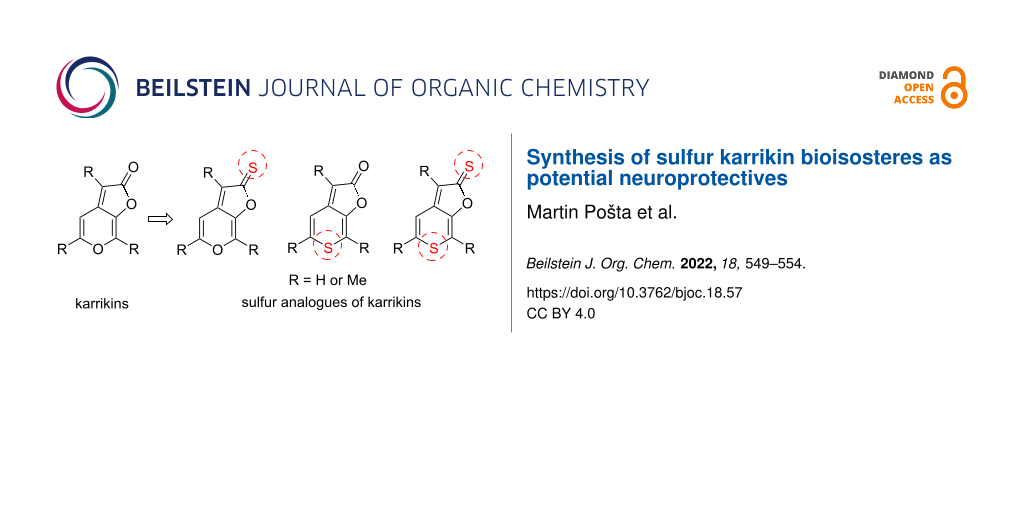
Abstract
The only known sulfur-containing karrikin, 3-methyl-2H-thiopyrano[3,4-b]furan-2-one, has been recently identified as an extremely efficient neuroprotective butenolide. Herein, we report the targeted synthesis of this compound as well as new synthetic protocols toward a class of compounds derived from 2H-furo[2,3-c]pyran-2-ones (karrikins) via bioisosteric exchange of oxygen with sulfur. In particular, we present synthetic procedures toward bioisosteres of karrikins with one or two sulfur heteroatoms incorporated into the core backbone together with evaluation of their biological activity in inhibition of acetylcholinesterase.

Graphical Abstract
Introduction
Neurological disorders, especially the Alzheimer’s and the Parkinson’s diseases represent a serious problem for elderly populations worldwide. Studies of these neurodegenerative diseases led to the discovery of a deficit of acetylcholine and dopamine levels in the brain of patients suffering with the Parkinson’s and the Alzheimer’s disease, respectively. Therefore, research is focused on the discovery of new drugs protecting acetylcholine and dopamine levels via inhibition of acetylcholinesterase (AChE) and monoamine oxidase (MAO). A promising source of such novel drugs could be the smoke of burning plants that contains more than 4000 chemical species [1] and among them numerous compounds possess biological activities in humans. The psychotropic effect of smoke is well known for centuries and its inhalation has different effects depending on plant material used. For example, smoking of Cannabis sativa has a range of mental and physical effects including a number of therapeutic applications [2-4]. Currently, the most popular is the use of tobacco products as antidepressants based on the MAO inhibitory properties of tobacco smoke and its components [5,6]. However, tobacco and other plant-derived smoke include toxic and carcinogenic compounds which are according to WHO responsible for the death of millions of people every year [7,8]. Nevertheless, plant-derived smoke is an attractive resource containing many small molecules, many of which are unknown, that could have untapped potential in medicine. The study of the relationship between the components of plant-derived smoke and MAO and/or AChE can lead to safe and efficient drugs and drug scaffolds from a yet unexploited resource.
One very important bioactive constituent of smoke of burning vegetation are karrikins (Figure 1) discovered in 2004 independently by Flematti [9] and Van Staden [10]. This class of molecules has been identified as an extremely potent seed germination stimulant, promoting germination and the development of early seedling in many plant species at sub-nanomolar concentrations [11-14]. Structurally, the karrikin backbone consists of a fused pyran and a furanone ring. Each particular molecule differs in the number of methyl groups in positions C3, C5 and C7. Structures of the four most active and abundant karrikins (KAR1–KAR4) are depicted in Figure 1. The synthesis of these heterocycles is rather challenging, because the fused pyran and furanone system cannot be easily prepared by standard cyclization methods [15-17].
![[1860-5397-18-57-1]](/bjoc/content/figures/1860-5397-18-57-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Structures of naturally occurring karrikins.
Figure 1: Structures of naturally occurring karrikins.
Although karrikins are extremely active plant growth regulators [14,18,19], their biological activity in humans was not investigated until recently. In 2019 a study of Naidoo et al. [20] reported naturally occurring karrikins as compounds with moderate inhibitory activity against both types of monoamine oxidases (MAO-A and MAO-B) and acetylcholinesterase, while the sulfur bioiostere 3-methyl-2H-thiopyrano[3,4-b]furan-2-one (8) [21] exhibited excellent activity. No targeted synthesis of 8 exists; it was isolated in low yield as a side-product in the synthesis of KAR1 (1) [21]. In this reaction sequence, thiopyranthione 6b is the side-product in the pyranthione 6a synthesis and both analogues were progressed to the final furan ring closure (Scheme 1). A plausible mechanism for the cyclization of compounds 7 is the Darzens reaction to episulfide, followed by Barton–Kellogg-type reaction with triphenylphosphine and elimination of triphenylphosphine sulfide. Compound 8 showed lower germination activity than KAR1 [22], but achieved IC50 values of 30 nM for MAO-B and 80 nM for AChE, respectively [20], and therefore, it represents a promising therapeutic potential against Alzheimer’s and Parkinson’s diseases.
![[1860-5397-18-57-i1]](/bjoc/content/inline/1860-5397-18-57-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: i) P4S10, THF; ii) 2-chloropropionyl chloride, Et3N; iii) Ph3P, NaOAc, Ac2O.
Scheme 1: i) P4S10, THF; ii) 2-chloropropionyl chloride, Et3N; iii) Ph3P, NaOAc, Ac2O.
The goal of this work is the development of a targeted synthesis of 8 and other sulfur analogues of karrikins in order to study the effect of bioisosteric exchange together with the effect of substitution along the karrikin backbone on the inhibitory activity against AChE. The target sulfur analogues of karrikins are shown on Figure 2.
![[1860-5397-18-57-2]](/bjoc/content/figures/1860-5397-18-57-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Target compounds with highlighted positions of oxygen to sulfur exchange.
Figure 2: Target compounds with highlighted positions of oxygen to sulfur exchange.
Results and Discussion
Synthesis of KAR analogues with sulfur in position C2
A series of thiones, derived from the 2H-furo[2,3-c]pyran-2-thione core, differing in the number and the position of methyl groups (9–12), has been prepared from appropriate karrikins. Starting materials, karrikins KAR1 (1), KAR3 (3) and KAR4 (4) (Figure 1) were synthesized from pyromeconic acid (5), allomaltol (13) or maltol (14), respectively, following a published procedure [21,23], while karrikin KAR2 (2) was synthesized from ᴅ-xylose [24].
The conversion of karrikins 1–4 to the corresponding C2 thiones 9–12 was accomplished using microwave-assisted heating with Lawesson’s reagent and hexamethyldisiloxane (HMDO) (Scheme 2) and provided the target compounds in good to high yields.
![[1860-5397-18-57-i2]](/bjoc/content/inline/1860-5397-18-57-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: i) Lawesson’s reagent, HMDO, toluene, MW irradiation, 120 °C, 60 min.
Scheme 2: i) Lawesson’s reagent, HMDO, toluene, MW irradiation, 120 °C, 60 min.
Synthesis of KAR analogues with sulfur in position 6
Two different synthetic approaches were used for the preparation of C6 sulfur bioisosteres bearing a thiopyran moiety. For the synthesis of KAR1, KAR3 and KAR4 analogues, we proposed a synthetic strategy based on a procedure [21] employing the cyclization of appropriate esters of thiopyranthiones (Scheme 3). We expected that heterocyclic atom exchange used in the literature [25] for the synthesis of dithiomaltol 16b directly from commercially available maltol (14) would proceed also with pyromeconic acid (5) [26] and allomaltol (13) [27]. Unfortunately, the treatment of hydroxypyranones 5, 13 and 14 with Lawesson’s reagent resulted in an inseparable mixture (approx. 1:1) of pyranthiones 6a, 15a, 16a and thiopyranthiones 6b, 15b, 16b in low yields up to 30% in all the cases.
![[1860-5397-18-57-i3]](/bjoc/content/inline/1860-5397-18-57-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: i) P4S10 or Lawesson’s reagent, see table for conditions; ii) 2-chloropropionyl chloride, Et3N, DCM, 0 °C; iii) AcONa, Ph3P, Ac2O, reflux. aReported yields are 1H NMR yields. bIsolated yield.
Scheme 3: i) P4S10 or Lawesson’s reagent, see table for conditions; ii) 2-chloropropionyl chloride, Et3N, DCM...
In order to increase the selectivity and the yields, an improved procedure [28] has been tested. An additive, HMDO applied together with Lawesson’s reagent improved significantly the yields of thiopyranthiones 6b, 15b, 16b over pyranthiones 6a, 15a, 16a. In contrast to previous reports on related systems [28-31], microwave heating did not improve the yields; therefore, conventional heating was used in our case.
Generally, 5 is the most reactive among the substrates and the experiments proved that only 5 and not 13 or 14 can be partially converted to thiopyranthione 6b in the presence of P4S10 without heating. This observation explains the formation of side-product 8 in the original Flematti’s synthesis [21] (Scheme 1).
With thiopyranthiones 6b, 15b, 16b in hand, we carried out the esterification step with 2-chloropropionyl chloride, followed by intramolecular cyclization which provided the desired compounds 8, 20 and 21 albeit in low yields.
Due to the extremely low isolated yield of compounds 20 and 21, we looked for an alternative more efficient procedure. In 2008 Sun et al. [32] described a method of karrikin alkylation in position 7 via direct metalation with lithium bis(trimethylsilyl)amide (LiHMDS), followed by the addition of an alkyl halide. Application of this method to 8 provided the target molecule 21 in good yield (Scheme 4). It has to be mentioned that the metalation proceeds exclusively at C7, and thus cannot be used for the preparation of 20 via alkylation at C5.
![[1860-5397-18-57-i4]](/bjoc/content/inline/1860-5397-18-57-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Our attempt to prepare the desmethyl analogue 26 through esterification of 6b with chloroacetyl chloride followed by intramolecular cyclization stayed unrewarded due to the lack of reactivity in the cyclization step.
In order to overcome this problem a new strategy was developed. We combined the synthetic protocol of Goddard-Borger [24] for the preparation of 2 using the Xavier’s procedure [33] towards 5-thiopyranose-fused butenolides and the reaction pathway is outlined in Scheme 5.
![[1860-5397-18-57-i5]](/bjoc/content/inline/1860-5397-18-57-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: i) a) NaOH, MeOH/H2O, rt, Amberlyst 15 [H+], b) AcOH 70% aq, reflux, 2 h; ii) a) EtOCOCl, pyridine, b) Et3N, CH2Cl2; iii) (Ph3P)4Pd, BSA, THF.
Scheme 5: i) a) NaOH, MeOH/H2O, rt, Amberlyst 15 [H+], b) AcOH 70% aq, reflux, 2 h; ii) a) EtOCOCl, pyridine,...
The synthesis of the key intermediate butenolide 23 was accomplished starting from easily available ᴅ-xylose, following a published multistep procedure [33]. The synthesis of target compound 26 was accomplished in two steps by double esterification and elimination of 23. Dicarbonate 24 partially eliminated over C7–C7a to give 25 upon aqueous workup. In order to maximize the yield, the organic fraction containing a mixture of 24 and 25 was treated with triethylamine in order to obtain the eliminated product exclusively. The second elimination over C4–C5 resulted in a low yield of 26 (41%) (not shown in Scheme 5) and a high (Ph3P)4Pd catalyst load (10%) was necessary, despite the fact that allylic carbonates are good substrates in these Tsuji–Trost eliminations [34]. However, in the presence of N,O-bis(trimethylsilyl)acetamide (BSA) a standard catalyst loading (4 mol %) was sufficient to obtain 26 in a good yield (74%, Scheme 5).
Synthesis of KAR analogues with sulfur in positions C2 and 6
2H-Thiopyrano[3,4-b]furan-2-thione derivatives 27–30 were prepared from 8, 20, 21, and 26 using microwave heating with Lawesson’s reagent and HMDO (Scheme 6). Thionation provided the title compounds in good to high yields.
![[1860-5397-18-57-i6]](/bjoc/content/inline/1860-5397-18-57-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: i) Lawesson’s reagent, HMDO, toluene, MW irradiation (120 °C), 60 min.
Scheme 6: i) Lawesson’s reagent, HMDO, toluene, MW irradiation (120 °C), 60 min.
Biochemical study – AchE inhibition
The compounds were further tested for their ability to inhibit AChE in vitro [24]. Based on the data by Naidoo et al. [20], where KAR3 and sulfur-substituted karrikin (8) showed significant (submicromolar) inhibition effects on AChE, comparable activity was expected in the current series of compounds. However, no significant activity was observed. At 10 µM screening concentration the most active analogues (28 and 29) only reached approx. 15% inhibition which corresponds to the expected IC50 values in high micromolar range (Table 1).
Table 1: Inhibition of acetylcholine esterase by the synthesized analogues.
| Compound | AChE residual activity at 10 µM (% of untreated) | AChE IC50 (µM) |
| none | 100 ± 3 | |
| galanthamine | 16 ± 1 | 1.5 ± 0.5 |
| 1 | – | 477 ± 0.12 [20] |
| 3 | – | 0.37 ± 0.02 [20] |
| 8 | – | 0.08 ± 0.006 [20] |
| 9 | 95 ± 8 | n.d. |
| 10 | 99 ± 3 | n.d. |
| 11 | 93 ± 3 | n.d. |
| 12 | 91 ± 2 | n.d. |
| 20 | 90 ± 2 | n.d. |
| 21 | 91 ± 2 | n.d. |
| 26 | 99 ± 0.4 | n.d. |
| 27 | 97 ± 5 | n.d. |
| 28 | 85 ± 1 | n.d. |
| 29 | 86 ± 2 | n.d. |
| 30 | 92 ± 1 | n.d. |
| n.d. - not determined, IC50 >> 10 µM | ||
Conclusion
In conclusion, efficient synthetic protocols for the synthesis of sulfur analogues of karrikin were developed. The original synthetic methodology for the preparation of analogues of karrikins with one sulfur atom in position C2 or position 6, or two sulfur atoms in both positions was improved; however, observed biological activities towards AChE were rather low.
Supporting Information
| Supporting Information File 1: Experimental part, compound characterization and copies of NMR spectra. | ||
| Format: PDF | Size: 1.2 MB | Download |
References
-
Andreoli, C.; Gigante, D.; Nunziata, A. Toxicol. In Vitro 2003, 17, 587–594. doi:10.1016/s0887-2333(03)00091-2
Return to citation in text: [1] -
Braithwaite, I.; Bhagavan, C.; Doppen, M.; Kung, S.; Oldfield, K.; Newton-Howes, G. Curr. Opin. Psychol. 2021, 38, 1–10. doi:10.1016/j.copsyc.2020.06.002
Return to citation in text: [1] -
Allan, G. M.; Finley, C. R.; Ton, J.; Perry, D.; Ramji, J.; Crawford, K.; Lindblad, A. J.; Korownyk, C.; Kolber, M. R. Can. Fam. Physician 2018, 64, e78–e94.
Return to citation in text: [1] -
Volkow, N. D.; Baler, R. D.; Compton, W. M.; Weiss, S. R. B. N. Engl. J. Med. 2014, 370, 2219–2227. doi:10.1056/nejmra1402309
Return to citation in text: [1] -
Fowler, J. S.; Volkow, N. D.; Wang, G.-J.; Pappas, N.; Logan, J.; MacGregor, R.; Alexoff, D.; Shea, C.; Schlyer, D.; Wolf, A. P.; Warner, D.; Zezulkova, I.; Cilento, R. Nature 1996, 379, 733–736. doi:10.1038/379733a0
Return to citation in text: [1] -
Yu, P. H.; Boulton, A. A. Life Sci. 1987, 41, 675–682. doi:10.1016/0024-3205(87)90446-2
Return to citation in text: [1] -
https://www.who.int/news-room/fact-sheets/detail/tobacco.
Return to citation in text: [1] -
GBD 2019 Tobacco Collaborators. Lancet 2021, 397, 2337–2360. doi:10.1016/s0140-6736(21)01169-7
Return to citation in text: [1] -
Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Science 2004, 305, 977. doi:10.1126/science.1099944
Return to citation in text: [1] -
Van Staden, J.; Jäger, A. K.; Light, M. E.; Burger, B. V. S. Afr. J. Bot. 2004, 70, 654–659. doi:10.1016/s0254-6299(15)30206-4
Return to citation in text: [1] -
Daws, M. I.; Davies, J.; Pritchard, H. W.; Brown, N. A. C.; Van Staden, J. Plant Growth Regul. 2007, 51, 73–82. doi:10.1007/s10725-006-9149-8
Return to citation in text: [1] -
Dixon, K. W.; Merritt, D. J.; Flematti, G. R.; Ghisalberti, E. L. Acta Hortic. 2009, 813, 155–170. doi:10.17660/actahortic.2009.813.20
Return to citation in text: [1] -
Jain, N.; Kulkarni, M. G.; van Staden, J. Plant Growth Regul. 2006, 49, 263–267. doi:10.1007/s10725-006-9136-0
Return to citation in text: [1] -
Meng, Y.; Shuai, H.; Luo, X.; Chen, F.; Zhou, W.; Yang, W.; Shu, K. Front. Plant Sci. 2017, 7, 2021. doi:10.3389/fpls.2016.02021
Return to citation in text: [1] [2] -
Belsky, I.; Dodiuk, H.; Shvo, Y. J. Org. Chem. 1974, 39, 989–995. doi:10.1021/jo00921a028
Return to citation in text: [1] -
Tanyeli, C.; Caliskan, Z. Z. Synth. Commun. 2000, 30, 2855–2862. doi:10.1080/00397910008087436
Return to citation in text: [1] -
Demir, A. S.; Gercek, Z.; Duygu, N.; Igdir, A. C.; Reis, O. Can. J. Chem. 1999, 77, 1336–1339. doi:10.1139/v99-136
Return to citation in text: [1] -
Waters, M. T.; Scaffidi, A.; Sun, Y. K.; Flematti, G. R.; Smith, S. M. Plant J. 2014, 79, 623–631. doi:10.1111/tpj.12430
Return to citation in text: [1] -
Waters, M. T.; Scaffidi, A.; Flematti, G. R.; Smith, S. M. Curr. Opin. Plant Biol. 2013, 16, 667–673. doi:10.1016/j.pbi.2013.07.005
Return to citation in text: [1] -
Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Tetrahedron Lett. 2005, 46, 5719–5721. doi:10.1016/j.tetlet.2005.06.077
Return to citation in text: [1] [2] [3] [4] [5] -
Nair, J. J.; Pošta, M.; Papenfus, H. B.; Munro, O. Q.; Beier, P.; Van Staden, J. S. Afr. J. Bot. 2014, 91, 53–57. doi:10.1016/j.sajb.2013.12.003
Return to citation in text: [1] -
Flematti, G. R.; Goddard-Borger, E. D.; Merritt, D. J.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. J. Agric. Food Chem. 2007, 55, 2189–2194. doi:10.1021/jf0633241
Return to citation in text: [1] -
Goddard-Borger, E. D.; Ghisalberti, E. L.; Stick, R. V. Eur. J. Org. Chem. 2007, 3925–3934. doi:10.1002/ejoc.200700334
Return to citation in text: [1] [2] [3] -
Brayton, D.; Jacobsen, F. E.; Cohen, S. M.; Farmer, P. J. Chem. Commun. 2006, 206–208. doi:10.1039/b511966a
Return to citation in text: [1] -
Liu, Z. D.; Khodr, H. H.; Liu, D. Y.; Lu, S. L.; Hider, R. C. J. Med. Chem. 1999, 42, 4814–4823. doi:10.1021/jm991080o
Return to citation in text: [1] -
Ma, Y.; Luo, W.; Quinn, P. J.; Liu, Z.; Hider, R. C. J. Med. Chem. 2004, 47, 6349–6362. doi:10.1021/jm049751s
Return to citation in text: [1] -
Bruner, B.; Walker, M. B.; Ghimire, M. M.; Zhang, D.; Selke, M.; Klausmeyer, K. K.; Omary, M. A.; Farmer, P. J. Dalton Trans. 2014, 43, 11548–11556. doi:10.1039/c4dt00961d
Return to citation in text: [1] [2] -
Filippi, J.-J.; Fernandez, X.; Lizzani-Cuvelier, L.; Loiseau, A.-M. Tetrahedron Lett. 2003, 44, 6647–6650. doi:10.1016/s0040-4039(03)01636-8
Return to citation in text: [1] -
Perreux, L.; Loupy, A. Tetrahedron 2001, 57, 9199–9223. doi:10.1016/s0040-4020(01)00905-x
Return to citation in text: [1] -
Rodríguez, A. M.; Prieto, P.; de la Hoz, A.; Díaz-Ortiz, Á.; Martín, D. R.; García, J. I. ChemistryOpen 2015, 4, 308–317. doi:10.1002/open.201402123
Return to citation in text: [1] -
Sun, K.; Chen, Y.; Wagerle, T.; Linnstaedt, D.; Currie, M.; Chmura, P.; Song, Y.; Xu, M. Tetrahedron Lett. 2008, 49, 2922–2925. doi:10.1016/j.tetlet.2008.03.024
Return to citation in text: [1] -
Xavier, N. M.; Madeira, P. J. A.; Florêncio, M. H.; Rauter, A. P. Eur. J. Org. Chem. 2009, 4983–4991. doi:10.1002/ejoc.200900573
Return to citation in text: [1] [2] -
Takahashi, T.; Nakagawa, N.; Minoshima, T.; Yamada, H.; Tsuji, J. Tetrahedron Lett. 1990, 31, 4333–4336. doi:10.1016/s0040-4039(00)97614-7
Return to citation in text: [1]
| 34. | Takahashi, T.; Nakagawa, N.; Minoshima, T.; Yamada, H.; Tsuji, J. Tetrahedron Lett. 1990, 31, 4333–4336. doi:10.1016/s0040-4039(00)97614-7 |
| 24. | Goddard-Borger, E. D.; Ghisalberti, E. L.; Stick, R. V. Eur. J. Org. Chem. 2007, 3925–3934. doi:10.1002/ejoc.200700334 |
| 20. | Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014 |
| 1. | Andreoli, C.; Gigante, D.; Nunziata, A. Toxicol. In Vitro 2003, 17, 587–594. doi:10.1016/s0887-2333(03)00091-2 |
| 9. | Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Science 2004, 305, 977. doi:10.1126/science.1099944 |
| 21. | Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Tetrahedron Lett. 2005, 46, 5719–5721. doi:10.1016/j.tetlet.2005.06.077 |
| 23. | Flematti, G. R.; Goddard-Borger, E. D.; Merritt, D. J.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. J. Agric. Food Chem. 2007, 55, 2189–2194. doi:10.1021/jf0633241 |
| 7. | https://www.who.int/news-room/fact-sheets/detail/tobacco. |
| 8. | GBD 2019 Tobacco Collaborators. Lancet 2021, 397, 2337–2360. doi:10.1016/s0140-6736(21)01169-7 |
| 24. | Goddard-Borger, E. D.; Ghisalberti, E. L.; Stick, R. V. Eur. J. Org. Chem. 2007, 3925–3934. doi:10.1002/ejoc.200700334 |
| 5. | Fowler, J. S.; Volkow, N. D.; Wang, G.-J.; Pappas, N.; Logan, J.; MacGregor, R.; Alexoff, D.; Shea, C.; Schlyer, D.; Wolf, A. P.; Warner, D.; Zezulkova, I.; Cilento, R. Nature 1996, 379, 733–736. doi:10.1038/379733a0 |
| 6. | Yu, P. H.; Boulton, A. A. Life Sci. 1987, 41, 675–682. doi:10.1016/0024-3205(87)90446-2 |
| 22. | Nair, J. J.; Pošta, M.; Papenfus, H. B.; Munro, O. Q.; Beier, P.; Van Staden, J. S. Afr. J. Bot. 2014, 91, 53–57. doi:10.1016/j.sajb.2013.12.003 |
| 2. | Braithwaite, I.; Bhagavan, C.; Doppen, M.; Kung, S.; Oldfield, K.; Newton-Howes, G. Curr. Opin. Psychol. 2021, 38, 1–10. doi:10.1016/j.copsyc.2020.06.002 |
| 3. | Allan, G. M.; Finley, C. R.; Ton, J.; Perry, D.; Ramji, J.; Crawford, K.; Lindblad, A. J.; Korownyk, C.; Kolber, M. R. Can. Fam. Physician 2018, 64, e78–e94. |
| 4. | Volkow, N. D.; Baler, R. D.; Compton, W. M.; Weiss, S. R. B. N. Engl. J. Med. 2014, 370, 2219–2227. doi:10.1056/nejmra1402309 |
| 20. | Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014 |
| 14. | Meng, Y.; Shuai, H.; Luo, X.; Chen, F.; Zhou, W.; Yang, W.; Shu, K. Front. Plant Sci. 2017, 7, 2021. doi:10.3389/fpls.2016.02021 |
| 18. | Waters, M. T.; Scaffidi, A.; Sun, Y. K.; Flematti, G. R.; Smith, S. M. Plant J. 2014, 79, 623–631. doi:10.1111/tpj.12430 |
| 19. | Waters, M. T.; Scaffidi, A.; Flematti, G. R.; Smith, S. M. Curr. Opin. Plant Biol. 2013, 16, 667–673. doi:10.1016/j.pbi.2013.07.005 |
| 21. | Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Tetrahedron Lett. 2005, 46, 5719–5721. doi:10.1016/j.tetlet.2005.06.077 |
| 20. | Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014 |
| 15. | Belsky, I.; Dodiuk, H.; Shvo, Y. J. Org. Chem. 1974, 39, 989–995. doi:10.1021/jo00921a028 |
| 16. | Tanyeli, C.; Caliskan, Z. Z. Synth. Commun. 2000, 30, 2855–2862. doi:10.1080/00397910008087436 |
| 17. | Demir, A. S.; Gercek, Z.; Duygu, N.; Igdir, A. C.; Reis, O. Can. J. Chem. 1999, 77, 1336–1339. doi:10.1139/v99-136 |
| 21. | Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Tetrahedron Lett. 2005, 46, 5719–5721. doi:10.1016/j.tetlet.2005.06.077 |
| 11. | Daws, M. I.; Davies, J.; Pritchard, H. W.; Brown, N. A. C.; Van Staden, J. Plant Growth Regul. 2007, 51, 73–82. doi:10.1007/s10725-006-9149-8 |
| 12. | Dixon, K. W.; Merritt, D. J.; Flematti, G. R.; Ghisalberti, E. L. Acta Hortic. 2009, 813, 155–170. doi:10.17660/actahortic.2009.813.20 |
| 13. | Jain, N.; Kulkarni, M. G.; van Staden, J. Plant Growth Regul. 2006, 49, 263–267. doi:10.1007/s10725-006-9136-0 |
| 14. | Meng, Y.; Shuai, H.; Luo, X.; Chen, F.; Zhou, W.; Yang, W.; Shu, K. Front. Plant Sci. 2017, 7, 2021. doi:10.3389/fpls.2016.02021 |
| 20. | Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014 |
| 10. | Van Staden, J.; Jäger, A. K.; Light, M. E.; Burger, B. V. S. Afr. J. Bot. 2004, 70, 654–659. doi:10.1016/s0254-6299(15)30206-4 |
| 20. | Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014 |
| 20. | Naidoo, D.; Pošta, M.; Roy, A.; Kulkarni, M.; Van Staden, J. Phytochemistry 2019, 163, 187–194. doi:10.1016/j.phytochem.2019.03.014 |
| 26. | Liu, Z. D.; Khodr, H. H.; Liu, D. Y.; Lu, S. L.; Hider, R. C. J. Med. Chem. 1999, 42, 4814–4823. doi:10.1021/jm991080o |
| 21. | Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Tetrahedron Lett. 2005, 46, 5719–5721. doi:10.1016/j.tetlet.2005.06.077 |
| 25. | Brayton, D.; Jacobsen, F. E.; Cohen, S. M.; Farmer, P. J. Chem. Commun. 2006, 206–208. doi:10.1039/b511966a |
| 33. | Xavier, N. M.; Madeira, P. J. A.; Florêncio, M. H.; Rauter, A. P. Eur. J. Org. Chem. 2009, 4983–4991. doi:10.1002/ejoc.200900573 |
| 33. | Xavier, N. M.; Madeira, P. J. A.; Florêncio, M. H.; Rauter, A. P. Eur. J. Org. Chem. 2009, 4983–4991. doi:10.1002/ejoc.200900573 |
| 32. | Sun, K.; Chen, Y.; Wagerle, T.; Linnstaedt, D.; Currie, M.; Chmura, P.; Song, Y.; Xu, M. Tetrahedron Lett. 2008, 49, 2922–2925. doi:10.1016/j.tetlet.2008.03.024 |
| 24. | Goddard-Borger, E. D.; Ghisalberti, E. L.; Stick, R. V. Eur. J. Org. Chem. 2007, 3925–3934. doi:10.1002/ejoc.200700334 |
| 28. | Bruner, B.; Walker, M. B.; Ghimire, M. M.; Zhang, D.; Selke, M.; Klausmeyer, K. K.; Omary, M. A.; Farmer, P. J. Dalton Trans. 2014, 43, 11548–11556. doi:10.1039/c4dt00961d |
| 29. | Filippi, J.-J.; Fernandez, X.; Lizzani-Cuvelier, L.; Loiseau, A.-M. Tetrahedron Lett. 2003, 44, 6647–6650. doi:10.1016/s0040-4039(03)01636-8 |
| 30. | Perreux, L.; Loupy, A. Tetrahedron 2001, 57, 9199–9223. doi:10.1016/s0040-4020(01)00905-x |
| 31. | Rodríguez, A. M.; Prieto, P.; de la Hoz, A.; Díaz-Ortiz, Á.; Martín, D. R.; García, J. I. ChemistryOpen 2015, 4, 308–317. doi:10.1002/open.201402123 |
| 21. | Flematti, G. R.; Ghisalberti, E. L.; Dixon, K. W.; Trengove, R. D. Tetrahedron Lett. 2005, 46, 5719–5721. doi:10.1016/j.tetlet.2005.06.077 |
| 27. | Ma, Y.; Luo, W.; Quinn, P. J.; Liu, Z.; Hider, R. C. J. Med. Chem. 2004, 47, 6349–6362. doi:10.1021/jm049751s |
| 28. | Bruner, B.; Walker, M. B.; Ghimire, M. M.; Zhang, D.; Selke, M.; Klausmeyer, K. K.; Omary, M. A.; Farmer, P. J. Dalton Trans. 2014, 43, 11548–11556. doi:10.1039/c4dt00961d |
© 2022 Pošta et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.