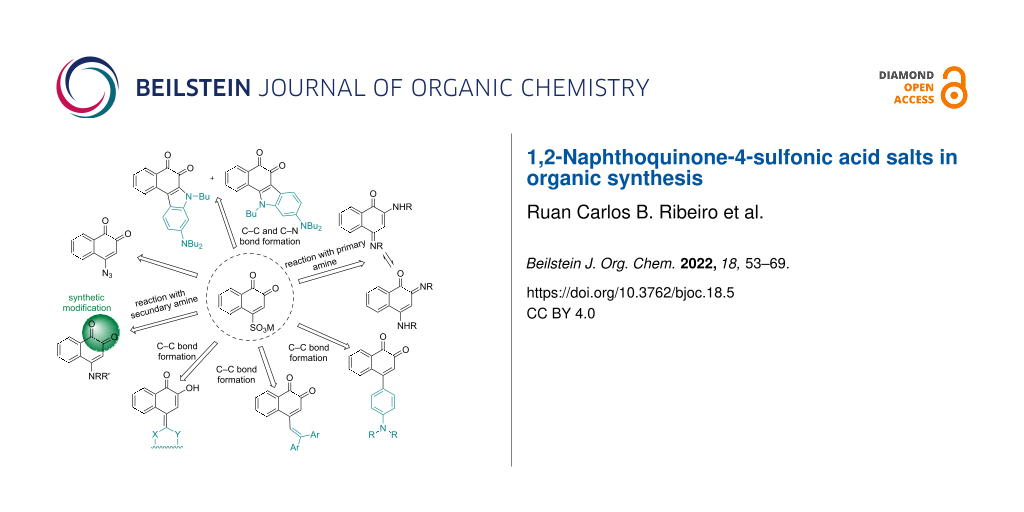
Abstract
Several low molecular weight naphthoquinones are very useful in organic synthesis. These compounds have given rise to thousands of other naphthoquinones that have been tested against various microorganisms and pharmacological targets, including being used in the preparation of several drugs that are on the pharmaceutical market. Among these naphthoquinones, the series of compounds prepared from 1,2-naphthoquinone-4-sulfonic acid salts (β-NQS) stands out. In addition to being used in organic synthesis, they are excellent analytical derivatization reagents to spectrophotometrically determine drugs containing primary and secondary amino groups. This review summarizes the literature involving β-NQS.

Graphical Abstract
Introduction
The general class of quinones is very important because their compounds show biological activities against several pathogens related to important diseases and are used for the production of special materials [1-4]. These compounds are biosynthesized by oxidative processes of catecholamines and other compounds, but they can also be ingested as exogenous products of air and water. The most common quinones, such as benzoquinone, naphthoquinone, anthraquinone, and phenanthrenequinone, can be formed by incomplete combustion or photooxidation of their respective polycyclic aromatic hydrocarbons (PAHs) [5,6].
Among all of the compounds in this class, 1,2- and 1,4-naphthoquinones stand out, as they are present in plants, fungi, lichens, bacteria, algae, viruses, insects, and higher organisms and perform several biochemical functions, such as defense, transference of electrons in various oxidative processes in aerobic metabolism, photosynthesis, oxidative phosphorylation, blood clotting, and other electron transport reactions [7,8]. These biochemical functions give them several biological activities, such as antibacterial, fungicidal, antimalarial, trypanocidal, and antitumor. For this reason, many plant extracts that are rich in naphthoquinones continue to be widely used in folk medicine in several countries. More than 350 naphthoquinones that have been isolated from nature are described in the literature, and it is the most abundant structural subunit in the quinone family. Through the synthesis of new naphthoquinones by innovative methods, this class is constantly expanding. Several hypotheses have been formulated and tested to explain the biological activity of these substances. In general, activities against microorganisms are related to the ability to accept one and/or two electrons through a redox cycle promoted by the 1,2- or 1,4-naphthoquinone system. In this cycle, transient reactive oxygen (ROS) and nitrogen (RNS) species are formed as free radicals, peroxides, superoxide anions, radical anions, or dianions. These species generated inside cells accelerate hypoxia and cause several damages to its components, such as carbohydrates, lipids, membrane components, and enzymes that are critical for DNA replication [9-12]. Most synthetic strategies toward naphthoquinones with potential biological activity start from natural and synthetic naphthoquinones, inserting new fragments in the general structure or modifying functional groups. In Figure 1, eight low molecular weight naphthoquinones are highlighted, which are the most commonly used in organic synthesis. Among them, quinones 1–5 are naturally occurring, are simple to prepare and are commercially available. Others are exclusively synthetic (6–8) and prepared from naphthoquinones 1 or 2. Except for naphthoquinone 8 (β-NQS), the applications in organic synthesis of all other in Figure 1 shown naphthoquinones are already summarized in reviews [13-20].
![[1860-5397-18-5-1]](/bjoc/content/figures/1860-5397-18-5-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Naphthoquinones are commonly used in organic synthesis.
Figure 1: Naphthoquinones are commonly used in organic synthesis.
Several natural naphthoquinones with antibacterial, fungicidal, antimalarial, antiviral, trypanocidal, leishmanicidal, and antitumor activity serve as inspiration for the pharmaceutical industry [21-24]. However, they are considered as Pan Assay Interference compounds (PAINS) because they display biological activity in many assays, but because of such reactivity it can be very difficult to advance to the clinic against drug targets [25,26]. Among the most prominent natural naphthoquinones are vitamin K1 (9), lapachol (10), and β-lapachone (11) (Figure 2A). All of these compounds have important characteristics, but it should be noted that in addition to their biological properties, these compounds have served as raw materials for the synthesis of new naphthoquinone derivatives [27]. Drugs belonging to the class of naphthoquinones are on the pharmaceutical market for the treatment of various diseases. In Figure 2B, three drugs are highlighted that continue to be used in medical practice. Atovaquone (12) is a drug that targets the elimination of the parasite Plasmodium spp. which is the etiological agent of malaria [28,29]. According to data from the World Health Organization (WHO), in 2018, there were approximately 228 million cases of malaria worldwide, with the majority of cases in Africa. It is a serious illness that can lead to death if not treated immediately. This medication has therapeutic use for the treatment or prevention of mild cases of Plasmodium vivae infection. Two other drugs structurally similar to lapachol, buparvaquone (13) and parvaquone (14), are used to treat animal diseases, such as bovine theileriosis (east coast fever, corridor disease, Zimbabwean theileriosis, and tropical theileriosis) [30-33]. Buparvaquone (13), phosphate prodrugs, and some formulations were evaluated in vitro and in vivo against Leishmania donovani, which causes visceral and cutaneous leishmaniasis. It has been observed that the prodrugs improved efficacy when compared to buparvaquone. Parvaquone (14) is a naphthoquinone with antitheilerial properties that is commercialized for the treatment of East Coast fever. This drug is effective in the treatment of cattle infected with Theileria annulata transmitted by the brown tick Rhipicephalus appendiculatans [34-36].
![[1860-5397-18-5-2]](/bjoc/content/figures/1860-5397-18-5-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Some important natural and synthetic naphthoquinones.
Figure 2: Some important natural and synthetic naphthoquinones.
As part of our research program on the synthesis of biologically active quinones, we are interested in the synthesis and biological evaluation of naphthoquinones obtained by short routes from readily available starting materials [37-39]. This review summarizes literature data involving 1,2-naphthoquinone-4-sulfonic acid salts (β-NQS), organized based on the general classification of reactions, and explores the possibility of providing practical guidance to synthetic chemists for further research on naphthoquinone compounds.
Review
Synthesis of 1,2-naphthoquinone-4-sulfonic acid salts
β-NQS are easily synthesized by traditional methods and commercialized by dozens of chemical companies, such as VladaChem, Ambeed, eNovation Chemicals, Tokyo Chemical Industry, Acros Organics, Abcr GmbH, Sigma-Aldrich, and others. Due to this wide commercial availability, these compounds have been widely used in several industrial applications as analytical reagents for the determination of amines and in the synthesis of other hybrid naphthoquinones [40-42].
β-NQS preparation methods generally employ β-naphthol or 1-amino-β-naphthol as starting materials. The first procedure was developed by Witt [43] in the late 19th century when he prepared 1,2-naphthoquinone-4-sulfonic acid ammonium salt (16) in a 60–75% yield from 1-amino-β-naphthol-4-sulfonic acid (15) by oxidation with nitric acid in an aqueous medium (Scheme 1A). In 1894, Böniger developed [44] the first reaction of β-NQS with phenylamines. He synthesized β-NQS using a modified method developed by Witt, which quickly reacted with different amines to form colored products with reddish hues. The reaction with aniline forms the substitution product of the sulfonic group with a phenylamino group in a 90% yield. In his study, he proposed that the structure of the nucleophilic addition product was tautomer 19 (Scheme 1B).
![[1860-5397-18-5-i1]](/bjoc/content/inline/1860-5397-18-5-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic studies of BNQs and reactions with amines.
Scheme 1: Synthetic studies of BNQs and reactions with amines.
In search of a reagent that could form stable adducts with amino acids to be used as colorimetric indicators, Folin [45] developed a method based on naphthoquinone since there were records in the literature that indicated that naphthoquinones reacted with amines and proteins to form colored products. Among the tested o-naphthoquinones, he found that 1,2-naphthoquinone-4-sulfonic acid sodium salt (β-NQSNa, 18) was very effective as a colorimetric indicator of blood amino acids. This compound came to be called Folin's reagent. To achieve 18 with adequate purity to be used in the tests, an elaborate large-scale synthetic route was developed. β-Naphthol (20) was transformed into α-nitroso-β-naphthol (17); then, in a single step, a sulfonic group was added, and the nitrous group was reduced, forming compound 15, which was transformed into β-NQSNa (18) after oxidation with nitric acid. Despite not knowing exactly the structure of the adduct, Folin speculated that the reaction probably occurs in the o-quinone moiety group. Subsequently, Obo [46] demonstrated that the reaction of β-NQSNa (18) and glycine ethyl ester form 19 in a 46% yield, indicating that the reaction occurred at C4 (Scheme 2). Fu and co-workers [47] prepared a new electrochemical sensor for the specific recognition of cholylglycine, which is a combination of cholic acid and glycine. The β-cyclodextrin/graphene oxide composite forms an inclusion complex with a β-NQS guest. The amino group of cholylglycine can bind to β-NQS by a nucleophilic substitution reaction, resulting in a decrease in the electrochemical signal. Danielson [48] found some errors in Folin’s analytical method and optimized it for better amino acid determination. Martin and Fieser [49] described an optimized method, analogous to Folin’s procedures, with temperature control, producing β-NQSNH4 16 and β-NQSK 20 in high yields (Scheme 2).
![[1860-5397-18-5-i2]](/bjoc/content/inline/1860-5397-18-5-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Methods described for the synthesis of β-NQS.
Scheme 2: Methods described for the synthesis of β-NQS.
It is possible to identify three alternatives for the functionalization of β-NQS with amines: a) substitution of sulfonate by secondary amines; b) substitution of sulfonate by primary amines, followed by isomerization; and c) double addition of primary amines. In addition, other nucleophiles can also be used. All cases will be covered throughout the text.
Synthesis of 4-amino-β-naphthoquinones and analogues from β-NQS
After the first discovery that β-NQS reacts quickly with amines to form colored products in good yields, this reagent became quite popular in quantitative analytical determinations of some drugs containing free primary and secondary amino groups [50,51]. As β-NQS is commercially available, it has become a widely used reagent for the chromogenic determination of pharmaceutical amines by spectrophotometry in pharmaceutical formulations [52]. This method, which uses a reagent to form a colored product and determine its concentration by a spectrophotometric method, is the most convenient, simple, and inexpensive method for analytical work. Hiyama [53] noted that little is known about the biological activities of sulfonic naphthalene derivatives, despite being important intermediates for the synthesis of dyes. Then, he prepared several naphthalene sulfonic derivatives and tested them for their effects on bacteria and viruses, but none of the compounds presented important activity.
Hashimoto and co-workers [54] were the first to apply β-NQSNa for the qualitative analysis of phenethylamine derivatives (amphetamine, methamphetamine, 2,5-dimethoxy-4-methylamphetamine, mescaline, ephedrine, and norephedrine). The reaction products were separated by thin-layer chromatography and analyzed by elemental analysis, nuclear magnetic resonance, infrared spectroscopy, and mass spectrometry. In addition, several authors concluded that this was a good method for analytically determining low levels of activated aromatic amines in drugs. This method continued to be used over the years and was subsequently optimized by a spectrophotometric determination technique coupled with continuous flow [55]. In addition to the sulfonic acid substitution reaction of position C4 of β-NQS, quinone can be involved in a redox process and, therefore, can be used as an electrode in electrochemical processes. Subsequently, Legua and co-workers [56,57] applied this method to determine amphetamines in urine. Figure 3 highlights some important drugs containing primary or secondary amino groups in capsules, tablets, powders, formulations, formulations of associated drugs, injection formulations, and biological fluids [42,58-69].
![[1860-5397-18-5-3]](/bjoc/content/figures/1860-5397-18-5-3.svg?scale=2.0&max-width=1024&background=FFFFFF)
β-NQS reacts with aliphatic and aromatic amines, secondary or primary, by substituting the sulfonic acid group at position C4. These reactions are dependent on the structures of the aliphatic amino reagents. In the case of secondary aliphatic amines, 4-alkyl- (or aryl-) amino-1,2-naphthoquinones 21 are formed (Scheme 3A), but using primary amines forms a product mixture (Scheme 3B), mainly due to a tautomeric equilibrium (Scheme 3C) [41]. Hartke and Lohmann [70] studied the reaction of β-NQS with secondary aliphatic amines in detail and observed that the 4-amino-1,2-naphthoquinone 21 products are yellow. However, reactions with primary aliphatic amines also form 4-amino-1,2-naphthoquinones 22, but they are violet in color. Structure 22 cannot be responsible for the violet color, and it is attributed to other byproducts. When the reaction with equimolar amounts of β-NQSNa with methyl-, ethyl- and isopropylamine takes place in water at room temperature, a mixture of products is formed, among which 22 and 23 are the majority. It is important to note that the complexity of the reaction of β-NQS with primary aliphatic amines has already been reported in the literature. Fieser and Fieser [71] studied the reduction potentials of various naphthoquinones and reported that they were unable to obtain 4-arylamino-1,2-naphthoquinones from β-NQSNa but that these derivatives can be readily prepared from 4-ethoxy-1,2-naphthoquinone. Similarly, Yano and co-workers [72] studied the tautomeric equilibrium of 4-arylamino-1,2-naphthoquinones in DMSO-d6, pyridine-d5, and NaOD solutions in D2O. In neutral solvents, the most stable tautomer is 4-arylamino-1,2-naphthoquinone A, while in weakly basic solvents, or ethanolic sulfuric acid, B is the most stable tautomer (Scheme 3C).
![[1860-5397-18-5-i3]](/bjoc/content/inline/1860-5397-18-5-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Reactions between β-NQS and amines.
Scheme 3: Reactions between β-NQS and amines.
Reactions employing equimolecular amounts of β-NQS and primary arylamines are cleaner and form the substitution product at position C4. Fieser and Fieser were the first to study tautomerism between 4-arylamino-1,2-naphthoquinone A and 2-hydroxy-1,4-naphthoquinone-4-arylimine B (Scheme 3C) using the redox potential compared to the pH of the medium.
It was observed that naphthoquinones A prevail in all pH regions except for extreme acidity, where there is a shift to the form of 2-hydroxy-1,4-naphthoquinone-4-arylimines [73,74]. However, in weakly acidic or alkaline solutions, A is the most stable tautomer (Scheme 3C) [75]. Fragoso and co-workers [76] studied the tautomeric equilibrium between A and B using semiempirical calculations (AM1 and PM3) and DFT (B3LYP/6-31G(d)) in the gas phase and water, where it was observed that in the gas phase B is the most stable, while in water A is formed, which is in agreement with the experimental results reported in the literature. There was no effect of the substituents on the phenyl group on the stability of the two tautomers.
A method to isomerize 22 to 26 was developed by Gornostaev and co-workers [77]. This method involves refluxing 22 in 85% aqueous acetic acid leading to 26 in 58–65% yield. The proposal to explain the isomerization involves two routes: one through the hydrolysis of 22 leading to lawsone (4) and the subsequent addition of arylamines in position C2, and the other involves the addition of arylamines at position C2 of 22, leading to 24 with two equivalents of arylamine, which after hydrolysis of the imine at position C4 provides 26 (Scheme 4).
![[1860-5397-18-5-i4]](/bjoc/content/inline/1860-5397-18-5-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Isomerization of 4-arylamino-1,2-naphthoquinones.
Scheme 4: Isomerization of 4-arylamino-1,2-naphthoquinones.
The same group developed reaction conditions using different hydrophilic solvents to synthesize unsymmetrical 2-amino-4-imino compounds 28 from 27 in good yields by employing primary aromatic amines (Scheme 5) [78].
![[1860-5397-18-5-i5]](/bjoc/content/inline/1860-5397-18-5-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of unsymmetrical 2-amino-4-imino compounds.
Scheme 5: Synthesis of unsymmetrical 2-amino-4-imino compounds.
Ortiz and co-workers [79] performed several reactions employing two equivalents of isoxazolylamines in aqueous solution under reflux with HCl catalysis, resulting in bis(isoxazolyl)naphthoquinones 29 (Scheme 6).
![[1860-5397-18-5-i6]](/bjoc/content/inline/1860-5397-18-5-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of bis(isoxazolyl)naphthoquinones from β-NQS.
Scheme 6: Synthesis of bis(isoxazolyl)naphthoquinones from β-NQS.
The development of synthetic methodologies for the preparation of many bioactive substances is still a challenge. The main issues in synthetic organic chemistry and medicinal chemistry of naphthoquinones are the diversification of the strategies to obtain derivatives [8,80]. β-NQS are excellent electrophiles and have been used for obtaining naphthoquinones substituted by alkyl- or arylamines.
The reactivity of aminopyrazolopyridine 30 with β-NQSNa (18) for the preparation of 32 with antioxidant properties was investigated by Gouda in 2012 [81]. Intermediate 18 was treated with two equivalents of 30 resulting in bispyrazolopyridine 31, which after treatment with refluxing acetic acid produced imidazopyrazole 32 (Scheme 7). Compounds 31 and 32 were evaluated against their antioxidant activity and exhibited promising activity.
![[1860-5397-18-5-i7]](/bjoc/content/inline/1860-5397-18-5-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 7: The reaction of β-NQS with 30 followed by cycle condensation.
Scheme 7: The reaction of β-NQS with 30 followed by cycle condensation.
Protein tyrosine phosphatase 1B (PTP1B) is essential in the dephosphorylation of the activated insulin receptor, and inhibition of this enzyme would be an excellent strategy for the treatment of type 2 diabetes. Ahn and co-workers [82] synthesized and evaluated several 1,2-naphthoquinones substituted at position C4 with alkyl- or arylamino groups for their inhibition of the PTP1B protein. Furthermore, to discover new effective anti-inflammatory and analgesic agents, Gouda and co-workers [83] synthesized various compounds in good yields from the reaction of β-NQS 18 with 2-amino-5-selenothiazoles, such as 33 and 34. The authors reported that most of the compounds tested had similar anti-inflammatory properties or greater activity than meloxicam (Scheme 8).
![[1860-5397-18-5-i8]](/bjoc/content/inline/1860-5397-18-5-i8.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 8: Synthesis of 4-(2-amino-5-selenothiazoles)-1,2-naphthoquinones.
Scheme 8: Synthesis of 4-(2-amino-5-selenothiazoles)-1,2-naphthoquinones.
β-Lapachone (11) is a potent reversible inhibitor of the liver enzyme human carboxylesterase (hCE1) that cleaves carboxylic esters. This enzyme functions in the detoxification metabolism of carcinogenic and mutagenic organic compounds, converting them into nontoxic metabolites. This compound served as inspiration for Hatfield and co-workers [84], who proposed the synthesis of a series of amino-N-methylated compounds 36 and phenoxy-1,2-naphthoquinones 35 with a carbon skeleton similar to β-lapachone (11), which could modulate hCE1 activity. Studies have shown that amino-N-methylated-1,2-naphthoquinones 36 are more selective and potent inhibitors than phenoxy-1,2-naphthoquinones 35 and β-lapachone (11) for hCE1 (Scheme 9). It is important to note that 35 can be obtained from other reagents, such as 2-naphthol, in a cascade of reactions involving oxidation to 1,2-naphthoquinones followed by Michael addition to the olefin and reoxidation [85]. In addition, Yang and co-workers synthesized other naphthoquinone derivatives 37 from β-NQSNa (18) [86]. These compounds were evaluated for their antiproliferative activities on human cancer cells, with three of them being the most active (37a–c). It has been shown that the mechanism of action passes through the production of intracellular ROS and includes inhibition of tubulin polymerization (Scheme 9).
![[1860-5397-18-5-i9]](/bjoc/content/inline/1860-5397-18-5-i9.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 9: Synthesis of amino- and phenoxy-1,2-naphthoquinones.
Scheme 9: Synthesis of amino- and phenoxy-1,2-naphthoquinones.
Semicarbazides and thiosemicarbazides are substances used to identify aldehydes and ketones, are very versatile in the synthesis of heterocycles, and have several applications in the preparation of important drugs. Nucleophilic nitrogen semicarbazides can easily add to carbon C4 of β-NQSNa (18) to produce 1,2-naphthoquinones containing these groups [87]. This is a reaction very similar to adding amines to 18. Yamada and co-workers [88] studied the preparation of several 4-semicarbazide- and 4-thiosemicarbazide-1,2-naphthoquinones by the reaction of 18 with semicarbazides and thiosemicarbazides to obtain new compounds with improved hemostatic activities. These compounds were obtained in moderate yields and were capable of reducing the bleeding time (Scheme 10).
![[1860-5397-18-5-i10]](/bjoc/content/inline/1860-5397-18-5-i10.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 10: Synthesis of 4-semicarbazide-1,2-naphthoquinone.
Scheme 10: Synthesis of 4-semicarbazide-1,2-naphthoquinone.
Synthesis of 4-azido-β-naphthoquinones from β-NQS
β-NQS can react with other nitrogenous nucleophiles, such as azide ions. The reaction of 18 in the presence of sodium azide and water produces 4-azido-1,2-naphthoquinone (39) in a 52% yield. Although 39 can produce many other derivatives, few reactions have been studied. The reaction of 39 with concentrated sulfuric acid at room temperature produces azepinedione 40 in an 82% yield. This compound can be transformed into several 3-substituted and 4-hydroxy derivatives. However, an interesting transformation results from the treatment of 40 with hot aqueous sodium hydroxide, resulting in 2-oxoquinoline 41 in an 80% yield (Scheme 11) [89].
![[1860-5397-18-5-i11]](/bjoc/content/inline/1860-5397-18-5-i11.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 11: Reactions of 4-azido-1,2-naphthoquinone.
Scheme 11: Reactions of 4-azido-1,2-naphthoquinone.
Modifications in β-carbonyls
1,2-Naphthoquinone derivatives can be obtained through modifications in the carbonyls, leading to the formation of new compounds containing different groups, such as hydroxylamines, oxiranes, hydrazones, and heterocycles. These modifications can be easily carried out from the products of β-NQS 8 reactions with substituted amines and phenols (Figure 4). The objective of these transformations is to search for new compounds that present new physicochemical and biological properties.
![[1860-5397-18-5-4]](/bjoc/content/figures/1860-5397-18-5-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: Modifications that can be easily carried out from the products of β-NQS 8.
Figure 4: Modifications that can be easily carried out from the products of β-NQS 8.
For the first time, Campos and co-workers [90] performed the synthesis of novel 4-amino-1,2-naphthoquinones 43a–c containing carbohydrates and evaluated their antitumor activity in vitro. These compounds were transformed into a series of hydrazones 44a–l with different substituted aryl groups, which were also evaluated against tumor cells. To prepare 43a–c, 18 was reacted with different amines 42a–c in the presence of an ethanol–water mixture under ultrasonication, followed by reaction with arylhydrazines, which led to hydrazones 44a–l according to the classical procedure that employs methanol at room temperature. All compounds were evaluated against different human cancer cell lines, including leukemia (HL-60), melanoma (MDA-MB-435), colon cancer (HCT-116), and central nervous system cancer (SF-295). 4-Amino-1,2-naphthoquinones 43a–c exhibited considerable cytotoxic activities, with 43a being the most active against HL-60 and MDA-MB-435 cells (Scheme 12).
![[1860-5397-18-5-i12]](/bjoc/content/inline/1860-5397-18-5-i12.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 12: Derivatives of 1,2-naphthoquinones obtained from β-NQS.
Scheme 12: Derivatives of 1,2-naphthoquinones obtained from β-NQS.
Other naphthoquinone derivatives were synthesized with modifications at one of the carbonyls. Tseng and co-workers [91] synthesized 4-arylamino-1,2-naphthoquinones 45a,b and 4-phenoxy-1,2-naphthoquinones 47a,b as potential anti-inflammatory agents capable of inhibiting the expression of nitric oxide (NO) and PGE2 in alveolar macrophages. Then, oximes 46a,b and 48b were obtained by condensation of 45a,b and 47b with hydroxylamine, respectively. Biological results indicated that 47b significantly attenuated the release of inflammatory mediators (NO, TNF-α, and MMP-9) in a concentration-dependent manner. These data indicated that 47b targets p38 kinase and NF-κB and may serve as an anti-inflammatory agent (Scheme 13).
![[1860-5397-18-5-i13]](/bjoc/content/inline/1860-5397-18-5-i13.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 13: Oximes as well as 4-amino- and 4-phenoxy-1,2-naphthoquinone as potential anti-inflammatory agents.
Scheme 13: Oximes as well as 4-amino- and 4-phenoxy-1,2-naphthoquinone as potential anti-inflammatory agents.
In 2020, Almeida and co-workers [92] synthesized naphthoquinone imines from β-NQSNa (18) with modifications in β-carbonyls. These compounds were obtained in a sequence of reactions involving the addition of arylamines to β-NQS 18 followed by N-alkynylation and then Cu(I)-catalyzed heterocyclization with tosyl azide in toluene at room temperature, leading to triazoles 50c–k in moderate to excellent yields (Scheme 14).
![[1860-5397-18-5-i14]](/bjoc/content/inline/1860-5397-18-5-i14.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 14: Synthesis of triazoles from β-NQS.
Scheme 14: Synthesis of triazoles from β-NQS.
The research group that most explored the formation of heterocycles from 1,2-naphthoquinones was Pinto’s group, which prepared several imidazolyl, oxazolyl, phenoxazinyl, and indolyl heterocycles that were evaluated against some pharmacological targets [93-96]. Many of the methods that are in use today have been developed by this group. Lee and co-workers [97] developed the synthesis of naphtho[1,2-d]oxazole heterocycles from β-NQS as potential antiviral agents capable of inhibiting the HCV virus. Compound 45 was obtained from β-NQSNa (18) as shown above and reacted with substituted benzaldehyde or furfuryl aldehyde to form naphthoxazoles 51a–i and 53a–c, respectively. These compounds were then N-methylated, leading to the corresponding compounds 52a–i and 54a–c. Naphthoxazol 53c was the most effective anti-HCV agent, exhibiting an IC50 value of 0.63 μM, higher than that of the standard drug (ribavirin, IC50 13 μM) (Scheme 15). Naphthoxazoles stand out for exhibiting solid-state fluorescence, although the fluorescence partially disappears in solution, and there is a large shift to red and blue [98,99].
![[1860-5397-18-5-i15]](/bjoc/content/inline/1860-5397-18-5-i15.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 15: Synthesis of naphtho[1,2-d]oxazoles from β-NQS.
Scheme 15: Synthesis of naphtho[1,2-d]oxazoles from β-NQS.
Carbon–carbon bond formation
The main steps in a synthesis usually involve C–C bond formation, which is usually the main reaction step, or functional group transformations. Organometallics are the most commonly used catalysts to promote C–C bond formation. In addition, other so-called classical reactions are also widely used, such as Friedel–Crafts alkylation and acylation, Wittig and Horner–Emmons reactions, carbonyl addition/substitution, α-alkylation, aldol reactions, and pericyclic reactions.
Yoshida and co-workers [100] demonstrated that some metal ions are capable of activating aromatic compounds by chelation and promoting nucleophilic additions. For instance, 1-aminoanthraquinone quickly reacts with butylamine under the influence of Lewis acid catalysts to give 1-amino-4-butylaminoanthraquinone. Similarly, quinoline-5,8-diones react with amines under catalysis with Ni(II) ions to selectively give substituted amino derivatives [101,102]. The same group demonstrated that the reactions between β-NQS 18 and N,N’-dialkylanilines or 1,1-bis[p(dimethylamino)phenyl]ethylene in acetic acid efficiently produced 4-vinyl-1,2-naphthoquinones 55 and 4-aryl-1,2-naphthoquinones 56a,b, respectively, under nickel(II) catalysis, forming a C–C bond (Scheme 16) [103]. When the reaction was carried out in 10% aqueous methanol solution at room temperature for 5 hours, 56b was produced in an 85% yield [104,105]. Then, Ooyama and co-workers [99] transformed the 1,2-dicarbonyl group into the fused imidazo[4,5-a] heterocycle via a reaction of 56b with 4-cyanobenzaldehyde and an NH3 source in a 75% yield. The crystal of 57 exhibits a sensitive color change and fluorescence enhancement behavior with a blueshift in the emission maximum upon enclathration of various types of organic solvents. It is important to note that other authors carried out the reaction of 18 with N,N-diethylaniline without the presence of Ni(II) [104,105].
![[1860-5397-18-5-i16]](/bjoc/content/inline/1860-5397-18-5-i16.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 16: A) Arylation and vinylation of β-NQS catalyzed by Ni(II) salts. B) Transformation of the 1,2-dicarbonyl group in the fused imidazo[4,5-a] heterocycle.
Scheme 16: A) Arylation and vinylation of β-NQS catalyzed by Ni(II) salts. B) Transformation of the 1,2-dicarb...
In the search for fluorophores of heterocyclic quinoid type to study their photophysical properties in solution and in the solid-state, Ooyama and co-workers [106] studied a synthetic route for the preparation of compounds with the tricyclic benzo[c]carbazol-6-one skeleton. The strategy used was through the reaction to β-NQS 18 with a bifunctional amine (3-amino-N,N-dibutylaniline) under NiOAc2 catalysis to obtain the 4-arylated compound. As expected, 4-arylated-benzo[c]carbazole-5,6-dione 58 and 4-amino-1,2-naphthoquinone 59 were formed in 5% and 35% yields, respectively. However, the reaction of 18 with 3-butylamino-N,N-dibutylaniline in DMF in the presence of CuCl2 formed two isomers of 4-arylated-N-butylbenzo[c]carbazole-5,6-dione 60 and 4-amino-benzo[a]carbazol-5,6-dione 61 in 39% and 13% yields, respectively. These two reactions demonstrate the importance of the catalyst in complex formation with carbonyls of 18 that promote nucleophilic desulfoamination or nucleophilic desulfoarylation at position C4, and the following intramolecular cyclization occurs to produce 1,2-naphthoquinones fused with the benzo[a]carbazole or benzo[c]carbazole system (Scheme 17).
![[1860-5397-18-5-i17]](/bjoc/content/inline/1860-5397-18-5-i17.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 17: Benzo[a]carbazole and benzo[c]carbazoles fused with 1,2-naphthoquinone.
Scheme 17: Benzo[a]carbazole and benzo[c]carbazoles fused with 1,2-naphthoquinone.
An interesting reaction for the formation of 4-cyanoethyl-l,2-naphthoquinone from β-NQS−M+ was developed by Gates and Newhall in 1948 [107]. Land and co-workers demonstrated that cyanomethyl derivatives of ortho-quinones undergo facile tautomerism to para-quinomethanes [108]. Villemin and co-workers summarized these reactions, which were expanded to several other condensation products with methylene acid compounds [109]. These authors developed two methods (A and B) to prepare 2-hydroxynaphthoquinomethanes 62 with diverse structures by condensation of 18 with active methylene compounds (Scheme 18). Method A involves the reaction promoted by sodium hydroxide in an ethanol/water mixture at 40 °C, and method B was carried out with t-BuOK in polyethylene glycol (PEG300) at room temperature. In both methods, the reaction conditions were mild and produced the product in moderate to good yields.
![[1860-5397-18-5-i18]](/bjoc/content/inline/1860-5397-18-5-i18.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 18: Synthesis of 1,2-naphthoquinones having a C=C bond from β-NQS. Method A: NaOH, EtOH/H2O, 40 °C, 2 h; Method B: t-BuOK, PEG-300, rt, 6–10 min.
Scheme 18: Synthesis of 1,2-naphthoquinones having a C=C bond from β-NQS. Method A: NaOH, EtOH/H2O, 40 °C, 2 h...
The same group investigated this reaction with substituted acetonitriles to obtain 2-hydroxynaphthoquinomethanes [110]. These reactions were carried out by method A described above, and the stereochemistry was attributed to the E-isomer (Scheme 19).
![[1860-5397-18-5-i19]](/bjoc/content/inline/1860-5397-18-5-i19.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 19: C=C bond formation from β-NQS and substituted acetonitriles.
Scheme 19: C=C bond formation from β-NQS and substituted acetonitriles.
Conclusion
Compounds containing the 1,2-naphthoquinone scaffold represent a class of natural and synthetic substances with important biological activities and are therefore relevant for studies in the field of medicinal chemistry. The salts of 1,2-naphthoquinone-4-sulfonic acid (NQS) are the reagents of choice for performing selective transformations at position C-4 and preparing new 1,2-naphthoquinones with promising pharmacological properties. β-NQS is also important in quantitative analytical determinations of drugs containing free primary and secondary amino groups, as they react quickly and in high yield with amines to form colored products in good yields.
This review reports several examples of syntheses of functionalized 1,2-naphthoquinones substituted at the C4 position of β-NQS. Despite great advances in the area, there are still many opportunities for the development of new bioactive compounds of great relevance to humanity. We hope this article will serve as a source of inspiration for current and future researchers in chemical, pharmaceutical, and biological sciences.
A lot of the literature in this area is quite old. Perhaps there are also opportunities in this area to apply modern chemistry methodology, and also in the development of more sophisticated sensors and dyes.
References
-
Thomson, R. H. Naturally Occurring Quinones IV, 4th ed.; Blackie Acad. and Professional: New York, NY, USA, 1997. doi:10.1007/978-94-009-1551-0
Return to citation in text: [1] -
O’Brien, P. J. Chem.-Biol. Interact. 1991, 80, 1–41.
Return to citation in text: [1] -
Powis, G. Pharmacol. Ther. 1987, 35, 57–162. doi:10.1016/0163-7258(87)90105-7
Return to citation in text: [1] -
Rinehart, K. L., Jr.; Shield, L. S. Fortschr. Chem. Org. Naturst. 1976, 33, 231–307. doi:10.1007/978-3-7091-3262-3_3
Return to citation in text: [1] -
Santos, L. O.; dos Anjos, J. P.; Ferreira, S. L. C.; de Andrade, J. B. Microchem. J. 2017, 133, 431–440. doi:10.1016/j.microc.2017.04.012
Return to citation in text: [1] -
Sousa, E. T.; Lopes, W. A.; de Andrade, J. B. Quim. Nova 2016, 39, 486–495.
Return to citation in text: [1] -
Pereyra, C. E.; Dantas, R. F.; Ferreira, S. B.; Gomes, L. P.; Silva-Jr, F. P. Cancer Cell Int. 2019, 19, 207. doi:10.1186/s12935-019-0925-8
Return to citation in text: [1] -
Qiu, H.-Y.; Wang, P.-F.; Lin, H.-Y.; Tang, C.-Y.; Zhu, H.-L.; Yang, Y.-H. Chem. Biol. Drug Des. 2018, 91, 681–690. doi:10.1111/cbdd.13141
Return to citation in text: [1] [2] -
Varghese, K. J.; Silvipriya, K. S.; Resmi, S.; Jolly, C. I. Inventi Impact: Cosmeceuticals 2010, 1, 1–5.
Return to citation in text: [1] -
Saeed, S. M. G.; Sayeed, S. A.; Ashraf, S.; Naz, S.; Siddiqi, R.; Ali, R.; Mesaik, M. A. Pak. J. Bot. 2013, 45, 1431–1436.
Return to citation in text: [1] -
Yusuf, M.; Ahmad, A.; Shahid, M.; Khan, M. I.; Khan, S. A.; Manzoor, N.; Mohammad, F. J. Cleaner Prod. 2012, 27, 42–50. doi:10.1016/j.jclepro.2012.01.005
Return to citation in text: [1] -
de Paiva, S. R.; Lima, L. A.; Figueiredo, M. R.; Kaplan, M. A. C. An. Acad. Bras. Cienc. 2004, 76, 499–504. doi:10.1590/s0001-37652004000300004
Return to citation in text: [1] -
de Carvalho da Silva, F.; Francisco Ferreira, V. Curr. Org. Synth. 2016, 13, 334–371. doi:10.2174/1570179412666150817220343
Return to citation in text: [1] -
Liao, C.-C.; Peddinti, R. K. Sci. Synth. 2006, 28, 323–324.
Return to citation in text: [1] -
Chaudhary, A.; Khurana, J. M. Curr. Org. Chem. 2016, 20, 1314–1344. doi:10.2174/1385272820666151125231522
Return to citation in text: [1] -
López, L. I. L.; Flores, S. D. N.; Belmares, S. Y. S.; Galindo, A. S. Vitae 2014, 21, 248–258.
Return to citation in text: [1] -
Sartori, M. F. Chem. Rev. 1963, 63, 279–296. doi:10.1021/cr60223a005
Return to citation in text: [1] -
Badave, K. D.; Khan, A. A.; Rane, S. Y. Anti-Cancer Agents Med. Chem. 2016, 16, 1017–1030. doi:10.2174/1871520616666160310143316
Return to citation in text: [1] -
Padhye, S.; Dandawate, P.; Yusufi, M.; Ahmad, A.; Sarkar, F. H. Med. Res. Rev. 2012, 32, 1131–1158. doi:10.1002/med.20235
Return to citation in text: [1] -
Lamoureux, G.; Perez, A. L.; Araya, M.; Agüero, C. J. Phys. Org. Chem. 2008, 21, 1022–1028. doi:10.1002/poc.1435
Return to citation in text: [1] -
Sharma, A.; Santos, I. O.; Gaur, P.; Ferreira, V. F.; Garcia, C. R. S.; da Rocha, D. R. Eur. J. Med. Chem. 2013, 59, 48–53. doi:10.1016/j.ejmech.2012.10.052
Return to citation in text: [1] -
García-Barrantes, P. M.; Lamoureux, G. V.; Pérez, A. L.; García-Sánchez, R. N.; Martínez, A. R.; San Feliciano, A. Eur. J. Med. Chem. 2013, 70, 548–557. doi:10.1016/j.ejmech.2013.10.011
Return to citation in text: [1] -
Lezama-Dávila, C. M.; Isaac-Márquez, A. P.; Kapadia, G.; Owens, K.; Oghumu, S.; Beverley, S.; Satoskar, A. R. Biol. Pharm. Bull. 2012, 35, 1761–1764. doi:10.1248/bpb.b12-00419
Return to citation in text: [1] -
Asche, C. Mini-Rev. Med. Chem. 2005, 5, 449–467. doi:10.2174/1389557053765556
Return to citation in text: [1] -
Baell, J. B.; Nissink, J. W. M. ACS Chem. Biol. 2018, 13, 36–44. doi:10.1021/acschembio.7b00903
Return to citation in text: [1] -
Aldrich, C.; Bertozzi, C.; Georg, G. I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K. M., Jr.; Schepartz, A.; Wang, S. ACS Med. Chem. Lett. 2017, 8, 379–382. doi:10.1021/acsmedchemlett.7b00056
Return to citation in text: [1] -
Combs, G. F., Jr.; McClung, J. P. Vitamin K. In The Vitamins; Combs, G. F., Jr.; McClung, J. P., Eds.; Academic Press, 2017; pp 243–265. doi:10.1016/b978-0-12-802965-7.00009-5
Return to citation in text: [1] -
Olliaro, P.; Wirth, D. J. Pharm. Pharmacol. 1997, 49 (Suppl. 2), 29–33. doi:10.1111/j.2042-7158.1997.tb06157.x
Return to citation in text: [1] -
França, T. C. C.; dos Santos, M. G.; Figueroa-Villar, J. D. Quim. Nova 2008, 31, 1271–1278. doi:10.1590/s0100-40422008000500060
Return to citation in text: [1] -
Watts, J.; Playford, M.; Hickey, K. N. Z. Vet. J. 2016, 64, 3–9. doi:10.1080/00480169.2015.1064792
Return to citation in text: [1] -
Dobbelaere, D.; Heussler, V. Annu. Rev. Microbiol. 1999, 53, 1–42. doi:10.1146/annurev.micro.53.1.1
Return to citation in text: [1] -
Mhadhbi, M.; Naouach, A.; Boumiza, A.; Chaabani, M. F.; BenAbderazzak, S.; Darghouth, M. A. Vet. Parasitol. 2010, 169, 241–247. doi:10.1016/j.vetpar.2010.01.013
Return to citation in text: [1] -
Sharifiyazdi, H.; Namazi, F.; Oryan, A.; Shahriari, R.; Razavi, M. Vet. Parasitol. 2012, 187, 431–435. doi:10.1016/j.vetpar.2012.01.016
Return to citation in text: [1] -
Hashemi-Fesharki, R. Res. Vet. Sci. 1991, 50, 204–207. doi:10.1016/0034-5288(91)90107-y
Return to citation in text: [1] -
Garnier, T.; Mantyla, A.; Jarvinen, T.; Lawrence, J.; Brown, M.; Croft, S. J. Antimicrob. Chemother. 2007, 60, 802–810. doi:10.1093/jac/dkm303
Return to citation in text: [1] -
Hawa, N.; Rae, D. G.; Younis, S.; Mahadi, W.; Ibrahim, R.; Al-Wahab, W. Trop. Anim. Health Prod. 1988, 20, 130–136. doi:10.1007/bf02240076
Return to citation in text: [1] -
Ferreira, V. F.; Ferreira, S. B.; de Carvalho da Silva, F. Org. Biomol. Chem. 2010, 8, 4793–4802. doi:10.1039/c0ob00277a
Return to citation in text: [1] -
da Rocha, D. R.; de Souza, A. C. G.; Resende, J. A. L. C.; Santos, W. C.; dos Santos, E. A.; Pessoa, C.; de Moraes, M. O.; Costa-Lotufo, L. V.; Montenegro, R. C.; Ferreira, V. F. Org. Biomol. Chem. 2011, 9, 4315–4322. doi:10.1039/c1ob05209h
Return to citation in text: [1] -
Moreira, C. d. S.; Nicoletti, C. D.; Pinheiro, D. P.; de Moraes, L. G. C.; Futuro, D. O.; Ferreira, V. F.; Pessoa, C. d. Ó.; da Rocha, D. R. Med. Chem. Res. 2019, 28, 2109–2117. doi:10.1007/s00044-019-02439-w
Return to citation in text: [1] -
Pradhan, R.; Dandawate, P.; Vyas, A.; Padhye, S.; Biersack, B.; Schobert, R.; Ahmad, A.; Sarkar, F. H. Curr. Drug Targets 2012, 13, 1777–1798. doi:10.2174/138945012804545588
Return to citation in text: [1] -
Gouda, M. A.; Eldien, H. F.; Girges, M. M.; Berghot, M. A. Med. Chem. 2013, 3, 228–232.
Return to citation in text: [1] [2] -
Nakahara, Y.; Ishigami, A.; Takeda, Y. J. Chromatogr. B: Biomed. Sci. Appl. 1989, 489, 371–376. doi:10.1016/s0378-4347(00)82916-3
Return to citation in text: [1] [2] -
Witt, O. N. Ber. Dtsch. Chem. Ges. 1891, 24, 3154–3157. doi:10.1002/cber.189102402156
Return to citation in text: [1] -
Böniger, M. Ber. Dtsch. Chem. Ges. 1894, 27, 23–30. doi:10.1002/cber.18940270107
Return to citation in text: [1] -
Folin, O.; Wu, H. J. Biol. Chem. 1922, 51, 377–391. doi:10.1016/s0021-9258(18)85880-x
Return to citation in text: [1] -
Obo, F. J. Biochem. 1941, 33, 231–235. doi:10.1093/oxfordjournals.jbchem.a126035
Return to citation in text: [1] -
Fu, B.; Liu, T.; Chen, J.; Li, K. Sens. Actuators, B 2018, 272, 598–604. doi:10.1016/j.snb.2018.05.068
Return to citation in text: [1] -
Danielson, I. S. J. Biol. Chem. 1933, 101, 505–522. doi:10.1016/s0021-9258(18)75897-3
Return to citation in text: [1] -
Martin, E. L.; Fieser, L. F. Org. Synth. 1941, 21, 91. doi:10.15227/orgsyn.021.0091
Return to citation in text: [1] -
Gummadi, S.; Kommoju, M. Am. J. PharmTech Res. 2019, 9, 14–37. doi:10.46624/ajptr.2019.v9.i1.002
Return to citation in text: [1] -
Adegoke, O. A. Int. J. Pharm. Sci. Rev. Res. 2012, 14, 6–24.
Return to citation in text: [1] -
Elbashir, A. A.; Ahmed, A. A.; Ali Ahmed, S. M.; Aboul-Enein, H. Y. Appl. Spectrosc. Rev. 2012, 47, 219–232. doi:10.1080/05704928.2011.639107
Return to citation in text: [1] -
Hiyama, M. Yakugaku Zasshi 1952, 72, 1367–1370. doi:10.1248/yakushi1947.72.10_1367
Return to citation in text: [1] -
Hashimoto, Y.; Endo, M.; Tominaga, K.; Inuzuka, S.; Moriyasu, M. Microchim. Acta 1978, 70, 493–504. doi:10.1007/bf01197101
Return to citation in text: [1] -
Saurina, J.; Hernández-Cassuo, S. Anal. Chim. Acta 1993, 283, 414–420. doi:10.1016/0003-2670(93)85252-f
Return to citation in text: [1] -
Legua, C. M.; Falcó, P. C.; Cabeza, A. S. Anal. Chim. Acta 1993, 283, 635–644. doi:10.1016/0003-2670(93)85276-p
Return to citation in text: [1] -
Molins Legua, C.; Campins Falcó, P.; Sevillano Cabeza, A. Fresenius' J. Anal. Chem. 1994, 349, 311–316. doi:10.1007/bf00323210
Return to citation in text: [1] -
Xu, L.; Wang, H.; Xiao, Y. Spectrochim. Acta, Part A 2004, 60, 3007–3012. doi:10.1016/j.saa.2004.02.018
Return to citation in text: [1] -
Kumar, C. H. A.; Kumar, T. A.; Gurupadayya, B. M.; Sloka, N.; Reddy, M. B. R. Arch. Appl. Sci. Res. 2010, 2, 278–287.
Return to citation in text: [1] -
Elbashir, A. A.; Elwagee, A. H. E. J. Assoc. Arab Univ. Basic Appl. Sci. 2012, 11, 32–36. doi:10.1016/j.jaubas.2011.12.003
Return to citation in text: [1] -
Osman, R. A. M.; Elbashir, A. A. Int. J. Bioanal. Methods Bioequival. Stud. 2019, 5, 82–92.
Return to citation in text: [1] -
Khalil, N. A.; Ibrahim, W. H. Tikrit J. Pure Sci. 2020, 25, 68–74. doi:10.25130/j.v25i1.938
Return to citation in text: [1] -
Awan, Z. A.; Hegazy, M. A.; Kammoun, A. K. Spectrochim. Acta, Part A 2020, 230, 118066. doi:10.1016/j.saa.2020.118066
Return to citation in text: [1] -
Darwish, I. A.; Al-Shehri, M. M.; El-Gendy, M. A. Chem. Cent. J. 2012, 6, 11. doi:10.1186/1752-153x-6-11
Return to citation in text: [1] -
Ahmed, S. M.; Elbashir, A. A. J. Anal. Bioanal. Tech. 2015, 6, 248.
Return to citation in text: [1] -
Altigani, A. M. N.; Elbashir, A. A. Austin J. Anal. Pharm. Chem. 2014, 1, 1019.
Return to citation in text: [1] -
Abdulrahman, S. A. M.; Basavaiah, K. Drug Test. Anal. 2011, 3, 748–754. doi:10.1002/dta.242
Return to citation in text: [1] -
Elbashir, A. A.; Awad, S. F. J. Pharmacovigilance 2013, 1, 1000105.
Return to citation in text: [1] -
Salman, B. I.; Hussein, S. A.; Ali, M. F. B.; Marzouq, M. A. Microchem. J. 2019, 145, 959–965. doi:10.1016/j.microc.2018.12.018
Return to citation in text: [1] -
Hartke, K.; Lohmann, U. Chem. Lett. 1983, 12, 693–696. doi:10.1246/cl.1983.693
Return to citation in text: [1] -
Fieser, L. F.; Fieser, M. J. Am. Chem. Soc. 1935, 57, 491–494. doi:10.1021/ja01306a031
Return to citation in text: [1] -
Yano, H.; Yamasaki, M.; Shimomura, Y.; Iwasaki, M.; Ohta, M.; Furuno, Y.; Kouno, K.; Onu, Y.; Ueda, Y. Chem. Pharm. Bull. 1980, 28, 1207–1213. doi:10.1248/cpb.28.1207
Return to citation in text: [1] -
Fieser, L. F.; Fieser, M. J. Am. Chem. Soc. 1934, 56, 1565–1578. doi:10.1021/ja01322a034
Return to citation in text: [1] -
Harmon, R. E.; Phipps, L. M.; Howell, J. A.; Gupta, S. K. Tetrahedron 1969, 25, 5807–5813. doi:10.1016/s0040-4020(01)83088-x
Return to citation in text: [1] -
Fernández, A. E.; De Bertorello, M. M.; Longhi, M. R. J. Liq. Chromatogr. 1984, 7, 2203–2217. doi:10.1080/01483918408068870
Return to citation in text: [1] -
Fragoso, T. P.; de Mesquita Carneiro, J. W.; Vargas, M. D. J. Mol. Model. 2010, 16, 825–830. doi:10.1007/s00894-009-0579-x
Return to citation in text: [1] -
Gornostaev, L. M.; Rukovets, T. A.; Lavrikova, T. I.; Khalyavina, Y. G.; Stashina, G. A. Russ. Chem. Bull. 2017, 66, 1007–1010. doi:10.1007/s11172-017-1847-z
Return to citation in text: [1] -
Gornostaev, L. M.; Rukovets, T. A.; Arnold, E. V.; Khalyavina, Y. G.; Gatilov, Y. V. Russ. J. Org. Chem. 2018, 54, 78–86. doi:10.1134/s1070428018010062
Return to citation in text: [1] -
Ortiz, C. S.; Longhi, M. R.; De Bertorello, M. M.; Briñon, M. C. Org. Prep. Proced. Int. 1991, 23, 181–185. doi:10.1080/00304949109458306
Return to citation in text: [1] -
Khalil, A. M.; Berghot, M. A.; Gouda, M. A. Eur. J. Med. Chem. 2010, 45, 1552–1559. doi:10.1016/j.ejmech.2009.12.064
Return to citation in text: [1] -
Gouda, M. A. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 155–162. doi:10.1002/ardp.201100171
Return to citation in text: [1] -
Ahn, J. H.; Cho, S. Y.; Ha, J. D.; Chu, S. Y.; Jung, S. H.; Jung, Y. S.; Baek, J. Y.; Choi, I. K.; Shin, E. Y.; Kang, S. K.; Kim, S. S.; Cheon, H. G.; Yang, S.-D.; Choi, J.-K. Bioorg. Med. Chem. Lett. 2002, 12, 1941–1946. doi:10.1016/s0960-894x(02)00331-1
Return to citation in text: [1] -
Gouda, M. A.; Sherif, Y. E.-S.; Elsherbini, M. S. Phosphorus, Sulfur Silicon Relat. Elem. 2014, 189, 1633–1643. doi:10.1080/10426507.2014.884091
Return to citation in text: [1] -
Hatfield, M. J.; Chen, J.; Fratt, E. M.; Chi, L.; Bollinger, J. C.; Binder, R. J.; Bowling, J.; Hyatt, J. L.; Scarborough, J.; Jeffries, C.; Potter, P. M. J. Med. Chem. 2017, 60, 1568–1579. doi:10.1021/acs.jmedchem.6b01849
Return to citation in text: [1] -
Takizawa, Y.; Munakata, T.; Iwasa, Y.; Suzuki, T.; Mitsuhashi, T. J. Org. Chem. 1985, 50, 4383–4386. doi:10.1021/jo00222a038
Return to citation in text: [1] -
Yang, H.; An, B.; Li, X.; Zeng, W. Bioorg. Med. Chem. Lett. 2018, 28, 3057–3063. doi:10.1016/j.bmcl.2018.07.047
Return to citation in text: [1] -
Carroll, F. I.; Miller, H. W.; Meck, R. J. Chem. Soc. C 1970, 1993–1996. doi:10.1039/j39700001993
Return to citation in text: [1] -
Yamada, T.; Yamashita, T.; Nakamura, M.; Shimamura, H.; Yamaguchi, A.; Takaya, M. Yakugaku Zasshi 1980, 100, 799–806. doi:10.1248/yakushi1947.100.8_799
Return to citation in text: [1] -
Moore, H. W.; Shelden, H. R.; Weyler, W., Jr. Tetrahedron Lett. 1969, 10, 1243–1246. doi:10.1016/s0040-4039(01)87853-9
Return to citation in text: [1] -
Campos, V. R.; dos Santos, E. A.; Ferreira, V. F.; Montenegro, R. C.; de Souza, M. C. B. V.; Costa-Lotufo, L. V.; de Moraes, M. O.; Regufe, A. K. P.; Jordão, A. K.; Pinto, A. C.; Resende, J. A. L. C.; Cunha, A. C. RSC Adv. 2012, 2, 11438–11448. doi:10.1039/c2ra21514d
Return to citation in text: [1] -
Tseng, C.-H.; Cheng, C.-M.; Tzeng, C.-C.; Peng, S.-I.; Yang, C.-L.; Chen, Y.-L. Bioorg. Med. Chem. 2013, 21, 523–531. doi:10.1016/j.bmc.2012.10.047
Return to citation in text: [1] -
Almeida, R. G.; Valença, W. O.; Rosa, L. G.; de Simone, C. A.; de Castro, S. L.; Barbosa, J. M. C.; Pinheiro, D. P.; Paier, C. R. K.; de Carvalho, G. G. C.; Pessoa, C.; Goulart, M. O. F.; Kharma, A.; da Silva Júnior, E. N. RSC Med. Chem. 2020, 11, 1145–1160. doi:10.1039/d0md00072h
Return to citation in text: [1] -
Pinto, A. V.; Pinto, C. N.; Pinto, C. F. R.; Rita, R. S.; Pezzella, C. A. C.; Castro, S. L. Arzneim. Forsch. 1997, 47, 74.
Return to citation in text: [1] -
Chaves, J. P.; Pinto, M. d. C. F. R.; Pinto, A. V. J. Braz. Chem. Soc. 1990, 1, 22–27. doi:10.5935/0103-5053.19900004
Return to citation in text: [1] -
Neves-Pinto, C.; Malta, V. R. S.; Pinto, M. d. C. F. R.; Santos, R. H. A.; de Castro, S. L.; Pinto, A. V. J. Med. Chem. 2002, 45, 2112–2115. doi:10.1021/jm010377v
Return to citation in text: [1] -
de Moura, K. C. G.; Emery, F. S.; Neves-Pinto, C.; Pinto, M. d. C. F. R.; Dantas, A. P.; Salomão, K.; de Castro, S. L.; Pinto, A. V. J. Braz. Chem. Soc. 2001, 12, 325–338. doi:10.1590/s0103-50532001000300003
Return to citation in text: [1] -
Tseng, C.-H.; Lin, C.-K.; Chen, Y.-L.; Tseng, C.-K.; Lee, J.-Y.; Lee, J.-C. Eur. J. Med. Chem. 2018, 143, 970–982. doi:10.1016/j.ejmech.2017.12.006
Return to citation in text: [1] -
Ooyama, Y.; Nonami, K.; Watanabe, S.; Yoshida, K. Dyes Pigm. 2008, 77, 315–322. doi:10.1016/j.dyepig.2007.05.018
Return to citation in text: [1] -
Ooyama, Y.; Nagano, S.; Okamura, M.; Yoshida, K. Eur. J. Org. Chem. 2008, 5899–5906. doi:10.1002/ejoc.200800832
Return to citation in text: [1] [2] -
Yoshida, K.; Matsuoka, M.; Yamashita, Y.; Kitao, T. Bull. Chem. Soc. Jpn. 1980, 53, 2552–2554. doi:10.1246/bcsj.53.2552
Return to citation in text: [1] -
Katsuhira, Y.; Miwa, I.; Hiroyuki, H.; Mayumi, Y.; Yuji, K. Bull. Chem. Soc. Jpn. 1988, 61, 4335–4340.
Return to citation in text: [1] -
Yoshida, K.; Yamamoto, M.; Ishiguro, M. Chem. Lett. 1986, 15, 1059–1062. doi:10.1246/cl.1986.1059
Return to citation in text: [1] -
Yoshida, K.; Koujiri, T.; Oga, N.; Ishiguro, M.; Kubo, Y. J. Chem. Soc., Chem. Commun. 1989, 708–710. doi:10.1039/c39890000708
Return to citation in text: [1] -
Rickwood, M.; Marsden, S. D.; Askew, V. E. Photochromic spiroxazine compounds. U.S. Patent US5446150A, Aug 29, 1995.
Return to citation in text: [1] [2] -
Tathe, A. B.; Sekar, N. J. Fluoresc. 2015, 25, 1403–1415. doi:10.1007/s10895-015-1631-0
Return to citation in text: [1] [2] -
Ooyama, Y.; Nabeshima, S.; Mamura, T.; Ooyama, H. E.; Yoshida, K. Tetrahedron 2010, 66, 7954–7960. doi:10.1016/j.tet.2010.08.026
Return to citation in text: [1] -
Gates, M.; Newhall, W. F. J. Am. Chem. Soc. 1948, 70, 2261–2263. doi:10.1021/ja01186a079
Return to citation in text: [1] -
Land, E. J.; Ramsden, C. A.; Riley, P. A.; Yoganathan, G. Tetrahedron 2003, 59, 9547–9554. doi:10.1016/j.tet.2003.10.008
Return to citation in text: [1] -
Villemin, D.; Benabdallah, M.; Choukchou-Braham, N.; Mostefa-Kara, B. Synth. Commun. 2010, 40, 3109–3118. doi:10.1080/00397911003797916
Return to citation in text: [1] -
Villemin, D.; Benabdallah, M.; Rahmoun, N.; Jouannic, C.; Choukchou-Braham, N.; Mostefa-Kara, B. Synth. Commun. 2010, 40, 3514–3521. doi:10.1080/00397910903457340
Return to citation in text: [1]
| 41. | Gouda, M. A.; Eldien, H. F.; Girges, M. M.; Berghot, M. A. Med. Chem. 2013, 3, 228–232. |
| 70. | Hartke, K.; Lohmann, U. Chem. Lett. 1983, 12, 693–696. doi:10.1246/cl.1983.693 |
| 71. | Fieser, L. F.; Fieser, M. J. Am. Chem. Soc. 1935, 57, 491–494. doi:10.1021/ja01306a031 |
| 79. | Ortiz, C. S.; Longhi, M. R.; De Bertorello, M. M.; Briñon, M. C. Org. Prep. Proced. Int. 1991, 23, 181–185. doi:10.1080/00304949109458306 |
| 8. | Qiu, H.-Y.; Wang, P.-F.; Lin, H.-Y.; Tang, C.-Y.; Zhu, H.-L.; Yang, Y.-H. Chem. Biol. Drug Des. 2018, 91, 681–690. doi:10.1111/cbdd.13141 |
| 80. | Khalil, A. M.; Berghot, M. A.; Gouda, M. A. Eur. J. Med. Chem. 2010, 45, 1552–1559. doi:10.1016/j.ejmech.2009.12.064 |
| 77. | Gornostaev, L. M.; Rukovets, T. A.; Lavrikova, T. I.; Khalyavina, Y. G.; Stashina, G. A. Russ. Chem. Bull. 2017, 66, 1007–1010. doi:10.1007/s11172-017-1847-z |
| 78. | Gornostaev, L. M.; Rukovets, T. A.; Arnold, E. V.; Khalyavina, Y. G.; Gatilov, Y. V. Russ. J. Org. Chem. 2018, 54, 78–86. doi:10.1134/s1070428018010062 |
| 75. | Fernández, A. E.; De Bertorello, M. M.; Longhi, M. R. J. Liq. Chromatogr. 1984, 7, 2203–2217. doi:10.1080/01483918408068870 |
| 76. | Fragoso, T. P.; de Mesquita Carneiro, J. W.; Vargas, M. D. J. Mol. Model. 2010, 16, 825–830. doi:10.1007/s00894-009-0579-x |
| 72. | Yano, H.; Yamasaki, M.; Shimomura, Y.; Iwasaki, M.; Ohta, M.; Furuno, Y.; Kouno, K.; Onu, Y.; Ueda, Y. Chem. Pharm. Bull. 1980, 28, 1207–1213. doi:10.1248/cpb.28.1207 |
| 73. | Fieser, L. F.; Fieser, M. J. Am. Chem. Soc. 1934, 56, 1565–1578. doi:10.1021/ja01322a034 |
| 74. | Harmon, R. E.; Phipps, L. M.; Howell, J. A.; Gupta, S. K. Tetrahedron 1969, 25, 5807–5813. doi:10.1016/s0040-4020(01)83088-x |
| 81. | Gouda, M. A. Arch. Pharm. (Weinheim, Ger.) 2012, 345, 155–162. doi:10.1002/ardp.201100171 |
| 82. | Ahn, J. H.; Cho, S. Y.; Ha, J. D.; Chu, S. Y.; Jung, S. H.; Jung, Y. S.; Baek, J. Y.; Choi, I. K.; Shin, E. Y.; Kang, S. K.; Kim, S. S.; Cheon, H. G.; Yang, S.-D.; Choi, J.-K. Bioorg. Med. Chem. Lett. 2002, 12, 1941–1946. doi:10.1016/s0960-894x(02)00331-1 |
| 83. | Gouda, M. A.; Sherif, Y. E.-S.; Elsherbini, M. S. Phosphorus, Sulfur Silicon Relat. Elem. 2014, 189, 1633–1643. doi:10.1080/10426507.2014.884091 |
| 90. | Campos, V. R.; dos Santos, E. A.; Ferreira, V. F.; Montenegro, R. C.; de Souza, M. C. B. V.; Costa-Lotufo, L. V.; de Moraes, M. O.; Regufe, A. K. P.; Jordão, A. K.; Pinto, A. C.; Resende, J. A. L. C.; Cunha, A. C. RSC Adv. 2012, 2, 11438–11448. doi:10.1039/c2ra21514d |
| 91. | Tseng, C.-H.; Cheng, C.-M.; Tzeng, C.-C.; Peng, S.-I.; Yang, C.-L.; Chen, Y.-L. Bioorg. Med. Chem. 2013, 21, 523–531. doi:10.1016/j.bmc.2012.10.047 |
| 88. | Yamada, T.; Yamashita, T.; Nakamura, M.; Shimamura, H.; Yamaguchi, A.; Takaya, M. Yakugaku Zasshi 1980, 100, 799–806. doi:10.1248/yakushi1947.100.8_799 |
| 89. | Moore, H. W.; Shelden, H. R.; Weyler, W., Jr. Tetrahedron Lett. 1969, 10, 1243–1246. doi:10.1016/s0040-4039(01)87853-9 |
| 86. | Yang, H.; An, B.; Li, X.; Zeng, W. Bioorg. Med. Chem. Lett. 2018, 28, 3057–3063. doi:10.1016/j.bmcl.2018.07.047 |
| 87. | Carroll, F. I.; Miller, H. W.; Meck, R. J. Chem. Soc. C 1970, 1993–1996. doi:10.1039/j39700001993 |
| 84. | Hatfield, M. J.; Chen, J.; Fratt, E. M.; Chi, L.; Bollinger, J. C.; Binder, R. J.; Bowling, J.; Hyatt, J. L.; Scarborough, J.; Jeffries, C.; Potter, P. M. J. Med. Chem. 2017, 60, 1568–1579. doi:10.1021/acs.jmedchem.6b01849 |
| 85. | Takizawa, Y.; Munakata, T.; Iwasa, Y.; Suzuki, T.; Mitsuhashi, T. J. Org. Chem. 1985, 50, 4383–4386. doi:10.1021/jo00222a038 |
| 93. | Pinto, A. V.; Pinto, C. N.; Pinto, C. F. R.; Rita, R. S.; Pezzella, C. A. C.; Castro, S. L. Arzneim. Forsch. 1997, 47, 74. |
| 94. | Chaves, J. P.; Pinto, M. d. C. F. R.; Pinto, A. V. J. Braz. Chem. Soc. 1990, 1, 22–27. doi:10.5935/0103-5053.19900004 |
| 95. | Neves-Pinto, C.; Malta, V. R. S.; Pinto, M. d. C. F. R.; Santos, R. H. A.; de Castro, S. L.; Pinto, A. V. J. Med. Chem. 2002, 45, 2112–2115. doi:10.1021/jm010377v |
| 96. | de Moura, K. C. G.; Emery, F. S.; Neves-Pinto, C.; Pinto, M. d. C. F. R.; Dantas, A. P.; Salomão, K.; de Castro, S. L.; Pinto, A. V. J. Braz. Chem. Soc. 2001, 12, 325–338. doi:10.1590/s0103-50532001000300003 |
| 97. | Tseng, C.-H.; Lin, C.-K.; Chen, Y.-L.; Tseng, C.-K.; Lee, J.-Y.; Lee, J.-C. Eur. J. Med. Chem. 2018, 143, 970–982. doi:10.1016/j.ejmech.2017.12.006 |
| 92. | Almeida, R. G.; Valença, W. O.; Rosa, L. G.; de Simone, C. A.; de Castro, S. L.; Barbosa, J. M. C.; Pinheiro, D. P.; Paier, C. R. K.; de Carvalho, G. G. C.; Pessoa, C.; Goulart, M. O. F.; Kharma, A.; da Silva Júnior, E. N. RSC Med. Chem. 2020, 11, 1145–1160. doi:10.1039/d0md00072h |
| 1. | Thomson, R. H. Naturally Occurring Quinones IV, 4th ed.; Blackie Acad. and Professional: New York, NY, USA, 1997. doi:10.1007/978-94-009-1551-0 |
| 2. | O’Brien, P. J. Chem.-Biol. Interact. 1991, 80, 1–41. |
| 3. | Powis, G. Pharmacol. Ther. 1987, 35, 57–162. doi:10.1016/0163-7258(87)90105-7 |
| 4. | Rinehart, K. L., Jr.; Shield, L. S. Fortschr. Chem. Org. Naturst. 1976, 33, 231–307. doi:10.1007/978-3-7091-3262-3_3 |
| 13. | de Carvalho da Silva, F.; Francisco Ferreira, V. Curr. Org. Synth. 2016, 13, 334–371. doi:10.2174/1570179412666150817220343 |
| 14. | Liao, C.-C.; Peddinti, R. K. Sci. Synth. 2006, 28, 323–324. |
| 15. | Chaudhary, A.; Khurana, J. M. Curr. Org. Chem. 2016, 20, 1314–1344. doi:10.2174/1385272820666151125231522 |
| 16. | López, L. I. L.; Flores, S. D. N.; Belmares, S. Y. S.; Galindo, A. S. Vitae 2014, 21, 248–258. |
| 17. | Sartori, M. F. Chem. Rev. 1963, 63, 279–296. doi:10.1021/cr60223a005 |
| 18. | Badave, K. D.; Khan, A. A.; Rane, S. Y. Anti-Cancer Agents Med. Chem. 2016, 16, 1017–1030. doi:10.2174/1871520616666160310143316 |
| 19. | Padhye, S.; Dandawate, P.; Yusufi, M.; Ahmad, A.; Sarkar, F. H. Med. Res. Rev. 2012, 32, 1131–1158. doi:10.1002/med.20235 |
| 20. | Lamoureux, G.; Perez, A. L.; Araya, M.; Agüero, C. J. Phys. Org. Chem. 2008, 21, 1022–1028. doi:10.1002/poc.1435 |
| 44. | Böniger, M. Ber. Dtsch. Chem. Ges. 1894, 27, 23–30. doi:10.1002/cber.18940270107 |
| 104. | Rickwood, M.; Marsden, S. D.; Askew, V. E. Photochromic spiroxazine compounds. U.S. Patent US5446150A, Aug 29, 1995. |
| 105. | Tathe, A. B.; Sekar, N. J. Fluoresc. 2015, 25, 1403–1415. doi:10.1007/s10895-015-1631-0 |
| 9. | Varghese, K. J.; Silvipriya, K. S.; Resmi, S.; Jolly, C. I. Inventi Impact: Cosmeceuticals 2010, 1, 1–5. |
| 10. | Saeed, S. M. G.; Sayeed, S. A.; Ashraf, S.; Naz, S.; Siddiqi, R.; Ali, R.; Mesaik, M. A. Pak. J. Bot. 2013, 45, 1431–1436. |
| 11. | Yusuf, M.; Ahmad, A.; Shahid, M.; Khan, M. I.; Khan, S. A.; Manzoor, N.; Mohammad, F. J. Cleaner Prod. 2012, 27, 42–50. doi:10.1016/j.jclepro.2012.01.005 |
| 12. | de Paiva, S. R.; Lima, L. A.; Figueiredo, M. R.; Kaplan, M. A. C. An. Acad. Bras. Cienc. 2004, 76, 499–504. doi:10.1590/s0001-37652004000300004 |
| 45. | Folin, O.; Wu, H. J. Biol. Chem. 1922, 51, 377–391. doi:10.1016/s0021-9258(18)85880-x |
| 7. | Pereyra, C. E.; Dantas, R. F.; Ferreira, S. B.; Gomes, L. P.; Silva-Jr, F. P. Cancer Cell Int. 2019, 19, 207. doi:10.1186/s12935-019-0925-8 |
| 8. | Qiu, H.-Y.; Wang, P.-F.; Lin, H.-Y.; Tang, C.-Y.; Zhu, H.-L.; Yang, Y.-H. Chem. Biol. Drug Des. 2018, 91, 681–690. doi:10.1111/cbdd.13141 |
| 40. | Pradhan, R.; Dandawate, P.; Vyas, A.; Padhye, S.; Biersack, B.; Schobert, R.; Ahmad, A.; Sarkar, F. H. Curr. Drug Targets 2012, 13, 1777–1798. doi:10.2174/138945012804545588 |
| 41. | Gouda, M. A.; Eldien, H. F.; Girges, M. M.; Berghot, M. A. Med. Chem. 2013, 3, 228–232. |
| 42. | Nakahara, Y.; Ishigami, A.; Takeda, Y. J. Chromatogr. B: Biomed. Sci. Appl. 1989, 489, 371–376. doi:10.1016/s0378-4347(00)82916-3 |
| 104. | Rickwood, M.; Marsden, S. D.; Askew, V. E. Photochromic spiroxazine compounds. U.S. Patent US5446150A, Aug 29, 1995. |
| 105. | Tathe, A. B.; Sekar, N. J. Fluoresc. 2015, 25, 1403–1415. doi:10.1007/s10895-015-1631-0 |
| 5. | Santos, L. O.; dos Anjos, J. P.; Ferreira, S. L. C.; de Andrade, J. B. Microchem. J. 2017, 133, 431–440. doi:10.1016/j.microc.2017.04.012 |
| 6. | Sousa, E. T.; Lopes, W. A.; de Andrade, J. B. Quim. Nova 2016, 39, 486–495. |
| 43. | Witt, O. N. Ber. Dtsch. Chem. Ges. 1891, 24, 3154–3157. doi:10.1002/cber.189102402156 |
| 99. | Ooyama, Y.; Nagano, S.; Okamura, M.; Yoshida, K. Eur. J. Org. Chem. 2008, 5899–5906. doi:10.1002/ejoc.200800832 |
| 28. | Olliaro, P.; Wirth, D. J. Pharm. Pharmacol. 1997, 49 (Suppl. 2), 29–33. doi:10.1111/j.2042-7158.1997.tb06157.x |
| 29. | França, T. C. C.; dos Santos, M. G.; Figueroa-Villar, J. D. Quim. Nova 2008, 31, 1271–1278. doi:10.1590/s0100-40422008000500060 |
| 34. | Hashemi-Fesharki, R. Res. Vet. Sci. 1991, 50, 204–207. doi:10.1016/0034-5288(91)90107-y |
| 35. | Garnier, T.; Mantyla, A.; Jarvinen, T.; Lawrence, J.; Brown, M.; Croft, S. J. Antimicrob. Chemother. 2007, 60, 802–810. doi:10.1093/jac/dkm303 |
| 36. | Hawa, N.; Rae, D. G.; Younis, S.; Mahadi, W.; Ibrahim, R.; Al-Wahab, W. Trop. Anim. Health Prod. 1988, 20, 130–136. doi:10.1007/bf02240076 |
| 101. | Katsuhira, Y.; Miwa, I.; Hiroyuki, H.; Mayumi, Y.; Yuji, K. Bull. Chem. Soc. Jpn. 1988, 61, 4335–4340. |
| 102. | Yoshida, K.; Yamamoto, M.; Ishiguro, M. Chem. Lett. 1986, 15, 1059–1062. doi:10.1246/cl.1986.1059 |
| 27. | Combs, G. F., Jr.; McClung, J. P. Vitamin K. In The Vitamins; Combs, G. F., Jr.; McClung, J. P., Eds.; Academic Press, 2017; pp 243–265. doi:10.1016/b978-0-12-802965-7.00009-5 |
| 37. | Ferreira, V. F.; Ferreira, S. B.; de Carvalho da Silva, F. Org. Biomol. Chem. 2010, 8, 4793–4802. doi:10.1039/c0ob00277a |
| 38. | da Rocha, D. R.; de Souza, A. C. G.; Resende, J. A. L. C.; Santos, W. C.; dos Santos, E. A.; Pessoa, C.; de Moraes, M. O.; Costa-Lotufo, L. V.; Montenegro, R. C.; Ferreira, V. F. Org. Biomol. Chem. 2011, 9, 4315–4322. doi:10.1039/c1ob05209h |
| 39. | Moreira, C. d. S.; Nicoletti, C. D.; Pinheiro, D. P.; de Moraes, L. G. C.; Futuro, D. O.; Ferreira, V. F.; Pessoa, C. d. Ó.; da Rocha, D. R. Med. Chem. Res. 2019, 28, 2109–2117. doi:10.1007/s00044-019-02439-w |
| 103. | Yoshida, K.; Koujiri, T.; Oga, N.; Ishiguro, M.; Kubo, Y. J. Chem. Soc., Chem. Commun. 1989, 708–710. doi:10.1039/c39890000708 |
| 25. | Baell, J. B.; Nissink, J. W. M. ACS Chem. Biol. 2018, 13, 36–44. doi:10.1021/acschembio.7b00903 |
| 26. | Aldrich, C.; Bertozzi, C.; Georg, G. I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K. M., Jr.; Schepartz, A.; Wang, S. ACS Med. Chem. Lett. 2017, 8, 379–382. doi:10.1021/acsmedchemlett.7b00056 |
| 98. | Ooyama, Y.; Nonami, K.; Watanabe, S.; Yoshida, K. Dyes Pigm. 2008, 77, 315–322. doi:10.1016/j.dyepig.2007.05.018 |
| 99. | Ooyama, Y.; Nagano, S.; Okamura, M.; Yoshida, K. Eur. J. Org. Chem. 2008, 5899–5906. doi:10.1002/ejoc.200800832 |
| 21. | Sharma, A.; Santos, I. O.; Gaur, P.; Ferreira, V. F.; Garcia, C. R. S.; da Rocha, D. R. Eur. J. Med. Chem. 2013, 59, 48–53. doi:10.1016/j.ejmech.2012.10.052 |
| 22. | García-Barrantes, P. M.; Lamoureux, G. V.; Pérez, A. L.; García-Sánchez, R. N.; Martínez, A. R.; San Feliciano, A. Eur. J. Med. Chem. 2013, 70, 548–557. doi:10.1016/j.ejmech.2013.10.011 |
| 23. | Lezama-Dávila, C. M.; Isaac-Márquez, A. P.; Kapadia, G.; Owens, K.; Oghumu, S.; Beverley, S.; Satoskar, A. R. Biol. Pharm. Bull. 2012, 35, 1761–1764. doi:10.1248/bpb.b12-00419 |
| 24. | Asche, C. Mini-Rev. Med. Chem. 2005, 5, 449–467. doi:10.2174/1389557053765556 |
| 30. | Watts, J.; Playford, M.; Hickey, K. N. Z. Vet. J. 2016, 64, 3–9. doi:10.1080/00480169.2015.1064792 |
| 31. | Dobbelaere, D.; Heussler, V. Annu. Rev. Microbiol. 1999, 53, 1–42. doi:10.1146/annurev.micro.53.1.1 |
| 32. | Mhadhbi, M.; Naouach, A.; Boumiza, A.; Chaabani, M. F.; BenAbderazzak, S.; Darghouth, M. A. Vet. Parasitol. 2010, 169, 241–247. doi:10.1016/j.vetpar.2010.01.013 |
| 33. | Sharifiyazdi, H.; Namazi, F.; Oryan, A.; Shahriari, R.; Razavi, M. Vet. Parasitol. 2012, 187, 431–435. doi:10.1016/j.vetpar.2012.01.016 |
| 100. | Yoshida, K.; Matsuoka, M.; Yamashita, Y.; Kitao, T. Bull. Chem. Soc. Jpn. 1980, 53, 2552–2554. doi:10.1246/bcsj.53.2552 |
| 48. | Danielson, I. S. J. Biol. Chem. 1933, 101, 505–522. doi:10.1016/s0021-9258(18)75897-3 |
| 46. | Obo, F. J. Biochem. 1941, 33, 231–235. doi:10.1093/oxfordjournals.jbchem.a126035 |
| 47. | Fu, B.; Liu, T.; Chen, J.; Li, K. Sens. Actuators, B 2018, 272, 598–604. doi:10.1016/j.snb.2018.05.068 |
| 108. | Land, E. J.; Ramsden, C. A.; Riley, P. A.; Yoganathan, G. Tetrahedron 2003, 59, 9547–9554. doi:10.1016/j.tet.2003.10.008 |
| 109. | Villemin, D.; Benabdallah, M.; Choukchou-Braham, N.; Mostefa-Kara, B. Synth. Commun. 2010, 40, 3109–3118. doi:10.1080/00397911003797916 |
| 106. | Ooyama, Y.; Nabeshima, S.; Mamura, T.; Ooyama, H. E.; Yoshida, K. Tetrahedron 2010, 66, 7954–7960. doi:10.1016/j.tet.2010.08.026 |
| 107. | Gates, M.; Newhall, W. F. J. Am. Chem. Soc. 1948, 70, 2261–2263. doi:10.1021/ja01186a079 |
| 56. | Legua, C. M.; Falcó, P. C.; Cabeza, A. S. Anal. Chim. Acta 1993, 283, 635–644. doi:10.1016/0003-2670(93)85276-p |
| 57. | Molins Legua, C.; Campins Falcó, P.; Sevillano Cabeza, A. Fresenius' J. Anal. Chem. 1994, 349, 311–316. doi:10.1007/bf00323210 |
| 42. | Nakahara, Y.; Ishigami, A.; Takeda, Y. J. Chromatogr. B: Biomed. Sci. Appl. 1989, 489, 371–376. doi:10.1016/s0378-4347(00)82916-3 |
| 58. | Xu, L.; Wang, H.; Xiao, Y. Spectrochim. Acta, Part A 2004, 60, 3007–3012. doi:10.1016/j.saa.2004.02.018 |
| 59. | Kumar, C. H. A.; Kumar, T. A.; Gurupadayya, B. M.; Sloka, N.; Reddy, M. B. R. Arch. Appl. Sci. Res. 2010, 2, 278–287. |
| 60. | Elbashir, A. A.; Elwagee, A. H. E. J. Assoc. Arab Univ. Basic Appl. Sci. 2012, 11, 32–36. doi:10.1016/j.jaubas.2011.12.003 |
| 61. | Osman, R. A. M.; Elbashir, A. A. Int. J. Bioanal. Methods Bioequival. Stud. 2019, 5, 82–92. |
| 62. | Khalil, N. A.; Ibrahim, W. H. Tikrit J. Pure Sci. 2020, 25, 68–74. doi:10.25130/j.v25i1.938 |
| 63. | Awan, Z. A.; Hegazy, M. A.; Kammoun, A. K. Spectrochim. Acta, Part A 2020, 230, 118066. doi:10.1016/j.saa.2020.118066 |
| 64. | Darwish, I. A.; Al-Shehri, M. M.; El-Gendy, M. A. Chem. Cent. J. 2012, 6, 11. doi:10.1186/1752-153x-6-11 |
| 65. | Ahmed, S. M.; Elbashir, A. A. J. Anal. Bioanal. Tech. 2015, 6, 248. |
| 66. | Altigani, A. M. N.; Elbashir, A. A. Austin J. Anal. Pharm. Chem. 2014, 1, 1019. |
| 67. | Abdulrahman, S. A. M.; Basavaiah, K. Drug Test. Anal. 2011, 3, 748–754. doi:10.1002/dta.242 |
| 68. | Elbashir, A. A.; Awad, S. F. J. Pharmacovigilance 2013, 1, 1000105. |
| 69. | Salman, B. I.; Hussein, S. A.; Ali, M. F. B.; Marzouq, M. A. Microchem. J. 2019, 145, 959–965. doi:10.1016/j.microc.2018.12.018 |
| 54. | Hashimoto, Y.; Endo, M.; Tominaga, K.; Inuzuka, S.; Moriyasu, M. Microchim. Acta 1978, 70, 493–504. doi:10.1007/bf01197101 |
| 55. | Saurina, J.; Hernández-Cassuo, S. Anal. Chim. Acta 1993, 283, 414–420. doi:10.1016/0003-2670(93)85252-f |
| 52. | Elbashir, A. A.; Ahmed, A. A.; Ali Ahmed, S. M.; Aboul-Enein, H. Y. Appl. Spectrosc. Rev. 2012, 47, 219–232. doi:10.1080/05704928.2011.639107 |
| 53. | Hiyama, M. Yakugaku Zasshi 1952, 72, 1367–1370. doi:10.1248/yakushi1947.72.10_1367 |
| 49. | Martin, E. L.; Fieser, L. F. Org. Synth. 1941, 21, 91. doi:10.15227/orgsyn.021.0091 |
| 110. | Villemin, D.; Benabdallah, M.; Rahmoun, N.; Jouannic, C.; Choukchou-Braham, N.; Mostefa-Kara, B. Synth. Commun. 2010, 40, 3514–3521. doi:10.1080/00397910903457340 |
| 50. | Gummadi, S.; Kommoju, M. Am. J. PharmTech Res. 2019, 9, 14–37. doi:10.46624/ajptr.2019.v9.i1.002 |
| 51. | Adegoke, O. A. Int. J. Pharm. Sci. Rev. Res. 2012, 14, 6–24. |
© 2022 Ribeiro et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.