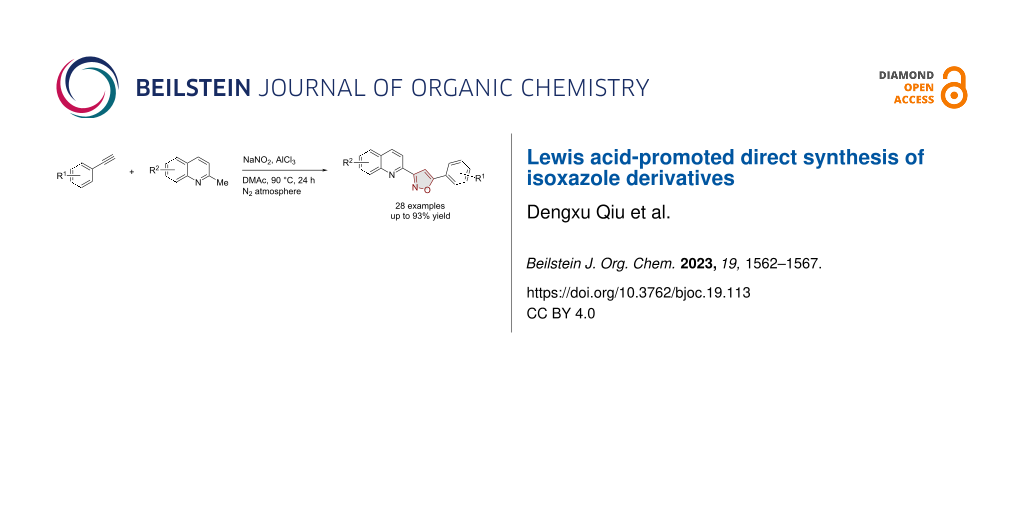
Abstract
Isoxazole derivatives were synthesized via a one-pot method utilizing 2-methylquinoline derivatives as template substrates, sodium nitrite as a nitrogen-oxygen source, and solely using aluminum trichloride as the additive. This approach circumvents the need for costly or highly toxic transition metals and presents a novel pathway for the synthesis of isoxazole derivatives.

Graphical Abstract
Introduction
The isoxazole derivatives not only exist in many natural products [1-3] and pharmaceutical intermediates [4-7], but also have great application values in organic synthesis [8,9] (Figure 1). In the past decades, many methods have been developed to prepare isoxazole derivatives [10-13]. However, most of the starting materials for these methods are oximes and hydroximinoyl chlorides [4,13-15]. Recently, the sp3 C–H bond functional group transformation of 2-methylquinoline derivatives into isoxazole derivatives has been reported [16]. In 2015, Yang’s group [10,17] reported the copper-catalyzed conversion of methylarenes into isoxazole derivatives with KNO3 as the source of nitrile oxide (Scheme 1, reaction 1). In 2019, Deng’s group [18] developed a three-component synthesis method of isoxazole derivatives using TBN as nitrogen source (Scheme 1, reaction 2). In 2017, Xu and co-workers [19] developed a copper-mediated annulation reaction to synthesize isoxazoles from two different alkynes. In fact, most methods mostly used highly toxic transition-metal catalysts such as copper metals. In order to develop cheaper and more environmentally friendly catalysts, our laboratory recently developed an alternative approach to the synthesis of isooxazoles starting from 2-methylquinoline and alkynes mediated by Brønsted acids in good yields (Scheme 1, reaction 3) [20].
![[1860-5397-19-113-1]](/bjoc/content/figures/1860-5397-19-113-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Natural products and drug molecules containing isoxazole moieties.
Figure 1: Natural products and drug molecules containing isoxazole moieties.
![[1860-5397-19-113-i1]](/bjoc/content/inline/1860-5397-19-113-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Traditional methods for the synthesis of isoxazoles and the current approach.
Scheme 1: Traditional methods for the synthesis of isoxazoles and the current approach.
The utilization of main element metal aluminum salts in organic synthesis holds significant potential due to their cost-effectiveness as compared to heavy metals. This renders them highly valuable for various applications in the field. Herein, we successfully developed a method that uses sodium nitrite as the source of nitrile oxide, and only applies aluminium trichloride as the Lewis acid to realize the sp3 C–H-bond activation of nitrogen heterocycles to synthesize isoxazole derivatives.
Results and Discussion
At the outset of this study, we chose the reaction of 2-methylquinoline (2a) with phenylacetylene (1a) in the presence of AlCl3 (3 equiv) and sodium nitrite (10 equiv) in DMAc at 90 °C under a nitrogen atmosphere. The desired isoxazole product 3a was isolated in 92% yield (Table 1, entry 1). It was found that no product was formed in the absence of AlCl3 and 64% yield were obtained when the reaction was performed using 2 equiv AlCl3 (Table 1, entries 2 and 3). When 5 equiv sodium nitrite were used, the corresponding yield was also decreased (Table 1, entry 4). Other Lewis acids employed in the reaction were less effective than AlCl3 (Table 1, entries 5–7). Furthermore, solvent screening showed that DMAc was the best reaction medium for this cycloaddition compared with DMSO and DMF (Table 1, entries 8 and 9). The reaction yield was decreased to 21% when increasing the temperature to 140 °C under standard conditions (Table 1, entry 10). Finally, the nitrogen atmosphere was essential since the yield substantially decreased under air atmosphere (Table 1, entry 11).
Table 1: Optimization of reaction conditions for the synthesis of isoxazolesa.
![[Graphic 1]](/bjoc/content/inline/1860-5397-19-113-i6.svg?max-width=637&scale=1.0)
|
||
| Entry | Variation from standard conditions | Yield (%)b |
| 1 | none | 92 |
| 2 | without AlCl3 | n.r. |
| 3 | 2 equiv of AlCl3 | 64 |
| 4 | 5 equiv of NaNO2 | 77 |
| 5 | FeCl3 instead of AlCl3 | 55 |
| 6 | TiCl4 instead of AlCl3 | 67 |
| 7 | BF3 instead of AlCl3 | n.r. |
| 8 | DMF instead of DMAc | 72 |
| 9 | DMSO instead of DMAc | n.r. |
| 10 | at 140 °C | 21 |
| 11 | under air | 26 |
aStandard reaction conditions: 1a (0.1 mmol, 1 equiv), 2a (0.2 mmol, 2 equiv), AlCl3 (0.3 mmol, 3 equiv), NaNO2 (1 mmol, 10.0 equiv), DMAc (1.0 mL), N2 atmosphere, 90 °C, 24 h. bIsolated yield; n.r., no reaction.
With the optimal reaction conditions in hand, various alkynes were examined as dipolarophiles (Scheme 2). A range of functional groups were tolerated in this reaction, such as alkyl, methoxy, halo, and heterocycles. It was found that electron-deficient groups in the phenyl ring (3g–i) were more beneficial to the reaction outcome than electron-rich groups in the phenyl ring (3a–f). The crystal structure of product 3i is shown in Figure 2. Also, substituents in different positions of the phenyl ring in acetylene 1 smoothly reacted with NaNO2 under the reaction conditions affording the products in good to excellent yields, which showed that the steric hindrance has little effect on the reaction (3j–n). Furthermore, some heteroaromatic and aliphatic alkynes were also utilized, and the corresponding products 3o and 3p were isolated in good yields. We also tried 1,2-diphenylethyne as sustrate, which is an internal alkyne instead of a terminal alkyne, but no desired product was obtained.
![[1860-5397-19-113-i2]](/bjoc/content/inline/1860-5397-19-113-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Reaction scope of alkynes. Conditions: 1 (0.1 mmol, 1 equiv), 2a (0.2 mmol, 2 equiv), AlCl3 (0.3 mmol, 3 equiv), NaNO2 (1 mmol, 10.0 equiv), DMAc (1.0 mL), N2 atmosphere, 90 °C, 24 h.
Scheme 2: Reaction scope of alkynes. Conditions: 1 (0.1 mmol, 1 equiv), 2a (0.2 mmol, 2 equiv), AlCl3 (0.3 mm...
![[1860-5397-19-113-2]](/bjoc/content/figures/1860-5397-19-113-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Next, we explored the substrate scope of 2-methylquinolines under the standard conditions. 2-Methylquinoline bearing different substituents at various positions gave the corresponding products with moderate to good yields (Scheme 3). It was observed that 2-methylquinoline with electron-deficient functional groups afforded the corresponding products in excellent yields of up to 92% (4a–c). Likewise, 2-methylquinoline substituted with electron-rich functional groups were suitable substrates and achieved good results (4d and 4e). Fortunately, various functional groups in different positions were also tolerated in the reaction (4f–k). Moreover, this reaction could be carried out with 1-methylisoquinoline as substrate, which afforded product 4l in 93% yield. Besides, we also tried 2-methylpyridine and 4-methylquinoline as substrates, but no reaction was detected under the standard conditions (4m and 4n). In addition, there was no product formed when 2-formylquinoline was used as the substrate.
![[1860-5397-19-113-i3]](/bjoc/content/inline/1860-5397-19-113-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reaction substrate scope of quinolines. Conditions: 1a (0.1 mmol, 1 equiv), 2 (0.2 mmol, 2 equiv), AlCl3 (0.3 mmol, 3 equiv), NaNO2 (1 mmol, 10.0 equiv), DMAc (1.0 mL), N2 atmosphere, 90 °C, 24 h.
Scheme 3: Reaction substrate scope of quinolines. Conditions: 1a (0.1 mmol, 1 equiv), 2 (0.2 mmol, 2 equiv), ...
To further demonstrate the synthetic versatility of this developed method, we carried out the reaction in a gram scale. It was found that the desired product could be obtained in 87% yield (Scheme 4).
![[1860-5397-19-113-i4]](/bjoc/content/inline/1860-5397-19-113-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Next, some control experiments were carried out to study the reaction mechanism. We found that the reaction of compound 3a could not be inhibited by TEMPO and BHT under the standard conditions. Therefore, it is assumed that the reaction is not a free radical reaction.
Based on the control experiments and previous literature [21], we propose the following possible mechanism, which is shown in Scheme 5. Aluminum trichloride reacts with sodium nitrite to form an intermediate aluminum complex A, which is further complexed with the starting material 2a to generate intermediate B and HONO [22]. Then, the intermediate B conjugates with HONO to generate intermediate C [22]. Next, the intermediate D is produced by the same progress. The intermediate D then undergoes elimination of nitroxylic acid to produce nitrile oxide E [23], which can be converted to the desired isoxazole with 1a through a 1,3-dipolar cycloaddition.
![[1860-5397-19-113-i5]](/bjoc/content/inline/1860-5397-19-113-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Control experiments and possible reaction mechanism.
Scheme 5: Control experiments and possible reaction mechanism.
Conclusion
In conclusion, we have developed an efficient and concise synthesis of isoxazole nitrogen heterocycles by direct C–H-bond activation of methyl heteroaromatics. The method avoids using toxic transition metals and provides a new way to synthesize isoxazole molecules. Further related transformations of products and application of this method are currently developed in our laboratory.
Experimental
Representative procedure for the synthesis of compound 3a. To a flame-dried 15 mL Schlenk tube filled with nitrogen, 2-methylquinoline (2a, 28.6 mg, 0.2 mmol), phenylacetylene (1a, 10.2 mg, 0.1 mmol), AlCl3 (40.0 mg, 0.3 mmol), sodium nitrite (35.0 mg, 1.0 mmol), and absolute dry DMAc (1.0 mL) were added under nitrogen. The formed mixture was stirred at 90 °C under nitrogen for 24 h with TLC monitoring. Upon completion, the solution was cooled to room temperature and the solvent was removed under vacuum directly. The crude residue was purified by column chromatography on silica gel (ethyl acetate/petroleum ether 40:1) to afford product 3a with 87% yield.
Supporting Information
| Supporting Information File 1: Characterization data and copies of spectra. | ||
| Format: PDF | Size: 4.1 MB | Download |
Funding
We are grateful for the financial support from the China Postdoctoral Science Foundation (2018T110533), the Natural Science Foundation of Jiangsu Province (BK20161328, BK20210789), and the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
-
Li, J.; Hu, M.; Li, C.; Li, C.; Li, J.; Wu, W.; Jiang, H. Adv. Synth. Catal. 2018, 360, 2707–2719. doi:10.1002/adsc.201800444
Return to citation in text: [1] -
Chalyk, B. A.; Hrebeniuk, K. V.; Gavrilenko, K. S.; Kulik, I. B.; Rozhenko, A. B.; Volochnyuk, D. M.; Liashuk, O. S.; Grygorenko, O. O. Eur. J. Org. Chem. 2018, 5585–5595. doi:10.1002/ejoc.201800753
Return to citation in text: [1] -
Duan, M.; Hou, G.; Zhao, Y.; Zhu, C.; Song, C. J. Org. Chem. 2022, 87, 11222–11225. doi:10.1021/acs.joc.2c00896
Return to citation in text: [1] -
Oancea, A.; Georgescu, E.; Georgescu, F.; Nicolescu, A.; Oprita, E. I.; Tudora, C.; Vladulescu, L.; Vladulescu, M.-C.; Oancea, F.; Deleanu, C. Beilstein J. Org. Chem. 2017, 13, 659–664. doi:10.3762/bjoc.13.65
Return to citation in text: [1] [2] -
Chen, Y.; Li, L.; He, X.; Li, Z. ACS Catal. 2019, 9, 9098–9102. doi:10.1021/acscatal.9b03189
Return to citation in text: [1] -
Tu, K. N.; Hirner, J. J.; Blum, S. A. Org. Lett. 2016, 18, 480–483. doi:10.1021/acs.orglett.5b03530
Return to citation in text: [1] -
Kumar, P.; Kapur, M. Org. Lett. 2020, 22, 5855–5860. doi:10.1021/acs.orglett.0c01935
Return to citation in text: [1] -
Kandula, V.; Bandaru, S. K.; Balakrishna, C.; Chatterjee, A.; Yennam, S.; Saunders, G. C.; Behera, M. Eur. J. Org. Chem. 2020, 6440–6446. doi:10.1002/ejoc.202001103
Return to citation in text: [1] -
Fu, M.; Li, H.; Su, M.; Cao, Z.; Liu, Y.; Liu, Q.; Guo, C. Adv. Synth. Catal. 2019, 361, 3420–3429. doi:10.1002/adsc.201900323
Return to citation in text: [1] -
Wang, G.-W.; Li, S.-X.; Wu, Q.-X.; Yang, S.-D. Org. Chem. Front. 2015, 2, 569–573. doi:10.1039/c5qo00053j
Return to citation in text: [1] [2] -
Sun, Y.; Abdukader, A.; Zhang, H.; Yang, W.; Liu, C. RSC Adv. 2017, 7, 55786–55789. doi:10.1039/c7ra11436b
Return to citation in text: [1] -
Hossain, M. I.; Khan, M. I. H.; Kim, S. J.; Le, H. V. Beilstein J. Org. Chem. 2022, 18, 446–458. doi:10.3762/bjoc.18.47
Return to citation in text: [1] -
Wu, W.; Chen, Q.; Tian, Y.; Xu, Y.; Huang, Y.; You, Y.; Weng, Z. Org. Chem. Front. 2020, 7, 1878–1883. doi:10.1039/d0qo00243g
Return to citation in text: [1] [2] -
Poh, J.-S.; García-Ruiz, C.; Zúñiga, A.; Meroni, F.; Blakemore, D. C.; Browne, D. L.; Ley, S. V. Org. Biomol. Chem. 2016, 14, 5983–5991. doi:10.1039/c6ob00970k
Return to citation in text: [1] -
Ledovskaya, M. S.; Rodygin, K. S.; Ananikov, V. P. Org. Chem. Front. 2018, 5, 226–231. doi:10.1039/c7qo00705a
Return to citation in text: [1] -
Duc, D. X.; Dung, V. C. Curr. Org. Chem. 2021, 25, 2938–2989. doi:10.2174/1385272825666211118104213
Return to citation in text: [1] -
Wang, G.-W.; Cheng, M.-X.; Ma, R.-S.; Yang, S.-D. Chem. Commun. 2015, 51, 6308–6311. doi:10.1039/c5cc01004g
Return to citation in text: [1] -
Wang, D.; Zhang, F.; Xiao, F.; Deng, G.-J. Org. Biomol. Chem. 2019, 17, 9163–9168. doi:10.1039/c9ob01909j
Return to citation in text: [1] -
Li, Y.; Gao, M.; Liu, B.; Xu, B. Org. Chem. Front. 2017, 4, 445–449. doi:10.1039/c6qo00704j
Return to citation in text: [1] -
Meng, L.; Zhang, S.; Jia, X.; Lv, L.; Yuan, Y. Tetrahedron Lett. 2019, 60, 1443–1447. doi:10.1016/j.tetlet.2019.04.047
Return to citation in text: [1] -
Ge, J.-J.; Yao, C.-Z.; Wang, M.-M.; Zheng, H.-X.; Kang, Y.-B.; Li, Y. Org. Lett. 2016, 18, 228–231. doi:10.1021/acs.orglett.5b03367
Return to citation in text: [1] -
Das, D.; Jain, P.; Pal, S.; Avasare, V. J. Phys. Chem. C 2019, 123, 23439–23445. doi:10.1021/acs.jpcc.9b02529
Return to citation in text: [1] [2] -
Baidya, M.; Yamamoto, H. J. Am. Chem. Soc. 2011, 133, 13880–13882. doi:10.1021/ja206736q
Return to citation in text: [1]
| 1. | Li, J.; Hu, M.; Li, C.; Li, C.; Li, J.; Wu, W.; Jiang, H. Adv. Synth. Catal. 2018, 360, 2707–2719. doi:10.1002/adsc.201800444 |
| 2. | Chalyk, B. A.; Hrebeniuk, K. V.; Gavrilenko, K. S.; Kulik, I. B.; Rozhenko, A. B.; Volochnyuk, D. M.; Liashuk, O. S.; Grygorenko, O. O. Eur. J. Org. Chem. 2018, 5585–5595. doi:10.1002/ejoc.201800753 |
| 3. | Duan, M.; Hou, G.; Zhao, Y.; Zhu, C.; Song, C. J. Org. Chem. 2022, 87, 11222–11225. doi:10.1021/acs.joc.2c00896 |
| 4. | Oancea, A.; Georgescu, E.; Georgescu, F.; Nicolescu, A.; Oprita, E. I.; Tudora, C.; Vladulescu, L.; Vladulescu, M.-C.; Oancea, F.; Deleanu, C. Beilstein J. Org. Chem. 2017, 13, 659–664. doi:10.3762/bjoc.13.65 |
| 13. | Wu, W.; Chen, Q.; Tian, Y.; Xu, Y.; Huang, Y.; You, Y.; Weng, Z. Org. Chem. Front. 2020, 7, 1878–1883. doi:10.1039/d0qo00243g |
| 14. | Poh, J.-S.; García-Ruiz, C.; Zúñiga, A.; Meroni, F.; Blakemore, D. C.; Browne, D. L.; Ley, S. V. Org. Biomol. Chem. 2016, 14, 5983–5991. doi:10.1039/c6ob00970k |
| 15. | Ledovskaya, M. S.; Rodygin, K. S.; Ananikov, V. P. Org. Chem. Front. 2018, 5, 226–231. doi:10.1039/c7qo00705a |
| 10. | Wang, G.-W.; Li, S.-X.; Wu, Q.-X.; Yang, S.-D. Org. Chem. Front. 2015, 2, 569–573. doi:10.1039/c5qo00053j |
| 11. | Sun, Y.; Abdukader, A.; Zhang, H.; Yang, W.; Liu, C. RSC Adv. 2017, 7, 55786–55789. doi:10.1039/c7ra11436b |
| 12. | Hossain, M. I.; Khan, M. I. H.; Kim, S. J.; Le, H. V. Beilstein J. Org. Chem. 2022, 18, 446–458. doi:10.3762/bjoc.18.47 |
| 13. | Wu, W.; Chen, Q.; Tian, Y.; Xu, Y.; Huang, Y.; You, Y.; Weng, Z. Org. Chem. Front. 2020, 7, 1878–1883. doi:10.1039/d0qo00243g |
| 8. | Kandula, V.; Bandaru, S. K.; Balakrishna, C.; Chatterjee, A.; Yennam, S.; Saunders, G. C.; Behera, M. Eur. J. Org. Chem. 2020, 6440–6446. doi:10.1002/ejoc.202001103 |
| 9. | Fu, M.; Li, H.; Su, M.; Cao, Z.; Liu, Y.; Liu, Q.; Guo, C. Adv. Synth. Catal. 2019, 361, 3420–3429. doi:10.1002/adsc.201900323 |
| 22. | Das, D.; Jain, P.; Pal, S.; Avasare, V. J. Phys. Chem. C 2019, 123, 23439–23445. doi:10.1021/acs.jpcc.9b02529 |
| 4. | Oancea, A.; Georgescu, E.; Georgescu, F.; Nicolescu, A.; Oprita, E. I.; Tudora, C.; Vladulescu, L.; Vladulescu, M.-C.; Oancea, F.; Deleanu, C. Beilstein J. Org. Chem. 2017, 13, 659–664. doi:10.3762/bjoc.13.65 |
| 5. | Chen, Y.; Li, L.; He, X.; Li, Z. ACS Catal. 2019, 9, 9098–9102. doi:10.1021/acscatal.9b03189 |
| 6. | Tu, K. N.; Hirner, J. J.; Blum, S. A. Org. Lett. 2016, 18, 480–483. doi:10.1021/acs.orglett.5b03530 |
| 7. | Kumar, P.; Kapur, M. Org. Lett. 2020, 22, 5855–5860. doi:10.1021/acs.orglett.0c01935 |
| 23. | Baidya, M.; Yamamoto, H. J. Am. Chem. Soc. 2011, 133, 13880–13882. doi:10.1021/ja206736q |
| 19. | Li, Y.; Gao, M.; Liu, B.; Xu, B. Org. Chem. Front. 2017, 4, 445–449. doi:10.1039/c6qo00704j |
| 21. | Ge, J.-J.; Yao, C.-Z.; Wang, M.-M.; Zheng, H.-X.; Kang, Y.-B.; Li, Y. Org. Lett. 2016, 18, 228–231. doi:10.1021/acs.orglett.5b03367 |
| 18. | Wang, D.; Zhang, F.; Xiao, F.; Deng, G.-J. Org. Biomol. Chem. 2019, 17, 9163–9168. doi:10.1039/c9ob01909j |
| 22. | Das, D.; Jain, P.; Pal, S.; Avasare, V. J. Phys. Chem. C 2019, 123, 23439–23445. doi:10.1021/acs.jpcc.9b02529 |
| 10. | Wang, G.-W.; Li, S.-X.; Wu, Q.-X.; Yang, S.-D. Org. Chem. Front. 2015, 2, 569–573. doi:10.1039/c5qo00053j |
| 17. | Wang, G.-W.; Cheng, M.-X.; Ma, R.-S.; Yang, S.-D. Chem. Commun. 2015, 51, 6308–6311. doi:10.1039/c5cc01004g |
| 16. | Duc, D. X.; Dung, V. C. Curr. Org. Chem. 2021, 25, 2938–2989. doi:10.2174/1385272825666211118104213 |
| 20. | Meng, L.; Zhang, S.; Jia, X.; Lv, L.; Yuan, Y. Tetrahedron Lett. 2019, 60, 1443–1447. doi:10.1016/j.tetlet.2019.04.047 |
© 2023 Qiu et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.