intensifying chemical reactions, reducing solvent and energy consumption, and replacing stoichiometric reagents with light and electricity to drive chemical processes [20-22]. Though all these features are highly desirable, significant efforts are needed to realize powerful and fit-for-purpose flow processes. To aid in controlling these often-complex scenarios chemists and chemical engineers commonly exploit reaction automation [23-25] in combination with various inline analysis techniques to generate data-rich processes with immediate feedback loops [26-30]. Researchers must embrace all available options at the outset of a given project especially if a highly robust flow process that combines several unit operations is targeted [31-33]. This is always a challenging undertaking as decisions on solvents, concentrations and preferred reagents and reaction conditions (e.g., thermal, photo- or electrochemical, biocatalyzed) will inevitably affect the resulting telescoped process. One question which is thereby of great importance is whether inline purification is required to improve the overall process. This is an important question as the removal of impurities and spent reagents typically facilitates subsequent chemical reactions in a telescoped multistep sequence. In addition, the successful integration of effective inline purification tools can mitigate safety risks during work-up (i.e., quenching of left-over reactive species) and reduce cost and time associated with traditional batch mode purifications, especially at larger scale (>100 g) [34-36]. Different approaches for integrating inline purification into flow processes have been reported over the years by scientists from academic and industrial laboratories and this perspective article aims to share these whilst highlighting further opportunities and remaining challenges.
Perspective
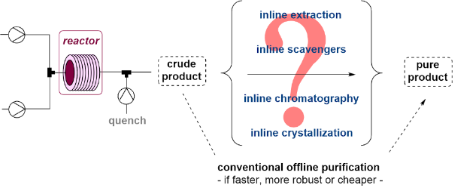
As outlined above, chemists can choose from a variety of tools when developing continuous flow experiments, however, more sophisticated flow processes are more complex in nature and will require additional tools and control points to ensure the success of a given campaign. The integration of inline analysis techniques is often coupled with inline purification tools to remove undesired materials that may hamper a subsequent downstream operation. By now literally all types of purification available for batch mode processing have been implemented in continuous flow mode. In the following sections, this perspective will highlight and discuss selected case studies exploiting SiO2 chromatography, extractive workups and phase separations, scavenger cartridges, precipitations/crystallizations, and filtrations/nanofiltrations.
Inline SiO2 chromatography
Although automated silica gel column chromatography systems are popular and routinely employed in both industry and academia, examples of such systems integrated in continuous flow reactions are scarce. Recently, two protocols were reported that use an Advion puriFlash® 5.250 system for continuous purifications [37,38]. The puriFlash® system is an automated liquid chromatography system for purifying mixtures in a continuous fashion by using alternating sample loops and chromatography columns (Scheme 1).
![[1860-5397-18-182-i1]](/bjoc/content/inline/1860-5397-18-182-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Automated in-line chromatography with the Advion puriFlash® system. The rightmost part of the scheme was adapted from [38], © 2022 K. Donnelly and M. Baumann, published by Beilstein-Institut, distributed under the terms of the Creative Common Attribution 4.0 International License, (https://creativecommons.org/licenses/by/4.0).
Scheme 1: Automated in-line chromatography with the Advion puriFlash® system. The rightmost part of the schem...
This system thus acts as a scale-up preparative HPLC. Two different sample loops are loaded with the crude reaction mixture, and therefore, the crude material is injected into two columns respectively. Automatically, the systems run different methods for sample separation, alternating columns so that one is engaged with the chromatography method while the other is being loaded. Depending on the synthesis flow rate and method duration, the injected volume of crude mixture can be manipulated. Furthermore, additional steps to re-equilibrate the stationary phase and reuse the cartridges are part of this process. The reported examples show the power of this system for different scenarios including photochemical reactions, use of gases, in-line extractions and both normal and reversed-phase chromatography (Table 1).
Table 1: Inline purifications with SiO2-based chromatography.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-182-i16.svg?max-width=637&scale=1.0)
|
||||
| Entry | Conditions | Purification | Outcome | Other observations |
| 1 |
420 nm LED
25–30 °C G–L photochemistry |
inline reversed phase |
82% yield
97.2% purity 0.23 mmol/h |
53% of photocatalyst recovery,
gas is released after the reactor, no separation of diastereomers achieved |
| 2 |
white LED modules
G–L heterogenous photochemistry |
inline reversed phase |
23% yield
>99% purity 0.18 mmol/h |
the presence of isopropanol as solvent did not afford good separation in normal phase,
gas is released after the reactor |
| 3 | 180 °C, 10 mL reactor coil + 10 mL coil for quench mixing | inline normal phase |
a) 37%, b) 28% yield
a), b) >99% purity a) 9.9 mmol/h, b) 7.6 mmol/h |
inline quench with L–L phase separation,
molecular sieves included before the purification system to avoid water traces, gram scale tested |
| 4 | heterogeneous K2CO3 at 100 °C | inline normal phase |
67–82% yield
1–2 mmol scale |
inline quench with L–L phase separation,
1–2 mmol scale, concentration needed to be adjusted (from 0.4 M to 0.2 M), short equilibration period between runs was required |
Optimization of conditions regarding the purification step is needed such as adjusting the concentration of the crude mixture, or the addition of a drying cartridge with molecular sieves. At the end, products with excellent purity are obtained (97–99% by LCMS). Although the productivity using these systems is relatively high and the target product is obtained after simple evaporation of the eluent, the purification step is the rate-limiting step of the overall process.
Besides obtaining the desired product in high purity and the wide variety of products that can be purified by SiO2 column chromatography, other noteworthy advantages are the possibility of isolating side products that may aid in mechanistic studies, and the recyclability and robustness of the SiO2 columns. One of the main issues that can be found with this purification technique is that prior manipulations are often needed to overcome purification problems such as solvent incompatibility, fouling of the column, removal of particulates or the elution of highly polar contaminants.
Further challenges may arise from fast flow reactions (tres < 1 min), when very dilute solutions are used, or if co-polar species are encountered. Under these circumstances the use of automated off-line chromatography systems may be a more advisable path to success. However, as more studies of these systems will be published for continuous flow operations, remaining bottlenecks will be overcome, and lower equipment cost will lead to an increasing number of users in academic and industrial laboratories where rapid delivery of high-quality samples of advanced structures is pivotal. At the same time, applications of these inline chromatography systems for the scaled synthesis of building blocks (>100 g) seems less likely. Reasons for this would be the need to readjust chromatography conditions and pre-treat crude samples by filtration and dilution. At this scale processes tend to be robust enough to facilitate product purification by trituration or recrystallization, however, under certain circumstances inline chromatography may still be a valuable tool that is competitive with other methods, i.e., for lipophilic compounds that cannot be crystallized easily. Additional reports on utilizing chromatography-like systems for in line purifications showcase further potential application areas for these tools (Table 2).
Table 2: Inline purifications using alternative chromatography techniques.
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-182-i17.svg?max-width=637&scale=1.0)
|
||||
| Entry | Technology | Purification | Outcome | Other observations |
| 1 |
SMB:
simulating bed chromatography |
Counter current between mobile and stationary phase. Four zones of columns connected end to end in series. Feed and eluent enter the system in opposite ways |
89% yield in steady state
99% purity |
Product has to be the first or last to elute,
limited to binary separations, crystallization was needed for artemisinin |
| 2 | capture-SMB technology | Countercurrent movement between mobile and stationary phase. Two twin interconnected columns (C18). Breakthrough curves of product-resin binding affinity. Two phases of purification: |
a) 81%, b) 71%, c) 93% yield
a) >99%, b) 92%, c)93% purity a) 22.7 g/hL, b) 7.1 g/hL, c) 7.5 g/hL |
Higher resin utilization and process efficiency; Columns are regenerated through the process,
less solvent use; simple reactions with known side products |
| 3 |
CFC:
continuous centrifugal chromatography |
Quasi continuous. Two immiscible liquid phases (one stationary, one mobile)
Inside a rotor with centrifugal forces |
59% yield
99.9% purity 2.27 g/ h |
Allows separations of by-products,
no need for column replacement/recycling, applicable to industrial process, inline extraction included |
| 4 |
SFC:
supercritical fluid chromatography |
Multiple DEAP (diethylaminopropyl polymer) columns in parallel with UV–vis detector with six-position valve to collect samples |
97% recovery from crude
>99% purity |
Semicontinuous approach, easier to work offline,
inline infrared detector before entering the SFC, CO2 supercritical conditions: harsh and non-anhydrous |
For instance, Seeberger and co-workers reported a simulated moving-bed (SMB) chromatography module based on a series of columns [39,40] for binary separations where the desired product is either the first or last to elute (Table 2, entry 1). A counter-current between the mobile and stationary phases is simulated by periodically shifting the two inlet and outlet ports in the direction of the mobile phase flow after a certain time. This technique was also applied to isolate artemisinin from a continuously produced reaction mixture. However, an offline crystallization had to be added to obtain the pure compound that met the requirements (>99.5%) [41]. Another very recent report which couples a Suzuki–Miyaura reaction with inline purification, uses capture-SMB technology as its purification approach [42] (Table 2, entry 2). The crude material is directed into two twin C-18 columns that work in a counter-current chromatography-based protocol, i.e., one is being loaded as the other one is being washed. Breakthrough curves of the affinity of the product to the resin are needed to optimize the process. The authors demonstrated this process for three non-complex compounds resulting in high purity and productivity (22.7 gh−1L−1 vs 17.2 gh−1L−1) with reduced solvent consumption (0.3 L gh−1L−1 vs 1.4 gh−1L−1) when compared to batch purification. Automation of the process is also described to avoid offline operations.
Örkényi and co-workers reported a centrifugal partition chromatography system (CFC) [43,44] (Table 1, entry 3). Here, the system is based on a quasi-continuous multiple dual mode that uses buffer flasks and instead of pumps two non-miscible phases are employed; one of them acts as the mobile phase and the other one as the stationary phase, which is maintained inside the rotating column by centrifugal forces. This technique resembles a liquid–liquid preparative chromatography technique, where both the stationary and mobile phases are liquids, and separation is based on the partitioning of the solutes between the immiscible liquid phases. The crude product solution is introduced in one phase and is pumped into the mobile phase which is contained inside a rotor by a strong centrifugal force. Finally, the product is obtained with high purity (>99% by GCMS), and the impurities are discarded.
Furthermore, Ley and co-workers reported a supercritical fluid chromatography system which utilizes multiple columns in parallel with a series of valves to stagger the injection of crude material to each of the column lines. The columns are filled with a diethylaminopropyl-functionalized polymer, followed by an UV–vis detector to identify and separate the different peaks. As different injection valves are used, a series of columns can be employed for a continuous stream of crude material being purified. The system was applied to separate an intermediate during the synthesis of isoniazid [45] (Table 2, entry 4).
In the field of biocatalysis, Raston, Weiss and co-workers developed a vortex fluidic device with an immobilized metal affinity chromatography (IMAC) resin to purify proteins in continuous flow mode [46]. This technology is highly efficient (run time ca. 10 minutes), reproducible, and scalable (stable over 5 days of processing) for soluble proteins containing a fused polyhistidine tag.
The preceding examples clearly showcase the diversity of applications that have been developed by academic laboratories to enable a particular chromatographic separation. While early versions may still be complex and not trialed by many users outside these groups, it is expected that future refinements will lead to more robust and frequently exploited technologies.
Inline extractions/separations
Inline extractions are the most common type of workup performed in both industry and academia. They can be divided into two groups: those performed with semipermeable membranes (e.g., Zaiput technology) and those based on bespoke gravity separators.
Since its initial description and commercialization in 2012 [47,48] numerous publications have showcased the utility and robustness of using semipermeable membranes for the separation of immiscible liquid–liquid phases. Most of the examples involve the separation of an organic phase from an aqueous one. However, two immiscible organic phases might be separated, like in the case of methanol and dodecane [49]. Solvent properties such as viscosity, interfacial tension, or salt concentrations in case of aqueous mixtures are some factors to consider [50]. In addition, these membranes are available in different sizes, and they can operate in series to improve separation. Some examples using them in a counter-current way and their application as solvent swap technology are also discussed below.
Though examples of using membrane-based inline extractions as the final purification step are rare, many applications exploit this approach as part of telescoped flow syntheses. This clearly shows the value of this approach for purifying reaction intermediates to remove spent reagents and unreacted reactants or remaining catalysts prior to another step. At this stage it appears that almost all reports use membrane separators at lab scale, which is a limitation of these membranes. Industrial applications often seek to intensify the workup process whereby an aqueous quench might be combined with an extraction, which necessitates robust tools that can handle large quantities of the reaction mixture for prolonged periods of time. Oftentimes companies avail of in-house knowledge for creating suitable extraction modules that can be adjusted from one campaign to another whilst exploiting common features.
Amongst the benefits of membrane-based liquid–liquid separators the ability to gradually adjust the pH of the crude material stands out as an option to enable continuous purification. Though this approach does not apply to all substrates, it allows for tuning of parameters and sequential removal of impurities. Thorough optimization as well as a set of pumps and several membranes are required, and scalability is often limited. In this context, Kappe and co-workers achieved the effective synthesis of methyl oximino acetoacetate using a multistage separation platform, including inline monitoring as depicted in Scheme 2 [51]. Moreover, a high process mass intensity, good space time yield and high product purity were obtained, proving these inline separation techniques can be used as a green and effective purification tool. Previously, Jamison and co-workers performed a total synthesis of atropine in flow using a similar setup [52]. After three sequential liquid–liquid extractions and a passage through a scavenger resin, atropine was obtained in >98% purity, after solvent evaporation as the only off-line operation. This was achieved due to careful pH control that enabled the separation of all byproducts, some of which having very similar structures.
![[1860-5397-18-182-i2]](/bjoc/content/inline/1860-5397-18-182-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Purification via pH tuning and several Zaiput membranes. Redrawn from [51].
Scheme 2: Purification via pH tuning and several Zaiput membranes. Redrawn from [51].
To purify a mixture by tuning its pH, the target compound must be ionizable. Recently, Blacker and co-workers developed an autonomous system for screening liquid–liquid extraction conditions using a Zaiput membrane [53]. The system is monitored by both online and inline analysis to control the acid concentration. Good extraction efficiency and purification were achieved for an amine as a test substrate that was contaminated with a minor impurity. Remaining challenges such as time-consuming optimization and the need for several pumps can be overcome through careful optimization of such autonomous processes.
A desirable feature of these membranes is that they can be applied in syntheses where all the impurities remain in one liquid phase while the desired product resides in the other liquid phase which means that the downstream step consists of offline solvent evaporation only. This is facilitated when using heterogeneous catalysts or immobilized enzymes that are retained in cartridge reactors. For instance, Paradisi and co-workers reported an N-acetylation approach to produce melatonin analogs, where the pure product is obtained in the organic phase after evaporation of the ethyl acetate solvent (used as acetyl donor) whereas the recovered aqueous phase contains the unreacted amine that can be recirculated in the system (Scheme 3) [54].
![[1860-5397-18-182-i3]](/bjoc/content/inline/1860-5397-18-182-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Two-phase recirculating system for purifications of an immobilized enzyme-based reaction. Redrawn from [54].
Scheme 3: Two-phase recirculating system for purifications of an immobilized enzyme-based reaction. Redrawn f...
Amongst the commonly cited challenges of implementing these membranes are their high cost and the perceived difficulties regarding scalability. The first issue is largely a problem for academic researchers where the cost to perform a traditional gravity separation is far lower than the price of these commercial membranes. The latter is a problem for industrial applications, as fouling and related issues may arise for prolonged periods of use that would impede the overall product quality. To solve the scalability problem, larger Zaiput membrane separators are available, however, reports of their successful implementation are so far very scarce. These larger systems were however successfully applied in biphasic oxidations of alcohols using a phase transfer catalyst (PTC) [55]. In a scale-up run, a three-stage counter-current cascade was used downstream of the first separation to remove the PTC (Scheme 4). In this scenario more than 90% of the phase transfer catalyst was separated from the phase containing the desired organic product. In addition, this study reports a reduction of the amount of extraction solvent required, which is a significant improvement compared to the original batch protocol.
![[1860-5397-18-182-i4]](/bjoc/content/inline/1860-5397-18-182-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Countercurrent L–L purification using large Zaiput membranes in the presence of a phase transfer catalyst (PTC). Redrawn from [55].
Scheme 4: Countercurrent L–L purification using large Zaiput membranes in the presence of a phase transfer ca...
The presence of a PTC can become challenging in these separations as recently reported by Kappe and co-workers. This study concerned the separation of an O-alkylation product where a PTC was needed as part of the process prior to the next synthetic step [56]. Interestingly, for small scale examples anisole worked well as a solvent for both telescoped reactions. However, in a long run a gravity separator was favored because of membrane fouling after 1 hour.
An automated fill-empty gravity separator was also used for the separation of phases in a scale up of an N-alkylation reaction reported by Eli-Lilly as an alternative to the Zaiput membranes that were better used for rapid screening of conditions [57].
The use of these membranes in telescoping reactions is a more widespread, accessible, and easier to use approach for both industry and academia. These are utilized to separate the organic phase (containing the desired product) from the aqueous phase that might be used to quench the reaction or to remove any reagent/byproduct as shown in Scheme 5. Finally, the desired product is purified offline, but telescoping two or more steps is only possible because of the initial phase separation. This saves time, money and presents a greener alternative, without the need for intermediate manipulations. What is more, as the final purification is offline, a high purity product is obtained without an extensive study of isolation conditions or expensive and specialized equipment. This approach is not unique but rather it can be observed in many published examples, such as the syntheses of lomustine [58], buclizine derivatives [59], the 3-step synthesis of captopril [60], or atazanavir [61]. In the last example, the authors solve a clogging issue of the membrane by using a guard column ahead of the extraction unit.
![[1860-5397-18-182-i5]](/bjoc/content/inline/1860-5397-18-182-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: General scheme of a telescoped flow process using L–L separators.
Scheme 5: General scheme of a telescoped flow process using L–L separators.
A dual application of aqueous quenching in addition to solvent swap is an interesting implementation of these membranes in reactions with water-miscible solvents. Leadbeater and co-workers reported a Grignard reaction in THF, whereby an acidic aqueous phase and a mixture of DCM and hexanes were added to the mixture before phase separation with a Zaiput membrane [62]. The aqueous phase was used to quench the mixture and to dissolve the magnesium salts, and the organic mixture was the appropriate solvent for the next step.
Recently, a regenerated cellulose membrane to purify micelles via inline dialysis was reported by Junkers and co-workers [63]. This semicontinuous closed-loop setup can remove the organic solvent (THF) to a concentration of less than 1% within four hours, and water-soluble monomers. In addition, the integration of this setup into an inline synthesis step starting from block copolymer solutions, and rhodamine encapsulation produced micelles without affecting the particle size is also demonstrated.
The growing uptake of continuous flow synthesis can also be seen in the development of less frequently exploited extraction technologies. This is the case for a multi-jet oscillating disc reactor (MJOD) [64], where a continuous flow iodination reaction was coupled with a telescoped continuous flow work-up section, which was assembled by a series of hold-up tanks and two MJOD flow reactors as liquid–liquid extractors. Finally, a semicontinuous step for solvent evaporation produced multigram quantities of product with good productivity and purity. Other bespoke alternatives that still need to pass further applications by synthetic chemists include porous capillary separators to separate different mixtures of organic and aqueous solvents [65], a concentric annular liquid–liquid phase separation approach for continuous processing [66] and a single-stage extraction device based on a vertical apparatus with reservoirs where each phase enters from the opposite side in relation to density [67].
A somewhat different approach for phase separation has been presented in several studies by Ley and co-workers [68-71]. The authors developed a ‘computer-vision’ approach, where a computer program monitors the interface level between the organic and aqueous phase of a vertical cylinder using a simple webcam setup. Furthermore, the different pumps are controlled by the system to keep the interface in the defined limits. As a consequence, both phases are pumped separately either to collect for offline solvent evaporation, to waste, if it is the undesired phase, or to a further telescoped step. One example of this approach to separate hydrazones in dichloromethane after the aqueous quench is depicted in Scheme 6. This setup, although widely used in the above-mentioned group, has not been reported by other groups or in industrial applications which may indicate an initial barrier of transferring such academic solutions from one group to another.
![[1860-5397-18-182-i6]](/bjoc/content/inline/1860-5397-18-182-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Example of phase separation using a computer-vision approach. Redrawn from [68].
Scheme 6: Example of phase separation using a computer-vision approach. Redrawn from [68].
Heterogeneous scavenging
The versatility of flow technology allows researchers to use different immobilized reagents/catalysts in cartridges to perform reactions in a heterogeneous fashion. This oftentimes leads to improved green credentials, safety, and sometimes efficiency, especially when it is applied to the downstream process. Commercially available resins, activated charcoal, polymer-supported species that trap spent reagents or side-products as well as related techniques are discussed in the following section.
A comprehensive book chapter which details different heterogenous reagents and scavengers, with their applications, limitations and general basic considerations was recently published by Ley and co-workers [72]. To achieve the isolation of a pure product using only inline heterogeneous cartridges, a sequence of different scavengers and immobilized reagents are needed. What is more, these columns can foul easily and need to be replaced or regenerated in scaled applications, which is expensive and might require manual intervention. Furthermore, this kind of purification cannot differentiate similar compounds if these contain the same functional groups. An advantage, however, is that this purification technique is easily coupled with heterogeneous catalysis. As such immobilized reagents and scavengers are more suited to catalytic processes than stoichiometric transformations.
Many interesting examples have been reported by the Ley group [73,74]. Highlights include the early report of synthesizing of 4,5-disubstituted oxazoles, which includes a protocol to recycle columns. In this case, a packed cartridge containing a base is used: PS-BEMP (tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine on polystyrene) which is valuable for reaction scale-ups [75] is used. Alternatively, a CuAAc (copper-catalyzed azide–alkyne cycloaddition) reaction has been demonstrated where the copper catalyst is supported on an Amberlist A-21 resin, which also improves safety issues as it traps toxic and explosive reactive intermediates (Scheme 7) [76]. Additional studies include a 3-step reaction to form triazoles in good yields [77], and the synthesis of the bisoxazole natural product siphonazole A using immobilized species [78]. The use of real-time IR and automated pump control to facilitate reagent addition and column robustness have been demonstrated in the synthesis of pyrazoles and other valuable building blocks [79].
![[1860-5397-18-182-i7]](/bjoc/content/inline/1860-5397-18-182-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: Example of an inline purification using heterogeneous scavenging. Redrawn from [76].
Scheme 7: Example of an inline purification using heterogeneous scavenging. Redrawn from [76].
In the same fashion as inline phase separations, the use of such cartridges is often considered to remove a reagent/catalyst/byproduct to telescope several reactions before performing the final purification step offline (Scheme 8) [80-83]. Notable applications of metal scavengers such as QuadraPure® resins involve the removal of leached metals as well as catalytic amounts of homogeneous metal catalysts as reported by Pitts and collaborators. This study achieves full removal of metal species after common homogenous catalytic reactions such as a Suzuki–Miyaura reaction, Sonogashira reaction or hydrogenation mediated by Wilkinson’s catalyst [84]. Other interesting examples to remove transition metals in continuous flow synthesis focus on palladium [85], cobalt [86], or copper (particularly useful for the widely used CuAAc) [87].
![[1860-5397-18-182-i8]](/bjoc/content/inline/1860-5397-18-182-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: General scheme of a telescoped process using heterogenous cartridges.
Scheme 8: General scheme of a telescoped process using heterogenous cartridges.
Nevertheless, the use of metal scavengers in large scale applications is limited as often discussed [88]. The use of a homogeneous scavenger as part of batch-based offline purifications can be an easier and more effective alternative. An efficient method to homogeneously scavenge a ruthenium complex used in a metathesis reaction was described by Grela and co-workers on a 60 g scale [89].
The use of heterogeneous scavenger columns in flow mode is sometimes criticized. If the final purification is offline, the use of cartridges can be perceived as an unnecessary use of resources. For example, this is the case in the flow synthesis of imatinib and its analogues, where several groups have demonstrated valuable syntheses. A report by the Ley group utilizes a series of cartridges to facilitate the overall process via several innovative and creative approaches to generate small amounts of novel analogs [90,91]. Nevertheless, an alternative 3-step telescoped synthesis with offline purification seems to be the simplest, most efficient, and industrially favorable approach [92] (Scheme 9). In another case, the authors opt to use stoichiometric quantities of reagents to avoid the use of cartridges, even if the performance is worse, to reduce cost and time [93].
![[1860-5397-18-182-i9]](/bjoc/content/inline/1860-5397-18-182-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Comparison of two strategies for flow-based imatinib syntheses. Redrawn from [91] and [92].
Scheme 9: Comparison of two strategies for flow-based imatinib syntheses. Redrawn from [91] and [92].
Catch and release strategy
An interesting approach regarding the use of resins is their application in catch and release strategies, where the product is temporarily trapped in a cartridge and then released in pure form using other conditions. Besides the common limitations previously mentioned for heterogeneous flow processes, it has the drawback that the solvent exchange may need to be performed manually and that the impurity might also contain the same functional group that has affinity to the resin (Scheme 10).
![[1860-5397-18-182-i10]](/bjoc/content/inline/1860-5397-18-182-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: General purification scheme using the catch and release strategy.
Scheme 10: General purification scheme using the catch and release strategy.
QP-SA is a commonly used resin which contains –SO3H groups and can sequester amines, whereby a solution of ammonia in MeOH can be used to release the product [94]. Amine-functionalized resins like A-900 can be used to trap carboxylic acids, from which the product can be released with diluted solutions of formic acid [95]. Another strategy that is applicable in the context of biocatalysis involves Ni–NTA resins used for protein purification which are commonly packed in cartridges to enable automated peptide purification via a catch and release strategy [96]. Additionally, functionalized residues can be used to perform a solid-state synthesis approach, such as in the synthesis of 2-aminopyridine derivatives starting from amino-iminium residues [97].
To improve on this technology, Baxendale and co-workers utilized an arrangement of ten columns with an automated system to facilitate solvent switching and column loading [98]. As the product is being trapped in one column, a secondary HPLC pump, which is monitored online, regenerates the previous column. However, in the example described, final offline purification was necessary. The authors also discuss an alternative route where only some heterogeneous scavengers are needed to remove impurities, being a more industrially feasible alternative.
One important application of this downstream strategy is in combination with flow biocatalysis. These reactions are generally slow but very specific with regards to functional groups present in complex molecular structures. In this context, some of the limitations of these transformations and the effective use of columns and liquid–liquid inline separations can be overcome by integrating them in continuous flow. These advantages can be seen in the works of Conti and Paradisi [99-104]. By using different cartridges to trap the desired product, the excess of reagents and starting materials can be recirculated thus improving efficiency and product formation. In this case, continuous synthesis offers an excellent route for the efficient and cost-effective preparation of different building blocks including nucleoside derivatives and ʟ-pipecolic acid. High efficiency was achieved with simple trapping columns downstream of the biocatalytic process, to separate the pure products from the mixture and recirculate the aqueous phase in a closed-loop system. An example for obtaining optically pure alcohols is depicted in Scheme 11 [105].
![[1860-5397-18-182-i11]](/bjoc/content/inline/1860-5397-18-182-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Exemplar catch and release purification of a stereoselective oxidation. Redrawn from [105].
Scheme 11: Exemplar catch and release purification of a stereoselective oxidation. Redrawn from [105].
The use of flow biocatalysis can also improve the purification of crude mixtures. This was recently demonstrated in a collaboration between Almac Sciences and the Baumann group where an immobilized lipase (e.g., CALB) facilitated the derivatization of high-boiling benzyl alcohol in scaled Curtius rearrangement reactions. Ultimately, this approach negated the use of column chromatography in favor of a simple trituration process to isolate pure carbamate products [106].
Another example for using this catch and release strategy would be when the product is trapped on silica due to its polarity prior to its release with a more polar solvent. This case is reported in the synthesis of iloperidone (Scheme 12) [107]. Though limited to applications with highly polar products, this strategy is attractive for rapid small-scale syntheses of such scaffolds and the optimization of the used solvents can broaden the uptake of this strategy.
![[1860-5397-18-182-i12]](/bjoc/content/inline/1860-5397-18-182-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Catch and release-type purification using conventional SiO2. Redrawn from [107].
Scheme 12: Catch and release-type purification using conventional SiO2. Redrawn from [107].
Inline precipitation and crystallization
One of the most widely acknowledged challenges within the field of flow chemistry is the necessity to operate under homogeneous conditions within the thin tubing that makes up the flow reactor components. The handling of slurries whether because of unintended precipitation or as a deliberate means to process insoluble materials presents the risk of clogging and pressure built-up which can severely compromise a flow run. Therefore, continuous flow crystallization and precipitation for the purification of a flow reaction mixture is not often reported and many of the protocols use a semi-continuous approach for instance via continuous stirred tank reactors to purify APIs. Wu and co-workers recently presented a detailed review on continuous crystallizations of pharmaceutical compounds which discusses these approaches [108].
It is in the field of crystallization where the biggest gap between industry and academia is visible. While industry has large downstream platforms to purify APIs, academia is at a very early stage for integrated purifications. Good examples from industry are Eli-Lilly's manufacturing platform used for the generation of a novel API under cGMP conditions currently in clinical trials (Scheme 13) [109], Novartis' report on the synthesis, purification, and final dosage preparation of aliskiren [110,111], or related examples of upstream and downstream processing of different APIs, such as lidocaine, diazepam, or diphenhydramine [29,30,112].
![[1860-5397-18-182-i13]](/bjoc/content/inline/1860-5397-18-182-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Schematic representation of an industrial continuous crystallization. Redrawn from [109].
Scheme 13: Schematic representation of an industrial continuous crystallization. Redrawn from [109].
Apart from the typical problems of handling solids such as fouling and blockages, polymorphism can be another challenge. Substantial knowledge for batch mode crystallization is available which requires a detailed understanding on the best solvent system, temperature gradient, and seeding point. Though these aspects can be reproduced potentially with higher control in flow mode, appropriate process control and steady state conditions are needed to successfully obtain a given polymorph. The work of Myerson and co-workers demonstrates this by examining polymorph dynamics to achieve a successful crystallization of ʟ-glutamic acid using a continuous mixed suspension mixed product removal crystallization (MSMPR) approach [113].
Recently, a kinetically regulated automated input crystallizer (KRAIC) was reported [114]. This segmented flow reactor consists of a controlled temperature gradient reactor where the reactor inlet is heated while the outlet is actively cooled. Temperature gradient curves can be generated by varying the inlet/outlet temperatures and the flow rate of the solution, allowing for conditions to be created to best approximate the ideal growth curve through the metastable zone (where controlled crystal growth occurs). Single crystals of paracetamol were obtained to prove this crystallization method.
The application of inline crystallizations or precipitation coupled to an organic reaction is uncommon and limited to certain substrates and/or reactor types. Furthermore, offline operations are often needed, as well as an in-depth solvent study to avoid reactor clogging. Some recent reports that use a crystallization as a purification technique after a synthetic transformation are highlighted in the following section (Scheme 14, Table 3).
![[1860-5397-18-182-i14]](/bjoc/content/inline/1860-5397-18-182-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: General scheme of an academic inline crystallization approach.
Scheme 14: General scheme of an academic inline crystallization approach.
Table 3: Crystallizations in continuous flow coupled to an organic reaction.
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-182-i18.svg?max-width=637&scale=1.0)
|
||||
| Entry | Reaction | Antisolvent | Purification | Other observations |
| 1 | MnO2-packed bed reactor, 80 °C | Air + carrier fluid (perfluoropolyether) mixed at 70 °C |
Trisegmented tubular crystallizer,
jacked tube at 10 °C and 3 FEP coils |
Control over crystal size and polymorphism,
rapid and robust 53% yield |
| 2 | Chemtrix reactor chip; 200 °C |
EtOAc
N2 |
Air-segmented flow crystallizer; −20 °C
PFA tubing |
Online MS reaction and crystal formation monitoring; 128 mg/h |
| 3 | Two PFA coils | MeOH/H2O | Mixed-suspension mixed-product-removal. Two vessels (50 °C and 25 °C) |
Avoids the degradation of the product in crude solution; scalable,
95% yield, 99.8% purity |
| 4 | Taylor vortex flow reactor; 70 °C | MeOH/H2O | Taylor vortex flow reactor; 5 °C |
Inline quench with phase separation,
scalable, 88% yield, 99.9% purity |
| 5 | Microreactor packed with Pd/Sn/Ce oxide catalyst; 90 °C | H2O | Two coiled FEP reactors at specific temperature (from 8 °C to 80 °C) in ultrasonic bath |
67–96% yield,
151–204 mg/h, scalable; no metal traces in products |
A continuous flow crystallization was performed using a trisegmented tubular crystallizer (KRAIC) coupled with a catalytic hydration of pyrazine carbonitrile [115]. After passing the reaction setup, the crude product is mixed with a carrier solvent and an air flow to generate a gas–liquid flow pattern. This is then directed into three different coils held at different temperatures where the solid product precipitates in a controlled manner prior to isolation by filtration. As the same solvent is used during the reaction and the crystallization stage, issues with incompatibilities are avoided. Despite optimizing the crystallization approach the authors mention a tendency of reactor blockages.
Hosoya and co-workers utilized a Taylor vortex flow reactor for continuous crystallizations coupled to an O-alkylation reaction which includes quenching, phase separation, and final filtration. After a thorough study of conditions to avoid clogging, a long run processing >100 g was achieved, giving the product in 95% yield and >99% purity [116]. The same authors also reported the crystallization of a simple dipeptide with the aim to overcome issues found in batch in relation to product stability and scale-up concerns. The peptide coupling reaction was performed continuously aided by a MSMPR crystallizer system. This system ultimately shortened the holding time for the quenched solution, leading to a robust crystallization procedure regardless of the manufacturing scale [117].
An air-segmented flow crystallizer was developed by Cooks and co-workers for the synthesis and purification of diphenhydramine [118]. In this study ethyl acetate was used as antisolvent and careful control of nitrogen flow (used to obtain segmented droplets) and the temperature were needed to obtain suitable crystals without reactor fouling. The droplets and crystals were monitored using an inline phototransistor and a video microscope. Finally, crystals were collected by filtration.
In another protocol, a continuous crystallization was successfully coupled to a Suzuki–Miyaura reaction [119]. Crystallization was identified as a suitable purification technique to separate products from unreacted starting materials while avoiding metal contamination. Water was used as antisolvent and two mixing units were needed in the crystallization process, each of them at a different temperature depending on the example (8 °C to 80 °C) in an ultrasonic bath.
In the field of flow biocatalysis, von Langermann and co-workers reported a three-part vessel system for the continuous synthesis and crystallization of (S)-1-(3-methoxyphenyl)ethylamine [120]. The system consists of a membrane reactor for the enzyme-mediated synthesis, a saturator vessel for the continuous supply of the amine donor, and a crystallizer where the desired product precipitates.
Flow reactions can be performed as neat reactions in certain circumstances allowing for the precipitation of the desired compound upon adding an appropriate solvent at the outlet of the reactor. In this case, the only manual interventions are the filtration and the washing of the solid. As the reaction is performed at elevated temperature there is reduced risk of clogging inside the reactor. These features can be observed in the flow syntheses of rufinamide [121] and diphenhydramine hydrochloride [122]. These protocols are notable for being green, atom efficient, and inexpensive due to the reduction in solvent use. Another similar scenario that is not fully explored relates to flow processes at elevated temperatures and high concentrations, followed by product precipitation upon cooling. This phenomenon is observed in the synthesis of (1H-imidazol-yl)cycloalkanols [123]. However, the authors preferred an offline chromatography step to obtain the pure product, but this precipitation approach can likely be explored for industrial interest.
Filtration and nanofiltration
Inline filtration devices can be coupled to other technologies to render a better overall purification performance. Working with continuous flow, the presence of solids is not recommended as precipitates or slurries can cause clogging or ineffective performance of pumps. A rotating sintered glass filter to remove solids before an inline separation was reported by Ley and co-workers [69]. In the same publication, a charcoal scavenger cartridge also acts as a particle filter. The incorporation of filtering membranes before chromatography is a solution to avoid particles in the chromatography system [37,124].
In relation to nanofiltration, several devices equipped with specific membranes have been reported. Hessel and co-workers published a detailed review about inline recycling and separation of homogeneous catalysts, which contains a detailed section on filtering metal catalysts [88]. One representative example was reported by Jensen and co-workers. The authors developed a nanofiltration device capable to retain and recycle the Hoveyda–Grubbs catalyst [125]. Interestingly, another vacuum Teflon membrane is used to remove the ethylene that is produced as byproduct in the metathesis reaction. Some subsequent publications include the Pd-complex removal after a Suzuki coupling [126], a Heck catalyst retention with reverse boiling-point-order solvent exchange [127], and a membrane for recovery of the photocatalyst TBADT (tetrabutylammonium decatungstate) [128]. Organic solvent inline nanofiltration is an interesting green approach for solvent recovery that also concentrates, and thus improves the purity of the target compounds. However, this technology is so far only applied in selected examples, such as a Michael addition [129], with continuous extraction of antioxidants from olive leaf waste [130], and of patchoulol from green algal cells [49]. In the last case, nanofiltration is coupled to an inline phase separation to both concentrate the compound and reuse the solvent. In another scenario, a two-stage membrane cascade was used as a purification method of roxithromycin from 4-dimethylaminopyridine [131].
On the other hand, vacuum-assisted filtrations for the inline collection and drying of the crystallized solid are rare, but also described. A representative example is the inline drying and filtration of ciprofloxacin [132] in a specific compact downstream device, which can be coupled with an inline crystallization module [133]. Another example is a vacuum-assisted rotating porous plate to aid filtering the slurry material obtained from the downstream module in the production of aliskiren [110]. However, the majority of the continuous crystallization procedures requires manual offline intervention to filter and dry the solid, and the reported inline filtrations require very specific industrial devices.
Summary and Outlook
As highlighted in this perspective, many different inline techniques and accompanying flow tools are available to enable the purification of reaction mixtures generated by various continuous transformations. Amongst these, continuous inline extractions are most widely exploited in both academic and industrial laboratories. In addition to liquid–liquid extractions, several studies exploit heterogeneous reagents and scavengers to facilitate liquid–solid separations inline. This approach has limitations when stoichiometric quantities of reagents or leftover reactants are to be removed, however, in case of catalytic amounts such scavengers are highly effective and widely applicable. The immobilization of catalysts including enzymes provides for robust flow protocols that avoid the need of separating the potentially unstable catalytic species from the reaction mixture. Inline filtration is a good option to remove small particles or specific metal catalysts. Organic solvent nanofiltration is not yet highly explored, but it could be a good alternative to remove and recycle solvents and highly valuable catalysts. However, the specificity of the membranes and the conditions required suggest that a general implementation in a flow process is not yet achieved. If small amounts of high-quality products are desired in an automated manner, inline column chromatography can be exploited, and several recent studies provide successful reports of implementing related tools. All these approaches have in common that diligent optimizations are required before a robust and reliable inline purification method is achieved. Though this is often accepted in academic case studies, industrial flow applications may only select inline purifications if they are critical, i.e., in case an inline quench can be coupled with a continuous extraction or a precious metal catalyst can be recycled. Final purification of the crude mixture can then be achieved using offline technologies such as HPLC or crystallizations. As such, optimization time and acceptance of new methods can be seen as reasons for non-uniform uptake of many inline purification tools amongst flow chemists in industrial settings. This may be compounded in flow campaigns where large quantities of drug-like structures are produced. In this scenario simple yet robust purification and isolation methods are needed that will include techniques such as crystallizations and precipitations that have thus gained traction amongst flow chemists. The relatively high cost and a lack of familiarity with new inline purification methods can be seen as initial obstacles for using tools developed in academic labs in industrial settings. However, a number of examples including commercially available membrane separators show that industry-based flow chemists use new tools if they suit a given timeline and process scale. At this stage, several decisions will determine if inline purification is used, and which tool is chosen. Besides experience, cost and timelines the availability and suitability of alternative offline purification options is crucial. As demonstrated in this perspective, it is possible to design effective multistep flow protocols that do not require inline purification tools and final offline purification can be achieved in a process-dependent fashion. As shown in the graphic below (Scheme 15), several factors and individual preferences impact the decision and choice of suitable options.
![[1860-5397-18-182-i15]](/bjoc/content/inline/1860-5397-18-182-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Simplified overview of purification options and selected criteria.
Scheme 15: Simplified overview of purification options and selected criteria.
It is, however, anticipated that with the advent of more sophisticated and robust automation tools the field of flow chemistry will continue to embrace inline purification as a valuable approach to remove incompatible species for subsequent steps or scavenge precious metals or biocatalysts in view of recycling. This will likely see applications for generating small volume drugs or other high value entities where fast access to a high quality product is key, or where the isolation and handling of intermediates is not viable due to their properties. Thus, it can be concluded that many opportunities remain for improved and widely applicable inline purification tools where interdisciplinary solutions will impact on the robustness and sustainability of future flow campaigns.
References
-
Gutmann, B.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed. 2015, 54, 6688–6728. doi:10.1002/anie.201409318
Return to citation in text: [1] -
Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183
Return to citation in text: [1] -
Porta, R.; Benaglia, M.; Puglisi, A. Org. Process Res. Dev. 2016, 20, 2–25. doi:10.1021/acs.oprd.5b00325
Return to citation in text: [1] -
García‐Lacuna, J.; Domínguez, G.; Pérez‐Castells, J. ChemSusChem 2020, 13, 5138–5163. doi:10.1002/cssc.202001372
Return to citation in text: [1] -
Colella, M.; Nagaki, A.; Luisi, R. Chem. – Eur. J. 2020, 26, 19–32. doi:10.1002/chem.201903353
Return to citation in text: [1] -
Movsisyan, M.; Delbeke, E. I. P.; Berton, J. K. E. T.; Battilocchio, C.; Ley, S. V.; Stevens, C. V. Chem. Soc. Rev. 2016, 45, 4892–4928. doi:10.1039/c5cs00902b
Return to citation in text: [1] -
Bogdan, A. R.; Dombrowski, A. W. J. Med. Chem. 2019, 62, 6422–6468. doi:10.1021/acs.jmedchem.8b01760
Return to citation in text: [1] -
Baumann, M.; Moody, T. S.; Smyth, M.; Wharry, S. Org. Process Res. Dev. 2020, 24, 1802–1813. doi:10.1021/acs.oprd.9b00524
Return to citation in text: [1] -
Domokos, A.; Nagy, B.; Szilágyi, B.; Marosi, G.; Nagy, Z. K. Org. Process Res. Dev. 2021, 25, 721–739. doi:10.1021/acs.oprd.0c00504
Return to citation in text: [1] -
Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707
Return to citation in text: [1] -
Politano, F.; Oksdath-Mansilla, G. Org. Process Res. Dev. 2018, 22, 1045–1062. doi:10.1021/acs.oprd.8b00213
Return to citation in text: [1] -
Sambiagio, C.; Noël, T. Trends Chem. 2020, 2, 92–106. doi:10.1016/j.trechm.2019.09.003
Return to citation in text: [1] -
Di Filippo, M.; Bracken, C.; Baumann, M. Molecules 2020, 25, 356. doi:10.3390/molecules25020356
Return to citation in text: [1] -
Atobe, M.; Tateno, H.; Matsumura, Y. Chem. Rev. 2018, 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353
Return to citation in text: [1] -
Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412
Return to citation in text: [1] -
Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497
Return to citation in text: [1] -
De Santis, P.; Meyer, L.-E.; Kara, S. React. Chem. Eng. 2020, 5, 2155–2184. doi:10.1039/d0re00335b
Return to citation in text: [1] -
Benítez-Mateos, A. I.; Contente, M. L.; Roura Padrosa, D.; Paradisi, F. React. Chem. Eng. 2021, 6, 599–611. doi:10.1039/d0re00483a
Return to citation in text: [1] -
Coloma, J.; Guiavarc’h, Y.; Hagedoorn, P.-L.; Hanefeld, U. Chem. Commun. 2021, 57, 11416–11428. doi:10.1039/d1cc04315c
Return to citation in text: [1] -
Newman, S. G.; Jensen, K. F. Green Chem. 2013, 15, 1456–1472. doi:10.1039/c3gc40374b
Return to citation in text: [1] -
Dallinger, D.; Kappe, C. O. Curr. Opin. Green Sustainable Chem. 2017, 7, 6–12. doi:10.1016/j.cogsc.2017.06.003
Return to citation in text: [1] -
Baumann, M.; Moody, T.; Smyth, M.; Wharry, S. Synthesis 2021, 53, 3963–3976. doi:10.1055/a-1541-1761
Return to citation in text: [1] -
Ley, S. V.; Fitzpatrick, D. E.; Ingham, R. J.; Myers, R. M. Angew. Chem., Int. Ed. 2015, 54, 3449–3464. doi:10.1002/anie.201410744
Return to citation in text: [1] -
Shukla, C. A.; Kulkarni, A. A. Beilstein J. Org. Chem. 2017, 13, 960–987. doi:10.3762/bjoc.13.97
Return to citation in text: [1] -
Gioiello, A.; Piccinno, A.; Lozza, A. M.; Cerra, B. J. Med. Chem. 2020, 63, 6624–6647. doi:10.1021/acs.jmedchem.9b01956
Return to citation in text: [1] -
Sans, V.; Cronin, L. Chem. Soc. Rev. 2016, 45, 2032–2043. doi:10.1039/c5cs00793c
Return to citation in text: [1] -
Sagmeister, P.; Williams, J. D.; Hone, C. A.; Kappe, C. O. React. Chem. Eng. 2019, 4, 1571–1578. doi:10.1039/c9re00087a
Return to citation in text: [1] -
Sagmeister, P.; Lebl, R.; Castillo, I.; Rehrl, J.; Kruisz, J.; Sipek, M.; Horn, M.; Sacher, S.; Cantillo, D.; Williams, J. D.; Kappe, C. O. Angew. Chem., Int. Ed. 2021, 60, 8139–8148. doi:10.1002/anie.202016007
Return to citation in text: [1] -
Adamo, A.; Beingessner, R. L.; Behnam, M.; Chen, J.; Jamison, T. F.; Jensen, K. F.; Monbaliu, J.-C. M.; Myerson, A. S.; Revalor, E. M.; Snead, D. R.; Stelzer, T.; Weeranoppanant, N.; Wong, S. Y.; Zhang, P. Science 2016, 352, 61–67. doi:10.1126/science.aaf1337
Return to citation in text: [1] [2] -
Bédard, A.-C.; Adamo, A.; Aroh, K. C.; Russell, M. G.; Bedermann, A. A.; Torosian, J.; Yue, B.; Jensen, K. F.; Jamison, T. F. Science 2018, 361, 1220–1225. doi:10.1126/science.aat0650
Return to citation in text: [1] [2] -
Britton, J.; Raston, C. L. Chem. Soc. Rev. 2017, 46, 1250–1271. doi:10.1039/c6cs00830e
Return to citation in text: [1] -
Britton, J.; Jamison, T. F. Nat. Protoc. 2017, 12, 2423–2446. doi:10.1038/nprot.2017.102
Return to citation in text: [1] -
Sagandira, C. R.; Nqeketo, S.; Mhlana, K.; Sonti, T.; Gaqa, S.; Watts, P. React. Chem. Eng. 2022, 7, 214–244. doi:10.1039/d1re00483b
Return to citation in text: [1] -
Bana, P.; Örkényi, R.; Lövei, K.; Lakó, Á.; Túrós, G. I.; Éles, J.; Faigl, F.; Greiner, I. Bioorg. Med. Chem. 2017, 25, 6180–6189. doi:10.1016/j.bmc.2016.12.046
Return to citation in text: [1] -
Weeranoppanant, N.; Adamo, A. ACS Med. Chem. Lett. 2020, 11, 9–15. doi:10.1021/acsmedchemlett.9b00491
Return to citation in text: [1] -
Meyer, L.-E.; Hobisch, M.; Kara, S. Curr. Opin. Biotechnol. 2022, 78, 102835. doi:10.1016/j.copbio.2022.102835
Return to citation in text: [1] -
Thomson, C. G.; Banks, C.; Allen, M.; Barker, G.; Coxon, C. R.; Lee, A.-L.; Vilela, F. J. Org. Chem. 2021, 86, 14079–14094. doi:10.1021/acs.joc.1c01151
Return to citation in text: [1] [2] -
Donnelly, K.; Baumann, M. Beilstein J. Org. Chem. 2022, 18, 232–239. doi:10.3762/bjoc.18.27
Return to citation in text: [1] [2] -
O'Brien, A. G.; Horváth, Z.; Lévesque, F.; Lee, J. W.; Seidel-Morgenstern, A.; Seeberger, P. H. Angew. Chem., Int. Ed. 2012, 51, 7028–7030. doi:10.1002/anie.201202795
Return to citation in text: [1] -
Lee, J. W.; Horváth, Z.; O’Brien, A. G.; Seeberger, P. H.; Seidel-Morgenstern, A. Chem. Eng. J. 2014, 251, 355–370. doi:10.1016/j.cej.2014.04.043
Return to citation in text: [1] -
Horváth, Z.; Horosanskaia, E.; Lee, J. W.; Lorenz, H.; Gilmore, K.; Seeberger, P. H.; Seidel-Morgenstern, A. Org. Process Res. Dev. 2015, 19, 624–634. doi:10.1021/acs.oprd.5b00048
Return to citation in text: [1] -
Sivo, A.; Kim, T. K.; Ruta, V.; Luisi, R.; Osorio-Tejada, J.; Escriba-Gelonch, M.; Hessel, V.; Sponchioni, M.; Vilé, G. React. Chem. Eng. 2022, 7, 2650–2658. doi:10.1039/d2re00242f
Return to citation in text: [1] -
Örkényi, R.; Éles, J.; Faigl, F.; Vincze, P.; Prechl, A.; Szakács, Z.; Kóti, J.; Greiner, I. Angew. Chem., Int. Ed. 2017, 56, 8742–8745. doi:10.1002/anie.201703852
Return to citation in text: [1] -
Lorántfy, L.; Rutterschmid, D.; Örkényi, R.; Bakonyi, D.; Faragó, J.; Dargó, G.; Könczöl, Á. Org. Process Res. Dev. 2020, 24, 2676–2688. doi:10.1021/acs.oprd.0c00338
Return to citation in text: [1] -
Fitzpatrick, D. E.; Mutton, R. J.; Ley, S. V. React. Chem. Eng. 2018, 3, 799–806. doi:10.1039/c8re00107c
Return to citation in text: [1] -
Britton, J.; Dyer, R. P.; Majumdar, S.; Raston, C. L.; Weiss, G. A. Angew. Chem., Int. Ed. 2017, 56, 2296–2301. doi:10.1002/anie.201610821
Return to citation in text: [1] -
Cervera-Padrell, A. E.; Morthensen, S. T.; Lewandowski, D. J.; Skovby, T.; Kiil, S.; Gernaey, K. V. Org. Process Res. Dev. 2012, 16, 888–900. doi:10.1021/op200242s
Return to citation in text: [1] -
Adamo, A.; Heider, P. L.; Weeranoppanant, N.; Jensen, K. F. Ind. Eng. Chem. Res. 2013, 52, 10802–10808. doi:10.1021/ie401180t
Return to citation in text: [1] -
Overmans, S.; Ignacz, G.; Beke, A. K.; Xu, J.; Saikaly, P. E.; Szekely, G.; Lauersen, K. J. Green Chem. 2022, 24, 5479–5489. doi:10.1039/d2gc00938b
Return to citation in text: [1] [2] -
For further technical details, pictures, and applications of Zaiput membranes, see refs [35,48] and/or visit: https://www.zaiput.com/product/liquid-liquid-gas-separators/
Return to citation in text: [1] -
Lebl, R.; Murray, T.; Adamo, A.; Cantillo, D.; Kappe, C. O. ACS Sustainable Chem. Eng. 2019, 7, 20088–20096. doi:10.1021/acssuschemeng.9b05954
Return to citation in text: [1] [2] -
Dai, C.; Snead, D. R.; Zhang, P.; Jamison, T. F. J. Flow Chem. 2015, 5, 133–138. doi:10.1556/1846.2015.00013
Return to citation in text: [1] -
Power, L. A.; Clayton, A. D.; Reynolds, W. R.; Hose, D. R. J.; Ainsworth, C.; Chamberlain, T. W.; Nguyen, B. N.; Bourne, R. A.; Kapur, N.; Blacker, A. J. React. Chem. Eng. 2021, 6, 1806–1810. doi:10.1039/d1re00205h
Return to citation in text: [1] -
Contente, M. L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Green Chem. 2019, 21, 3263–3266. doi:10.1039/c9gc01374a
Return to citation in text: [1] [2] -
Peer, M.; Weeranoppanant, N.; Adamo, A.; Zhang, Y.; Jensen, K. F. Org. Process Res. Dev. 2016, 20, 1677–1685. doi:10.1021/acs.oprd.6b00234
Return to citation in text: [1] [2] -
García-Lacuna, J.; Fleiß, T.; Munday, R.; Leslie, K.; O’Kearney-McMullan, A.; Hone, C. A.; Kappe, C. O. Org. Process Res. Dev. 2021, 25, 947–959. doi:10.1021/acs.oprd.1c00002
Return to citation in text: [1] -
Nieves-Remacha, M. J.; Torres, M.; Ruiz-Abad, M.; Rincón, J. A.; Cumming, G. R.; Garcia-Losada, P. React. Chem. Eng. 2019, 4, 334–345. doi:10.1039/c8re00203g
Return to citation in text: [1] -
Jaman, Z.; Sobreira, T. J. P.; Mufti, A.; Ferreira, C. R.; Cooks, R. G.; Thompson, D. H. Org. Process Res. Dev. 2019, 23, 334–341. doi:10.1021/acs.oprd.8b00387
Return to citation in text: [1] -
Borukhova, S.; Noël, T.; Hessel, V. ChemSusChem 2016, 9, 67–74. doi:10.1002/cssc.201501367
Return to citation in text: [1] -
De Vitis, V.; Dall'Oglio, F.; Pinto, A.; De Micheli, C.; Molinari, F.; Conti, P.; Romano, D.; Tamborini, L. ChemistryOpen 2017, 6, 668–673. doi:10.1002/open.201700082
Return to citation in text: [1] -
Dalla-Vechia, L.; Reichart, B.; Glasnov, T.; Miranda, L. S. M.; Kappe, C. O.; de Souza, R. O. M. A. Org. Biomol. Chem. 2013, 11, 6806–6813. doi:10.1039/c3ob41464g
Return to citation in text: [1] -
Hamlin, T. A.; Lazarus, G. M. L.; Kelly, C. B.; Leadbeater, N. E. Org. Process Res. Dev. 2014, 18, 1253–1258. doi:10.1021/op500190j
Return to citation in text: [1] -
Verstraete, K.; Buckinx, A.-L.; Zaquen, N.; Junkers, T. Macromolecules 2021, 54, 3865–3872. doi:10.1021/acs.macromol.1c00242
Return to citation in text: [1] -
Drageset, A.; Bjørsvik, H.-R. React. Chem. Eng. 2016, 1, 436–444. doi:10.1039/c6re00091f
Return to citation in text: [1] -
Phillips, T. W.; Bannock, J. H.; deMello, J. C. Lab Chip 2015, 15, 2960–2967. doi:10.1039/c5lc00430f
Return to citation in text: [1] -
Harding, M. J.; Feng, B.; Lopez-Rodriguez, R.; O'Connor, H.; Dowling, D.; Gibson, G.; Girard, K. P.; Ferguson, S. React. Chem. Eng. 2021, 6, 1635–1643. doi:10.1039/d1re00119a
Return to citation in text: [1] -
Day, C.; Saldarriaga, A.; Tilley, M.; Hunter, H.; Organ, M. G.; Wilson, D. J. Org. Process Res. Dev. 2016, 20, 1738–1743. doi:10.1021/acs.oprd.6b00226
Return to citation in text: [1] -
O’Brien, M.; Koos, P.; Browne, D. L.; Ley, S. V. Org. Biomol. Chem. 2012, 10, 7031–7036. doi:10.1039/c2ob25912e
Return to citation in text: [1] [2] -
Ingham, R. J.; Battilocchio, C.; Fitzpatrick, D. E.; Sliwinski, E.; Hawkins, J. M.; Ley, S. V. Angew. Chem., Int. Ed. 2015, 54, 144–148. doi:10.1002/anie.201409356
Return to citation in text: [1] [2] -
Hu, D. X.; O’Brien, M.; Ley, S. V. Org. Lett. 2012, 14, 4246–4249. doi:10.1021/ol301930h
Return to citation in text: [1] -
O'Brien, M.; Cooper, D. A.; Dolan, J. Tetrahedron Lett. 2017, 58, 829–834. doi:10.1016/j.tetlet.2017.01.029
Return to citation in text: [1] -
Ley, S. V.; Browne, D. L.; O’Brien, M. Immobilized reagents and Multistep Processes. In Flow Chemistry in Organic Chemistry; Jamison, T. F.; Koch, G., Eds.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2018; pp 273–312. doi:10.1055/sos-sd-228-00177
Return to citation in text: [1] -
Carter, C. F.; Baxendale, I. R.; Pavey, J. B. J.; Ley, S. V. Org. Biomol. Chem. 2010, 8, 1588–1595. doi:10.1039/b924309g
Return to citation in text: [1] -
Guetzoyan, L.; Nikbin, N.; Baxendale, I. R.; Ley, S. V. Chem. Sci. 2013, 4, 764–769. doi:10.1039/c2sc21850j
Return to citation in text: [1] -
Baumann, M.; Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Org. Lett. 2006, 8, 5231–5234. doi:10.1021/ol061975c
Return to citation in text: [1] -
Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k
Return to citation in text: [1] [2] -
Baxendale, I. R.; Ley, S. V.; Mansfield, A. C.; Smith, C. D. Angew. Chem., Int. Ed. 2009, 48, 4017–4021. doi:10.1002/anie.200900970
Return to citation in text: [1] -
Baumann, M.; Baxendale, I.; Brasholz, M.; Hayward, J.; Ley, S.; Nikbin, N. Synlett 2011, 1375–1380. doi:10.1055/s-0030-1260573
Return to citation in text: [1] -
Lange, H.; Carter, C. F.; Hopkin, M. D.; Burke, A.; Goode, J. G.; Baxendale, I. R.; Ley, S. V. Chem. Sci. 2011, 2, 765–769. doi:10.1039/c0sc00603c
Return to citation in text: [1] -
Nakano, Y.; Savage, G. P.; Saubern, S.; Scammells, P. J.; Polyzos, A. Aust. J. Chem. 2013, 66, 178–182. doi:10.1071/ch12463
Return to citation in text: [1] -
Brasholz, M.; Macdonald, J. M.; Saubern, S.; Ryan, J. H.; Holmes, A. B. Chem. – Eur. J. 2010, 16, 11471–11480. doi:10.1002/chem.201001435
Return to citation in text: [1] -
Hiebler, K.; Dertnig, C.; Soritz, S.; Maier, M. C.; Hörmann, T. R.; Grabner, B.; Gruber-Woelfler, H. J. Flow Chem. 2020, 10, 259–270. doi:10.1007/s41981-019-00058-5
Return to citation in text: [1] -
Correia, C. A.; Gilmore, K.; McQuade, D. T.; Seeberger, P. H. Angew. Chem., Int. Ed. 2015, 54, 4945–4948. doi:10.1002/anie.201411728
Return to citation in text: [1] -
Hinchcliffe, A.; Hughes, C.; Pears, D. A.; Pitts, M. R. Org. Process Res. Dev. 2007, 11, 477–481. doi:10.1021/op7000113
Return to citation in text: [1] -
Glasnov, T. N.; Kappe, C. O. Adv. Synth. Catal. 2010, 352, 3089–3097. doi:10.1002/adsc.201000646
Return to citation in text: [1] -
García-Lacuna, J.; Domínguez, G.; Blanco-Urgoiti, J.; Pérez-Castells, J. Org. Lett. 2018, 20, 5219–5223. doi:10.1021/acs.orglett.8b02168
Return to citation in text: [1] -
Ötvös, S. B.; Fülöp, F. Catal. Sci. Technol. 2015, 5, 4926–4941. doi:10.1039/c5cy00523j
Return to citation in text: [1] -
Vural Gürsel, I.; Noël, T.; Wang, Q.; Hessel, V. Green Chem. 2015, 17, 2012–2026. doi:10.1039/c4gc02160f
Return to citation in text: [1] [2] -
Toh, R. W.; Patrzałek, M.; Nienałtowski, T.; Piątkowski, J.; Kajetanowicz, A.; Wu, J.; Grela, K. ACS Sustainable Chem. Eng. 2021, 9, 16450–16458. doi:10.1021/acssuschemeng.1c06522
Return to citation in text: [1] -
Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Chem. Commun. 2010, 46, 2450–2452. doi:10.1039/c001550d
Return to citation in text: [1] -
Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/c2ob27002a
Return to citation in text: [1] [2] -
Fu, W. C.; Jamison, T. F. Org. Lett. 2019, 21, 6112–6116. doi:10.1021/acs.orglett.9b02259
Return to citation in text: [1] [2] -
Petersen, T. P.; Mirsharghi, S.; Rummel, P. C.; Thiele, S.; Rosenkilde, M. M.; Ritzén, A.; Ulven, T. Chem. – Eur. J. 2013, 19, 9343–9350. doi:10.1002/chem.201204350
Return to citation in text: [1] -
Qian, Z.; Baxendale, I. R.; Ley, S. V. Chem. – Eur. J. 2010, 16, 12342–12348. doi:10.1002/chem.201002147
Return to citation in text: [1] -
Polyzos, A.; O'Brien, M.; Petersen, T. P.; Baxendale, I. R.; Ley, S. V. Angew. Chem., Int. Ed. 2011, 50, 1190–1193. doi:10.1002/anie.201006618
Return to citation in text: [1] -
Britton, J.; Majumdar, S.; Weiss, G. A. Chem. Soc. Rev. 2018, 47, 5891–5918. doi:10.1039/c7cs00906b
Return to citation in text: [1] -
Ingham, R. J.; Riva, E.; Nikbin, N.; Baxendale, I. R.; Ley, S. V. Org. Lett. 2012, 14, 3920–3923. doi:10.1021/ol301673q
Return to citation in text: [1] -
Zak, J.; Ron, D.; Riva, E.; Harding, H. P.; Cross, B. C. S.; Baxendale, I. R. Chem. – Eur. J. 2012, 18, 9901–9910. doi:10.1002/chem.201201039
Return to citation in text: [1] -
Planchestainer, M.; Contente, M. L.; Cassidy, J.; Molinari, F.; Tamborini, L.; Paradisi, F. Green Chem. 2017, 19, 372–375. doi:10.1039/c6gc01780k
Return to citation in text: [1] -
Contente, M. L.; Paradisi, F. Nat. Catal. 2018, 1, 452–459. doi:10.1038/s41929-018-0082-9
Return to citation in text: [1] -
Roura Padrosa, D.; Benítez-Mateos, A. I.; Calvey, L.; Paradisi, F. Green Chem. 2020, 22, 5310–5316. doi:10.1039/d0gc01817a
Return to citation in text: [1] -
Benítez-Mateos, A. I.; Paradisi, F. ChemSusChem 2022, 15, e202102030. doi:10.1002/cssc.202102030
Return to citation in text: [1] -
Tamborini, L.; Romano, D.; Pinto, A.; Contente, M.; Iannuzzi, M. C.; Conti, P.; Molinari, F. Tetrahedron Lett. 2013, 54, 6090–6093. doi:10.1016/j.tetlet.2013.08.119
Return to citation in text: [1] -
Tamborini, L.; Romano, D.; Pinto, A.; Bertolani, A.; Molinari, F.; Conti, P. J. Mol. Catal. B: Enzym. 2012, 84, 78–82. doi:10.1016/j.molcatb.2012.02.008
Return to citation in text: [1] -
De Vitis, V.; Dall’Oglio, F.; Tentori, F.; Contente, M.; Romano, D.; Brenna, E.; Tamborini, L.; Molinari, F. Catalysts 2019, 9, 208. doi:10.3390/catal9030208
Return to citation in text: [1] [2] -
Baumann, M.; Leslie, A.; Moody, T. S.; Smyth, M.; Wharry, S. Org. Process Res. Dev. 2021, 25, 452–456. doi:10.1021/acs.oprd.0c00420
Return to citation in text: [1] -
Hartwig, J.; Kirschning, A. Chem. – Eur. J. 2016, 22, 3044–3052. doi:10.1002/chem.201504409
Return to citation in text: [1] [2] -
Ma, Y.; Wu, S.; Macaringue, E. G. J.; Zhang, T.; Gong, J.; Wang, J. Org. Process Res. Dev. 2020, 24, 1785–1801. doi:10.1021/acs.oprd.9b00362
Return to citation in text: [1] -
Johnson, M. D.; Burcham, C. L.; May, S. A.; Calvin, J. R.; McClary Groh, J.; Myers, S. S.; Webster, L. P.; Roberts, J. C.; Reddy, V. R.; Luciani, C. V.; Corrigan, A. P.; Spencer, R. D.; Moylan, R.; Boyse, R.; Murphy, J. D.; Stout, J. R. Org. Process Res. Dev. 2021, 25, 1284–1351. doi:10.1021/acs.oprd.0c00345
Return to citation in text: [1] [2] -
Mascia, S.; Heider, P. L.; Zhang, H.; Lakerveld, R.; Benyahia, B.; Barton, P. I.; Braatz, R. D.; Cooney, C. L.; Evans, J. M. B.; Jamison, T. F.; Jensen, K. F.; Myerson, A. S.; Trout, B. L. Angew. Chem., Int. Ed. 2013, 52, 12359–12363. doi:10.1002/anie.201305429
Return to citation in text: [1] [2] -
Quon, J. L.; Zhang, H.; Alvarez, A.; Evans, J.; Myerson, A. S.; Trout, B. L. Cryst. Growth Des. 2012, 12, 3036–3044. doi:10.1021/cg300253a
Return to citation in text: [1] -
Zhang, P.; Weeranoppanant, N.; Thomas, D. A.; Tahara, K.; Stelzer, T.; Russell, M. G.; O'Mahony, M.; Myerson, A. S.; Lin, H.; Kelly, L. P.; Jensen, K. F.; Jamison, T. F.; Dai, C.; Cui, Y.; Briggs, N.; Beingessner, R. L.; Adamo, A. Chem. – Eur. J. 2018, 24, 2776–2784. doi:10.1002/chem.201706004
Return to citation in text: [1] -
Lai, T.-T. C.; Ferguson, S.; Palmer, L.; Trout, B. L.; Myerson, A. S. Org. Process Res. Dev. 2014, 18, 1382–1390. doi:10.1021/op500171n
Return to citation in text: [1] -
Robertson, K.; Seeberger, P. H.; Gilmore, K. React. Chem. Eng. 2023, in press. doi:10.1039/d2re00183g
Return to citation in text: [1] -
Scott, C. D.; Labes, R.; Depardieu, M.; Battilocchio, C.; Davidson, M. G.; Ley, S. V.; Wilson, C. C.; Robertson, K. React. Chem. Eng. 2018, 3, 631–634. doi:10.1039/c8re00087e
Return to citation in text: [1] -
Hosoya, M.; Tanaka, M.; Manaka, A.; Nishijima, S.; Tsuno, N. Org. Process Res. Dev. 2022, 26, 1531–1544. doi:10.1021/acs.oprd.2c00088
Return to citation in text: [1] -
Tanaka, M.; Hosoya, M.; Manaka, A.; Tsuno, N. Chem. Eng. Res. Des. 2021, 175, 259–271. doi:10.1016/j.cherd.2021.09.013
Return to citation in text: [1] -
Loren, B. P.; Wleklinski, M.; Koswara, A.; Yammine, K.; Hu, Y.; Nagy, Z. K.; Thompson, D. H.; Cooks, R. G. Chem. Sci. 2017, 8, 4363–4370. doi:10.1039/c7sc00905d
Return to citation in text: [1] -
Lichtenegger, G. J.; Maier, M.; Khinast, J. G.; Gruber-Wölfler, H. J. Flow Chem. 2016, 6, 244–251. doi:10.1556/1846.2016.00021
Return to citation in text: [1] -
Hülsewede, D.; Temmel, E.; Kumm, P.; von Langermann, J. Crystals 2020, 10, 345. doi:10.3390/cryst10050345
Return to citation in text: [1] -
Borukhova, S.; Noël, T.; Metten, B.; de Vos, E.; Hessel, V. ChemSusChem 2013, 6, 2220–2225. doi:10.1002/cssc.201300684
Return to citation in text: [1] -
Snead, D. R.; Jamison, T. F. Chem. Sci. 2013, 4, 2822–2827. doi:10.1039/c3sc50859e
Return to citation in text: [1] -
Porcar, R.; Sans, V.; Ríos-Lombardía, N.; Gotor-Fernández, V.; Gotor, V.; Burguete, M. I.; García-Verdugo, E.; Luis, S. V. ACS Catal. 2012, 2, 1976–1983. doi:10.1021/cs300282w
Return to citation in text: [1] -
Gilmore, K.; Kopetzki, D.; Lee, J. W.; Horváth, Z.; McQuade, D. T.; Seidel-Morgenstern, A.; Seeberger, P. H. Chem. Commun. 2014, 50, 12652–12655. doi:10.1039/c4cc05098c
Return to citation in text: [1] -
O'Neal, E. J.; Jensen, K. F. ChemCatChem 2014, 6, 3004–3011. doi:10.1002/cctc.201402368
Return to citation in text: [1] -
Ormerod, D.; Lefevre, N.; Dorbec, M.; Eyskens, I.; Vloemans, P.; Duyssens, K.; Diez de la Torre, V.; Kaval, N.; Merkul, E.; Sergeyev, S.; Maes, B. U. W. Org. Process Res. Dev. 2016, 20, 911–920. doi:10.1021/acs.oprd.5b00418
Return to citation in text: [1] -
Peeva, L.; Da Silva Burgal, J.; Heckenast, Z.; Brazy, F.; Cazenave, F.; Livingston, A. Angew. Chem., Int. Ed. 2016, 55, 13576–13579. doi:10.1002/anie.201607795
Return to citation in text: [1] -
Wen, Z.; Pintossi, D.; Nuño, M.; Noël, T. Nat. Commun. 2022, 13, 6147. doi:10.1038/s41467-022-33821-9
Return to citation in text: [1] -
Fodi, T.; Didaskalou, C.; Kupai, J.; Balogh, G. T.; Huszthy, P.; Szekely, G. ChemSusChem 2017, 10, 3435–3444. doi:10.1002/cssc.201701120
Return to citation in text: [1] -
Voros, V.; Drioli, E.; Fonte, C.; Szekely, G. ACS Sustainable Chem. Eng. 2019, 7, 18444–18452. doi:10.1021/acssuschemeng.9b04245
Return to citation in text: [1] -
Peeva, L.; da Silva Burgal, J.; Valtcheva, I.; Livingston, A. G. Chem. Eng. Sci. 2014, 116, 183–194. doi:10.1016/j.ces.2014.04.022
Return to citation in text: [1] -
Capellades, G.; Neurohr, C.; Briggs, N.; Rapp, K.; Hammersmith, G.; Brancazio, D.; Derksen, B.; Myerson, A. S. Org. Process Res. Dev. 2021, 25, 1534–1546. doi:10.1021/acs.oprd.1c00117
Return to citation in text: [1] -
Capellades, G.; Neurohr, C.; Azad, M.; Brancazio, D.; Rapp, K.; Hammersmith, G.; Myerson, A. S. J. Pharm. Sci. 2020, 109, 1365–1372. doi:10.1016/j.xphs.2019.12.011
Return to citation in text: [1]
| 106. | Baumann, M.; Leslie, A.; Moody, T. S.; Smyth, M.; Wharry, S. Org. Process Res. Dev. 2021, 25, 452–456. doi:10.1021/acs.oprd.0c00420 |
| 107. | Hartwig, J.; Kirschning, A. Chem. – Eur. J. 2016, 22, 3044–3052. doi:10.1002/chem.201504409 |
| 105. | De Vitis, V.; Dall’Oglio, F.; Tentori, F.; Contente, M.; Romano, D.; Brenna, E.; Tamborini, L.; Molinari, F. Catalysts 2019, 9, 208. doi:10.3390/catal9030208 |
| 105. | De Vitis, V.; Dall’Oglio, F.; Tentori, F.; Contente, M.; Romano, D.; Brenna, E.; Tamborini, L.; Molinari, F. Catalysts 2019, 9, 208. doi:10.3390/catal9030208 |
| 99. | Planchestainer, M.; Contente, M. L.; Cassidy, J.; Molinari, F.; Tamborini, L.; Paradisi, F. Green Chem. 2017, 19, 372–375. doi:10.1039/c6gc01780k |
| 100. | Contente, M. L.; Paradisi, F. Nat. Catal. 2018, 1, 452–459. doi:10.1038/s41929-018-0082-9 |
| 101. | Roura Padrosa, D.; Benítez-Mateos, A. I.; Calvey, L.; Paradisi, F. Green Chem. 2020, 22, 5310–5316. doi:10.1039/d0gc01817a |
| 102. | Benítez-Mateos, A. I.; Paradisi, F. ChemSusChem 2022, 15, e202102030. doi:10.1002/cssc.202102030 |
| 103. | Tamborini, L.; Romano, D.; Pinto, A.; Contente, M.; Iannuzzi, M. C.; Conti, P.; Molinari, F. Tetrahedron Lett. 2013, 54, 6090–6093. doi:10.1016/j.tetlet.2013.08.119 |
| 104. | Tamborini, L.; Romano, D.; Pinto, A.; Bertolani, A.; Molinari, F.; Conti, P. J. Mol. Catal. B: Enzym. 2012, 84, 78–82. doi:10.1016/j.molcatb.2012.02.008 |
| 29. | Adamo, A.; Beingessner, R. L.; Behnam, M.; Chen, J.; Jamison, T. F.; Jensen, K. F.; Monbaliu, J.-C. M.; Myerson, A. S.; Revalor, E. M.; Snead, D. R.; Stelzer, T.; Weeranoppanant, N.; Wong, S. Y.; Zhang, P. Science 2016, 352, 61–67. doi:10.1126/science.aaf1337 |
| 30. | Bédard, A.-C.; Adamo, A.; Aroh, K. C.; Russell, M. G.; Bedermann, A. A.; Torosian, J.; Yue, B.; Jensen, K. F.; Jamison, T. F. Science 2018, 361, 1220–1225. doi:10.1126/science.aat0650 |
| 112. | Zhang, P.; Weeranoppanant, N.; Thomas, D. A.; Tahara, K.; Stelzer, T.; Russell, M. G.; O'Mahony, M.; Myerson, A. S.; Lin, H.; Kelly, L. P.; Jensen, K. F.; Jamison, T. F.; Dai, C.; Cui, Y.; Briggs, N.; Beingessner, R. L.; Adamo, A. Chem. – Eur. J. 2018, 24, 2776–2784. doi:10.1002/chem.201706004 |
| 109. | Johnson, M. D.; Burcham, C. L.; May, S. A.; Calvin, J. R.; McClary Groh, J.; Myers, S. S.; Webster, L. P.; Roberts, J. C.; Reddy, V. R.; Luciani, C. V.; Corrigan, A. P.; Spencer, R. D.; Moylan, R.; Boyse, R.; Murphy, J. D.; Stout, J. R. Org. Process Res. Dev. 2021, 25, 1284–1351. doi:10.1021/acs.oprd.0c00345 |
| 110. | Mascia, S.; Heider, P. L.; Zhang, H.; Lakerveld, R.; Benyahia, B.; Barton, P. I.; Braatz, R. D.; Cooney, C. L.; Evans, J. M. B.; Jamison, T. F.; Jensen, K. F.; Myerson, A. S.; Trout, B. L. Angew. Chem., Int. Ed. 2013, 52, 12359–12363. doi:10.1002/anie.201305429 |
| 111. | Quon, J. L.; Zhang, H.; Alvarez, A.; Evans, J.; Myerson, A. S.; Trout, B. L. Cryst. Growth Des. 2012, 12, 3036–3044. doi:10.1021/cg300253a |
| 107. | Hartwig, J.; Kirschning, A. Chem. – Eur. J. 2016, 22, 3044–3052. doi:10.1002/chem.201504409 |
| 108. | Ma, Y.; Wu, S.; Macaringue, E. G. J.; Zhang, T.; Gong, J.; Wang, J. Org. Process Res. Dev. 2020, 24, 1785–1801. doi:10.1021/acs.oprd.9b00362 |
| 116. | Hosoya, M.; Tanaka, M.; Manaka, A.; Nishijima, S.; Tsuno, N. Org. Process Res. Dev. 2022, 26, 1531–1544. doi:10.1021/acs.oprd.2c00088 |
| 35. | Weeranoppanant, N.; Adamo, A. ACS Med. Chem. Lett. 2020, 11, 9–15. doi:10.1021/acsmedchemlett.9b00491 |
| 48. | Adamo, A.; Heider, P. L.; Weeranoppanant, N.; Jensen, K. F. Ind. Eng. Chem. Res. 2013, 52, 10802–10808. doi:10.1021/ie401180t |
| 117. | Tanaka, M.; Hosoya, M.; Manaka, A.; Tsuno, N. Chem. Eng. Res. Des. 2021, 175, 259–271. doi:10.1016/j.cherd.2021.09.013 |
| 110. | Mascia, S.; Heider, P. L.; Zhang, H.; Lakerveld, R.; Benyahia, B.; Barton, P. I.; Braatz, R. D.; Cooney, C. L.; Evans, J. M. B.; Jamison, T. F.; Jensen, K. F.; Myerson, A. S.; Trout, B. L. Angew. Chem., Int. Ed. 2013, 52, 12359–12363. doi:10.1002/anie.201305429 |
| 114. | Robertson, K.; Seeberger, P. H.; Gilmore, K. React. Chem. Eng. 2023, in press. doi:10.1039/d2re00183g |
| 115. | Scott, C. D.; Labes, R.; Depardieu, M.; Battilocchio, C.; Davidson, M. G.; Ley, S. V.; Wilson, C. C.; Robertson, K. React. Chem. Eng. 2018, 3, 631–634. doi:10.1039/c8re00087e |
| 109. | Johnson, M. D.; Burcham, C. L.; May, S. A.; Calvin, J. R.; McClary Groh, J.; Myers, S. S.; Webster, L. P.; Roberts, J. C.; Reddy, V. R.; Luciani, C. V.; Corrigan, A. P.; Spencer, R. D.; Moylan, R.; Boyse, R.; Murphy, J. D.; Stout, J. R. Org. Process Res. Dev. 2021, 25, 1284–1351. doi:10.1021/acs.oprd.0c00345 |
| 131. | Peeva, L.; da Silva Burgal, J.; Valtcheva, I.; Livingston, A. G. Chem. Eng. Sci. 2014, 116, 183–194. doi:10.1016/j.ces.2014.04.022 |
| 113. | Lai, T.-T. C.; Ferguson, S.; Palmer, L.; Trout, B. L.; Myerson, A. S. Org. Process Res. Dev. 2014, 18, 1382–1390. doi:10.1021/op500171n |
| 49. | Overmans, S.; Ignacz, G.; Beke, A. K.; Xu, J.; Saikaly, P. E.; Szekely, G.; Lauersen, K. J. Green Chem. 2022, 24, 5479–5489. doi:10.1039/d2gc00938b |
| 133. | Capellades, G.; Neurohr, C.; Azad, M.; Brancazio, D.; Rapp, K.; Hammersmith, G.; Myerson, A. S. J. Pharm. Sci. 2020, 109, 1365–1372. doi:10.1016/j.xphs.2019.12.011 |
| 132. | Capellades, G.; Neurohr, C.; Briggs, N.; Rapp, K.; Hammersmith, G.; Brancazio, D.; Derksen, B.; Myerson, A. S. Org. Process Res. Dev. 2021, 25, 1534–1546. doi:10.1021/acs.oprd.1c00117 |
| 130. | Voros, V.; Drioli, E.; Fonte, C.; Szekely, G. ACS Sustainable Chem. Eng. 2019, 7, 18444–18452. doi:10.1021/acssuschemeng.9b04245 |
| 120. | Hülsewede, D.; Temmel, E.; Kumm, P.; von Langermann, J. Crystals 2020, 10, 345. doi:10.3390/cryst10050345 |
| 121. | Borukhova, S.; Noël, T.; Metten, B.; de Vos, E.; Hessel, V. ChemSusChem 2013, 6, 2220–2225. doi:10.1002/cssc.201300684 |
| 118. | Loren, B. P.; Wleklinski, M.; Koswara, A.; Yammine, K.; Hu, Y.; Nagy, Z. K.; Thompson, D. H.; Cooks, R. G. Chem. Sci. 2017, 8, 4363–4370. doi:10.1039/c7sc00905d |
| 119. | Lichtenegger, G. J.; Maier, M.; Khinast, J. G.; Gruber-Wölfler, H. J. Flow Chem. 2016, 6, 244–251. doi:10.1556/1846.2016.00021 |
| 79. | Lange, H.; Carter, C. F.; Hopkin, M. D.; Burke, A.; Goode, J. G.; Baxendale, I. R.; Ley, S. V. Chem. Sci. 2011, 2, 765–769. doi:10.1039/c0sc00603c |
| 76. | Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k |
| 78. | Baumann, M.; Baxendale, I.; Brasholz, M.; Hayward, J.; Ley, S.; Nikbin, N. Synlett 2011, 1375–1380. doi:10.1055/s-0030-1260573 |
| 1. | Gutmann, B.; Cantillo, D.; Kappe, C. O. Angew. Chem., Int. Ed. 2015, 54, 6688–6728. doi:10.1002/anie.201409318 |
| 2. | Plutschack, M. B.; Pieber, B.; Gilmore, K.; Seeberger, P. H. Chem. Rev. 2017, 117, 11796–11893. doi:10.1021/acs.chemrev.7b00183 |
| 14. | Atobe, M.; Tateno, H.; Matsumura, Y. Chem. Rev. 2018, 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353 |
| 15. | Noël, T.; Cao, Y.; Laudadio, G. Acc. Chem. Res. 2019, 52, 2858–2869. doi:10.1021/acs.accounts.9b00412 |
| 16. | Elsherbini, M.; Wirth, T. Acc. Chem. Res. 2019, 52, 3287–3296. doi:10.1021/acs.accounts.9b00497 |
| 89. | Toh, R. W.; Patrzałek, M.; Nienałtowski, T.; Piątkowski, J.; Kajetanowicz, A.; Wu, J.; Grela, K. ACS Sustainable Chem. Eng. 2021, 9, 16450–16458. doi:10.1021/acssuschemeng.1c06522 |
| 10. | Cambié, D.; Bottecchia, C.; Straathof, N. J. W.; Hessel, V.; Noël, T. Chem. Rev. 2016, 116, 10276–10341. doi:10.1021/acs.chemrev.5b00707 |
| 11. | Politano, F.; Oksdath-Mansilla, G. Org. Process Res. Dev. 2018, 22, 1045–1062. doi:10.1021/acs.oprd.8b00213 |
| 12. | Sambiagio, C.; Noël, T. Trends Chem. 2020, 2, 92–106. doi:10.1016/j.trechm.2019.09.003 |
| 13. | Di Filippo, M.; Bracken, C.; Baumann, M. Molecules 2020, 25, 356. doi:10.3390/molecules25020356 |
| 6. | Movsisyan, M.; Delbeke, E. I. P.; Berton, J. K. E. T.; Battilocchio, C.; Ley, S. V.; Stevens, C. V. Chem. Soc. Rev. 2016, 45, 4892–4928. doi:10.1039/c5cs00902b |
| 7. | Bogdan, A. R.; Dombrowski, A. W. J. Med. Chem. 2019, 62, 6422–6468. doi:10.1021/acs.jmedchem.8b01760 |
| 8. | Baumann, M.; Moody, T. S.; Smyth, M.; Wharry, S. Org. Process Res. Dev. 2020, 24, 1802–1813. doi:10.1021/acs.oprd.9b00524 |
| 9. | Domokos, A.; Nagy, B.; Szilágyi, B.; Marosi, G.; Nagy, Z. K. Org. Process Res. Dev. 2021, 25, 721–739. doi:10.1021/acs.oprd.0c00504 |
| 87. | Ötvös, S. B.; Fülöp, F. Catal. Sci. Technol. 2015, 5, 4926–4941. doi:10.1039/c5cy00523j |
| 3. | Porta, R.; Benaglia, M.; Puglisi, A. Org. Process Res. Dev. 2016, 20, 2–25. doi:10.1021/acs.oprd.5b00325 |
| 4. | García‐Lacuna, J.; Domínguez, G.; Pérez‐Castells, J. ChemSusChem 2020, 13, 5138–5163. doi:10.1002/cssc.202001372 |
| 5. | Colella, M.; Nagaki, A.; Luisi, R. Chem. – Eur. J. 2020, 26, 19–32. doi:10.1002/chem.201903353 |
| 88. | Vural Gürsel, I.; Noël, T.; Wang, Q.; Hessel, V. Green Chem. 2015, 17, 2012–2026. doi:10.1039/c4gc02160f |
| 26. | Sans, V.; Cronin, L. Chem. Soc. Rev. 2016, 45, 2032–2043. doi:10.1039/c5cs00793c |
| 27. | Sagmeister, P.; Williams, J. D.; Hone, C. A.; Kappe, C. O. React. Chem. Eng. 2019, 4, 1571–1578. doi:10.1039/c9re00087a |
| 28. | Sagmeister, P.; Lebl, R.; Castillo, I.; Rehrl, J.; Kruisz, J.; Sipek, M.; Horn, M.; Sacher, S.; Cantillo, D.; Williams, J. D.; Kappe, C. O. Angew. Chem., Int. Ed. 2021, 60, 8139–8148. doi:10.1002/anie.202016007 |
| 29. | Adamo, A.; Beingessner, R. L.; Behnam, M.; Chen, J.; Jamison, T. F.; Jensen, K. F.; Monbaliu, J.-C. M.; Myerson, A. S.; Revalor, E. M.; Snead, D. R.; Stelzer, T.; Weeranoppanant, N.; Wong, S. Y.; Zhang, P. Science 2016, 352, 61–67. doi:10.1126/science.aaf1337 |
| 30. | Bédard, A.-C.; Adamo, A.; Aroh, K. C.; Russell, M. G.; Bedermann, A. A.; Torosian, J.; Yue, B.; Jensen, K. F.; Jamison, T. F. Science 2018, 361, 1220–1225. doi:10.1126/science.aat0650 |
| 85. | Glasnov, T. N.; Kappe, C. O. Adv. Synth. Catal. 2010, 352, 3089–3097. doi:10.1002/adsc.201000646 |
| 23. | Ley, S. V.; Fitzpatrick, D. E.; Ingham, R. J.; Myers, R. M. Angew. Chem., Int. Ed. 2015, 54, 3449–3464. doi:10.1002/anie.201410744 |
| 24. | Shukla, C. A.; Kulkarni, A. A. Beilstein J. Org. Chem. 2017, 13, 960–987. doi:10.3762/bjoc.13.97 |
| 25. | Gioiello, A.; Piccinno, A.; Lozza, A. M.; Cerra, B. J. Med. Chem. 2020, 63, 6624–6647. doi:10.1021/acs.jmedchem.9b01956 |
| 86. | García-Lacuna, J.; Domínguez, G.; Blanco-Urgoiti, J.; Pérez-Castells, J. Org. Lett. 2018, 20, 5219–5223. doi:10.1021/acs.orglett.8b02168 |
| 20. | Newman, S. G.; Jensen, K. F. Green Chem. 2013, 15, 1456–1472. doi:10.1039/c3gc40374b |
| 21. | Dallinger, D.; Kappe, C. O. Curr. Opin. Green Sustainable Chem. 2017, 7, 6–12. doi:10.1016/j.cogsc.2017.06.003 |
| 22. | Baumann, M.; Moody, T.; Smyth, M.; Wharry, S. Synthesis 2021, 53, 3963–3976. doi:10.1055/a-1541-1761 |
| 80. | Nakano, Y.; Savage, G. P.; Saubern, S.; Scammells, P. J.; Polyzos, A. Aust. J. Chem. 2013, 66, 178–182. doi:10.1071/ch12463 |
| 81. | Brasholz, M.; Macdonald, J. M.; Saubern, S.; Ryan, J. H.; Holmes, A. B. Chem. – Eur. J. 2010, 16, 11471–11480. doi:10.1002/chem.201001435 |
| 82. | Hiebler, K.; Dertnig, C.; Soritz, S.; Maier, M. C.; Hörmann, T. R.; Grabner, B.; Gruber-Woelfler, H. J. Flow Chem. 2020, 10, 259–270. doi:10.1007/s41981-019-00058-5 |
| 83. | Correia, C. A.; Gilmore, K.; McQuade, D. T.; Seeberger, P. H. Angew. Chem., Int. Ed. 2015, 54, 4945–4948. doi:10.1002/anie.201411728 |
| 17. | De Santis, P.; Meyer, L.-E.; Kara, S. React. Chem. Eng. 2020, 5, 2155–2184. doi:10.1039/d0re00335b |
| 18. | Benítez-Mateos, A. I.; Contente, M. L.; Roura Padrosa, D.; Paradisi, F. React. Chem. Eng. 2021, 6, 599–611. doi:10.1039/d0re00483a |
| 19. | Coloma, J.; Guiavarc’h, Y.; Hagedoorn, P.-L.; Hanefeld, U. Chem. Commun. 2021, 57, 11416–11428. doi:10.1039/d1cc04315c |
| 84. | Hinchcliffe, A.; Hughes, C.; Pears, D. A.; Pitts, M. R. Org. Process Res. Dev. 2007, 11, 477–481. doi:10.1021/op7000113 |
| 93. | Petersen, T. P.; Mirsharghi, S.; Rummel, P. C.; Thiele, S.; Rosenkilde, M. M.; Ritzén, A.; Ulven, T. Chem. – Eur. J. 2013, 19, 9343–9350. doi:10.1002/chem.201204350 |
| 91. | Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/c2ob27002a |
| 90. | Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Chem. Commun. 2010, 46, 2450–2452. doi:10.1039/c001550d |
| 91. | Hopkin, M. D.; Baxendale, I. R.; Ley, S. V. Org. Biomol. Chem. 2013, 11, 1822–1839. doi:10.1039/c2ob27002a |
| 92. | Fu, W. C.; Jamison, T. F. Org. Lett. 2019, 21, 6112–6116. doi:10.1021/acs.orglett.9b02259 |
| 97. | Ingham, R. J.; Riva, E.; Nikbin, N.; Baxendale, I. R.; Ley, S. V. Org. Lett. 2012, 14, 3920–3923. doi:10.1021/ol301673q |
| 98. | Zak, J.; Ron, D.; Riva, E.; Harding, H. P.; Cross, B. C. S.; Baxendale, I. R. Chem. – Eur. J. 2012, 18, 9901–9910. doi:10.1002/chem.201201039 |
| 95. | Polyzos, A.; O'Brien, M.; Petersen, T. P.; Baxendale, I. R.; Ley, S. V. Angew. Chem., Int. Ed. 2011, 50, 1190–1193. doi:10.1002/anie.201006618 |
| 96. | Britton, J.; Majumdar, S.; Weiss, G. A. Chem. Soc. Rev. 2018, 47, 5891–5918. doi:10.1039/c7cs00906b |
| 92. | Fu, W. C.; Jamison, T. F. Org. Lett. 2019, 21, 6112–6116. doi:10.1021/acs.orglett.9b02259 |
| 94. | Qian, Z.; Baxendale, I. R.; Ley, S. V. Chem. – Eur. J. 2010, 16, 12342–12348. doi:10.1002/chem.201002147 |
| 54. | Contente, M. L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Green Chem. 2019, 21, 3263–3266. doi:10.1039/c9gc01374a |
| 55. | Peer, M.; Weeranoppanant, N.; Adamo, A.; Zhang, Y.; Jensen, K. F. Org. Process Res. Dev. 2016, 20, 1677–1685. doi:10.1021/acs.oprd.6b00234 |
| 55. | Peer, M.; Weeranoppanant, N.; Adamo, A.; Zhang, Y.; Jensen, K. F. Org. Process Res. Dev. 2016, 20, 1677–1685. doi:10.1021/acs.oprd.6b00234 |
| 62. | Hamlin, T. A.; Lazarus, G. M. L.; Kelly, C. B.; Leadbeater, N. E. Org. Process Res. Dev. 2014, 18, 1253–1258. doi:10.1021/op500190j |
| 63. | Verstraete, K.; Buckinx, A.-L.; Zaquen, N.; Junkers, T. Macromolecules 2021, 54, 3865–3872. doi:10.1021/acs.macromol.1c00242 |
| 60. | De Vitis, V.; Dall'Oglio, F.; Pinto, A.; De Micheli, C.; Molinari, F.; Conti, P.; Romano, D.; Tamborini, L. ChemistryOpen 2017, 6, 668–673. doi:10.1002/open.201700082 |
| 61. | Dalla-Vechia, L.; Reichart, B.; Glasnov, T.; Miranda, L. S. M.; Kappe, C. O.; de Souza, R. O. M. A. Org. Biomol. Chem. 2013, 11, 6806–6813. doi:10.1039/c3ob41464g |
| 58. | Jaman, Z.; Sobreira, T. J. P.; Mufti, A.; Ferreira, C. R.; Cooks, R. G.; Thompson, D. H. Org. Process Res. Dev. 2019, 23, 334–341. doi:10.1021/acs.oprd.8b00387 |
| 59. | Borukhova, S.; Noël, T.; Hessel, V. ChemSusChem 2016, 9, 67–74. doi:10.1002/cssc.201501367 |
| 56. | García-Lacuna, J.; Fleiß, T.; Munday, R.; Leslie, K.; O’Kearney-McMullan, A.; Hone, C. A.; Kappe, C. O. Org. Process Res. Dev. 2021, 25, 947–959. doi:10.1021/acs.oprd.1c00002 |
| 57. | Nieves-Remacha, M. J.; Torres, M.; Ruiz-Abad, M.; Rincón, J. A.; Cumming, G. R.; Garcia-Losada, P. React. Chem. Eng. 2019, 4, 334–345. doi:10.1039/c8re00203g |
| 64. | Drageset, A.; Bjørsvik, H.-R. React. Chem. Eng. 2016, 1, 436–444. doi:10.1039/c6re00091f |
| 65. | Phillips, T. W.; Bannock, J. H.; deMello, J. C. Lab Chip 2015, 15, 2960–2967. doi:10.1039/c5lc00430f |
| 66. | Harding, M. J.; Feng, B.; Lopez-Rodriguez, R.; O'Connor, H.; Dowling, D.; Gibson, G.; Girard, K. P.; Ferguson, S. React. Chem. Eng. 2021, 6, 1635–1643. doi:10.1039/d1re00119a |
| 76. | Smith, C. D.; Baxendale, I. R.; Lanners, S.; Hayward, J. J.; Smith, S. C.; Ley, S. V. Org. Biomol. Chem. 2007, 5, 1559–1561. doi:10.1039/b702995k |
| 77. | Baxendale, I. R.; Ley, S. V.; Mansfield, A. C.; Smith, C. D. Angew. Chem., Int. Ed. 2009, 48, 4017–4021. doi:10.1002/anie.200900970 |
| 73. | Carter, C. F.; Baxendale, I. R.; Pavey, J. B. J.; Ley, S. V. Org. Biomol. Chem. 2010, 8, 1588–1595. doi:10.1039/b924309g |
| 74. | Guetzoyan, L.; Nikbin, N.; Baxendale, I. R.; Ley, S. V. Chem. Sci. 2013, 4, 764–769. doi:10.1039/c2sc21850j |
| 75. | Baumann, M.; Baxendale, I. R.; Ley, S. V.; Smith, C. D.; Tranmer, G. K. Org. Lett. 2006, 8, 5231–5234. doi:10.1021/ol061975c |
| 68. | O’Brien, M.; Koos, P.; Browne, D. L.; Ley, S. V. Org. Biomol. Chem. 2012, 10, 7031–7036. doi:10.1039/c2ob25912e |
| 72. | Ley, S. V.; Browne, D. L.; O’Brien, M. Immobilized reagents and Multistep Processes. In Flow Chemistry in Organic Chemistry; Jamison, T. F.; Koch, G., Eds.; Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2018; pp 273–312. doi:10.1055/sos-sd-228-00177 |
| 67. | Day, C.; Saldarriaga, A.; Tilley, M.; Hunter, H.; Organ, M. G.; Wilson, D. J. Org. Process Res. Dev. 2016, 20, 1738–1743. doi:10.1021/acs.oprd.6b00226 |
| 68. | O’Brien, M.; Koos, P.; Browne, D. L.; Ley, S. V. Org. Biomol. Chem. 2012, 10, 7031–7036. doi:10.1039/c2ob25912e |
| 69. | Ingham, R. J.; Battilocchio, C.; Fitzpatrick, D. E.; Sliwinski, E.; Hawkins, J. M.; Ley, S. V. Angew. Chem., Int. Ed. 2015, 54, 144–148. doi:10.1002/anie.201409356 |
| 70. | Hu, D. X.; O’Brien, M.; Ley, S. V. Org. Lett. 2012, 14, 4246–4249. doi:10.1021/ol301930h |
| 71. | O'Brien, M.; Cooper, D. A.; Dolan, J. Tetrahedron Lett. 2017, 58, 829–834. doi:10.1016/j.tetlet.2017.01.029 |
| 125. | O'Neal, E. J.; Jensen, K. F. ChemCatChem 2014, 6, 3004–3011. doi:10.1002/cctc.201402368 |
| 126. | Ormerod, D.; Lefevre, N.; Dorbec, M.; Eyskens, I.; Vloemans, P.; Duyssens, K.; Diez de la Torre, V.; Kaval, N.; Merkul, E.; Sergeyev, S.; Maes, B. U. W. Org. Process Res. Dev. 2016, 20, 911–920. doi:10.1021/acs.oprd.5b00418 |
| 37. | Thomson, C. G.; Banks, C.; Allen, M.; Barker, G.; Coxon, C. R.; Lee, A.-L.; Vilela, F. J. Org. Chem. 2021, 86, 14079–14094. doi:10.1021/acs.joc.1c01151 |
| 124. | Gilmore, K.; Kopetzki, D.; Lee, J. W.; Horváth, Z.; McQuade, D. T.; Seidel-Morgenstern, A.; Seeberger, P. H. Chem. Commun. 2014, 50, 12652–12655. doi:10.1039/c4cc05098c |
| 88. | Vural Gürsel, I.; Noël, T.; Wang, Q.; Hessel, V. Green Chem. 2015, 17, 2012–2026. doi:10.1039/c4gc02160f |
| 123. | Porcar, R.; Sans, V.; Ríos-Lombardía, N.; Gotor-Fernández, V.; Gotor, V.; Burguete, M. I.; García-Verdugo, E.; Luis, S. V. ACS Catal. 2012, 2, 1976–1983. doi:10.1021/cs300282w |
| 69. | Ingham, R. J.; Battilocchio, C.; Fitzpatrick, D. E.; Sliwinski, E.; Hawkins, J. M.; Ley, S. V. Angew. Chem., Int. Ed. 2015, 54, 144–148. doi:10.1002/anie.201409356 |
| 122. | Snead, D. R.; Jamison, T. F. Chem. Sci. 2013, 4, 2822–2827. doi:10.1039/c3sc50859e |
| 41. | Horváth, Z.; Horosanskaia, E.; Lee, J. W.; Lorenz, H.; Gilmore, K.; Seeberger, P. H.; Seidel-Morgenstern, A. Org. Process Res. Dev. 2015, 19, 624–634. doi:10.1021/acs.oprd.5b00048 |
| 42. | Sivo, A.; Kim, T. K.; Ruta, V.; Luisi, R.; Osorio-Tejada, J.; Escriba-Gelonch, M.; Hessel, V.; Sponchioni, M.; Vilé, G. React. Chem. Eng. 2022, 7, 2650–2658. doi:10.1039/d2re00242f |
| 38. | Donnelly, K.; Baumann, M. Beilstein J. Org. Chem. 2022, 18, 232–239. doi:10.3762/bjoc.18.27 |
| 39. | O'Brien, A. G.; Horváth, Z.; Lévesque, F.; Lee, J. W.; Seidel-Morgenstern, A.; Seeberger, P. H. Angew. Chem., Int. Ed. 2012, 51, 7028–7030. doi:10.1002/anie.201202795 |
| 40. | Lee, J. W.; Horváth, Z.; O’Brien, A. G.; Seeberger, P. H.; Seidel-Morgenstern, A. Chem. Eng. J. 2014, 251, 355–370. doi:10.1016/j.cej.2014.04.043 |
| 34. | Bana, P.; Örkényi, R.; Lövei, K.; Lakó, Á.; Túrós, G. I.; Éles, J.; Faigl, F.; Greiner, I. Bioorg. Med. Chem. 2017, 25, 6180–6189. doi:10.1016/j.bmc.2016.12.046 |
| 35. | Weeranoppanant, N.; Adamo, A. ACS Med. Chem. Lett. 2020, 11, 9–15. doi:10.1021/acsmedchemlett.9b00491 |
| 36. | Meyer, L.-E.; Hobisch, M.; Kara, S. Curr. Opin. Biotechnol. 2022, 78, 102835. doi:10.1016/j.copbio.2022.102835 |
| 129. | Fodi, T.; Didaskalou, C.; Kupai, J.; Balogh, G. T.; Huszthy, P.; Szekely, G. ChemSusChem 2017, 10, 3435–3444. doi:10.1002/cssc.201701120 |
| 37. | Thomson, C. G.; Banks, C.; Allen, M.; Barker, G.; Coxon, C. R.; Lee, A.-L.; Vilela, F. J. Org. Chem. 2021, 86, 14079–14094. doi:10.1021/acs.joc.1c01151 |
| 38. | Donnelly, K.; Baumann, M. Beilstein J. Org. Chem. 2022, 18, 232–239. doi:10.3762/bjoc.18.27 |
| 127. | Peeva, L.; Da Silva Burgal, J.; Heckenast, Z.; Brazy, F.; Cazenave, F.; Livingston, A. Angew. Chem., Int. Ed. 2016, 55, 13576–13579. doi:10.1002/anie.201607795 |
| 31. | Britton, J.; Raston, C. L. Chem. Soc. Rev. 2017, 46, 1250–1271. doi:10.1039/c6cs00830e |
| 32. | Britton, J.; Jamison, T. F. Nat. Protoc. 2017, 12, 2423–2446. doi:10.1038/nprot.2017.102 |
| 33. | Sagandira, C. R.; Nqeketo, S.; Mhlana, K.; Sonti, T.; Gaqa, S.; Watts, P. React. Chem. Eng. 2022, 7, 214–244. doi:10.1039/d1re00483b |
| 128. | Wen, Z.; Pintossi, D.; Nuño, M.; Noël, T. Nat. Commun. 2022, 13, 6147. doi:10.1038/s41467-022-33821-9 |
| 46. | Britton, J.; Dyer, R. P.; Majumdar, S.; Raston, C. L.; Weiss, G. A. Angew. Chem., Int. Ed. 2017, 56, 2296–2301. doi:10.1002/anie.201610821 |
| 43. | Örkényi, R.; Éles, J.; Faigl, F.; Vincze, P.; Prechl, A.; Szakács, Z.; Kóti, J.; Greiner, I. Angew. Chem., Int. Ed. 2017, 56, 8742–8745. doi:10.1002/anie.201703852 |
| 44. | Lorántfy, L.; Rutterschmid, D.; Örkényi, R.; Bakonyi, D.; Faragó, J.; Dargó, G.; Könczöl, Á. Org. Process Res. Dev. 2020, 24, 2676–2688. doi:10.1021/acs.oprd.0c00338 |
| 45. | Fitzpatrick, D. E.; Mutton, R. J.; Ley, S. V. React. Chem. Eng. 2018, 3, 799–806. doi:10.1039/c8re00107c |
| 53. | Power, L. A.; Clayton, A. D.; Reynolds, W. R.; Hose, D. R. J.; Ainsworth, C.; Chamberlain, T. W.; Nguyen, B. N.; Bourne, R. A.; Kapur, N.; Blacker, A. J. React. Chem. Eng. 2021, 6, 1806–1810. doi:10.1039/d1re00205h |
| 54. | Contente, M. L.; Farris, S.; Tamborini, L.; Molinari, F.; Paradisi, F. Green Chem. 2019, 21, 3263–3266. doi:10.1039/c9gc01374a |
| 52. | Dai, C.; Snead, D. R.; Zhang, P.; Jamison, T. F. J. Flow Chem. 2015, 5, 133–138. doi:10.1556/1846.2015.00013 |
| 51. | Lebl, R.; Murray, T.; Adamo, A.; Cantillo, D.; Kappe, C. O. ACS Sustainable Chem. Eng. 2019, 7, 20088–20096. doi:10.1021/acssuschemeng.9b05954 |
| 50. | For further technical details, pictures, and applications of Zaiput membranes, see refs [35,48] and/or visit: https://www.zaiput.com/product/liquid-liquid-gas-separators/ |
| 51. | Lebl, R.; Murray, T.; Adamo, A.; Cantillo, D.; Kappe, C. O. ACS Sustainable Chem. Eng. 2019, 7, 20088–20096. doi:10.1021/acssuschemeng.9b05954 |
| 47. | Cervera-Padrell, A. E.; Morthensen, S. T.; Lewandowski, D. J.; Skovby, T.; Kiil, S.; Gernaey, K. V. Org. Process Res. Dev. 2012, 16, 888–900. doi:10.1021/op200242s |
| 48. | Adamo, A.; Heider, P. L.; Weeranoppanant, N.; Jensen, K. F. Ind. Eng. Chem. Res. 2013, 52, 10802–10808. doi:10.1021/ie401180t |
| 49. | Overmans, S.; Ignacz, G.; Beke, A. K.; Xu, J.; Saikaly, P. E.; Szekely, G.; Lauersen, K. J. Green Chem. 2022, 24, 5479–5489. doi:10.1039/d2gc00938b |
© 2022 García-Lacuna and Baumann; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.
