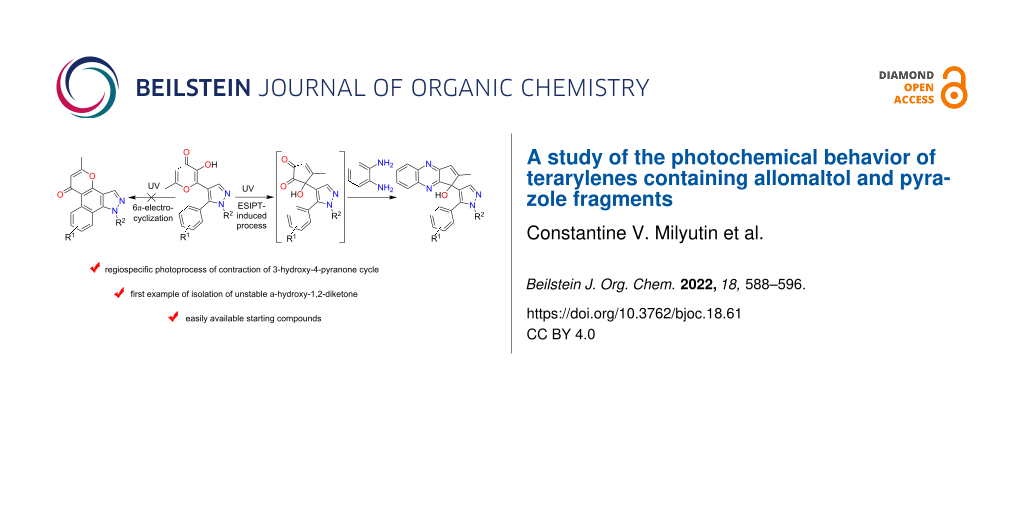
Abstract
The photochemical properties behavior of 3-hydroxy-4-pyranone containing terarylenes with a pyrazole bridge fragment were studied. It was shown that UV-induced 6π-electrocyclization of the 1,3,5-hexatriene system was not observed for the considered objects molecules. At the same time, the phototransformation of such systems proceeds exclusively in the direction of the contraction of the pyranone ring leading to unstable α-hydroxydiketones. For the first time the possibility of isolation of the resulting α-hydroxydiketones in pure form was demonstrated. Wherein, it was shown that relatively low stable α-hydroxydiketones can be trapped by reaction with 1,2-phenylenediamine. The general method for the preparation of the corresponding quinoxalines on the basis of the aforementioned condensation was implemented. It was demonstrated that the studied photoreaction does not depend on the type of pyrazole bridge. The structures of three of synthesized products were established by X-ray diffraction.

Graphical Abstract
Introduction
The investigation of the photochemical behavior of organic products is a significant part of modern materials science and technology. The UV-induced processes are extensively employed in synthetic organic chemistry [1-7]. The advantage of photochemical methods is the possibility of the preparation of diverse products that are problematic to obtain using other chemical approaches. It should be noted that light can be considered as a traceless agent, therefore, UV-induced processes are of considerable interest in the context of green chemistry [8,9]. At the same time in some cases UV irradiation of organic compounds leads to the formation of highly reactive intermediates. Such objects may, possessing a specific reactivity, which define their further application in organic synthesis. Thus, an actual problem is to study the photochemical behavior of various compounds capable of photogeneration of these unstable intermediates. Moreover, the results of such investigations can be used for the development of UV-promoted synthetic methods.
Among the photochemical processes used in organic synthesis, 6π-electrocyclization of 1,3,5-hexatriene systems is attracting considerable attention [10-17]. The photocyclization of terarylenes leading to the formation of polyheterocyclic compounds is one of the most widely studied processes of this type. Due to the wide variety of this products and their synthetic availability, terarylenes are convenient models for a detailed study of the correlation between the structure of such objects and their photochemical properties [18-21]. As a rule, the phototransformation of terarylenes proceeds in two stages: 6π-electrocyclization of 1,3,5-hexatriene systems and subsequent aromatization of the central benzene ring.
Among the significant diversity of terarylenes, compounds containing a 5-hydroxy-2-methyl-4H-pyran-4-one (allomaltol) fragment are of particular interest. For such systems two types of photoreactions can occur under UV irradiation: the classical photocyclization of the 1,3,5-hexatriene system and excited state intramolecular proton transfer (ESIPT) – promoted photoprocesses typical for a 3-hydroxypyran-4-one core. For the ESIPT-induced reactions the general direction is the contraction of the 4-pyranone cycle 1 leading to the formation of unstable α-hydroxy-1,2-diketones. These intermediates can be captured intramolecularly using various functional groups present in the molecule or by employment of additional reagents. For example, a convenient method for trapping of unstable α-hydroxy-1,2-diketones 2 is the condensation with 1,2-phenylenediamine resulting in the formation of fused quinoxalines 3 (Scheme 1A) [22]. Another variant of similar catching reaction is reduction by NaBH3CN with the formation of diols 4 (Scheme 1A) [23]. Thus, the introduction of additional reagents allows the use of photogenerated α-hydroxy-1,2-diketones in the synthesis of various products. An alternative approach for trapping of such unstable intermediates is the preliminary introduction of functional groups into the 3-hydroxypyran-4-one structure. In this case, the intermediate α-hydroxy-1,2-diketone is captured by intramolecular reaction with an active substituent in the side chain (Scheme 1B) [24,25]. Thus, the combination of ESIPT-induced photoreactions of 3-hydroxypyran-4-one derivatives and various trapping methods opens up significant synthetic possibilities. At the same time, such processes can be complicated if additional photosensitive fragments are presented in the structure. For example, we previously studied the photochemical behavior of oxazolone terarylenes 9 containing the allomaltol fragment (Scheme 1C) [26]. It has been shown that UV irradiation of such systems leads to a complex mixture of products. At the same time the blocking of ESIPT-processes by the modification of the hydroxy group allows one to direct the photoreaction exclusively to the pathway of 6π-electrocyclization of the 1,3,5-hexatriene system. Thus, the study of the photochemical properties of similar bifunctional objects provides access to novel UV irradiation-based synthetic methods.
![[1860-5397-18-61-i1]](/bjoc/content/inline/1860-5397-18-61-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Photochemical transformations of 3-hydroxypyran-4-one derivatives.
Scheme 1: Photochemical transformations of 3-hydroxypyran-4-one derivatives.
Continuing our studies devoted to the photochemistry of terarylenes [27-32], in this communication we studied the UV-promoted reaction of pyrazole derivatives 12 containing a 3-hydroxypyran-4-one fragment (Scheme 1D). In contrast to the previously described phototransformation of analogously substituted oxazolones 9, in the considered case, the photoreaction proceeds exclusively as a contraction of the pyranone ring, while 6π-electrocyclization of the hexatriene system was not observed.
Results and Discussion
The starting pyrazoles 12 containing the 3-hydroxypyran-4-one fragment were obtained by a literature method [33]. The methoxy derivative 16 was synthesized by methylation of the corresponding pyrazole 12a according to the standard procedure [24,26] (Scheme 2). Initially, based on the previously obtained results for terarylenes with an oxazolone bridge fragment, we chose pyrazole 16 containing a methoxy group in the allomaltol core as a model object for studying photochemical properties. As it was shown previously for similar oxazolone derivatives [26] the presence of a methoxyl substituent in the compound 16 must exclude the occurrence of ESIPT-promoted photoprocesses. Therefore, it could be expected that UV irradiation of such a compound would lead only to 6π-electrocyclization of the 1,3,5-hexatriene system. Initially, the photochemical behavior of pyrazole 16 was studied using NMR spectroscopy in a solution of DMSO-d6. UV irradiation at a wavelength of 365 nm for 48 h in an NMR tube did not lead to any transformations of the starting pyrazole 16. Our attempts to carry out the photocyclization of pyrazole 16 in various solvents (DMF, acetonitrile, toluene, ethanol, methylene chloride, dioxane) were also unsuccessful. In all cases, pyrazole 16 was isolated unchanged. At the same time similar results were obtained employing UV irradiation at a wavelength of 312 nm. Thus, based on the data presented above, we can conclude that for terarylenes containing a pyrazole bridge fragment and a pyran-4-one substituent, 6π-electrocyclization of 1,3,5-hexatriene system does not occur (Scheme 2).
![[1860-5397-18-61-i2]](/bjoc/content/inline/1860-5397-18-61-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Synthesis and study of the photochemical behavior of compound 16.
Scheme 2: Synthesis and study of the photochemical behavior of compound 16.
As it was demonstrated earlier [26], UV irradiation of terarylenes containing oxazolone and 3-hydroxypyran-4-one fragments 9 can lead to two types of photoprocesses. Based on the aforementioned results we can assume that the key difference of the related pyrazole derivatives considered in this report is the absence of a pathway associated with 6π-electrocyclization. Therefore, we supposed that for the corresponding pyrazoles 12 containing a hydroxy group in the allomaltol fragment only ESIPT-induced contraction of the pyranone ring will proceed under UV irradiation. At first, pyrazole 12a was selected as a model object for the investigation of this hypothesis. It was shown that the photoreaction of pyrazole 12a regiospecifically led to the formation of α-hydroxy-1,2-diketone 14a (Scheme 3).
![[1860-5397-18-61-i3]](/bjoc/content/inline/1860-5397-18-61-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
The phototransformation of compound 12a was studied using NMR monitoring. UV irradiation was carried out at a wavelength of 365 nm in a solution of DMSO-d6 (Figure 1). It should be noted that after UV irradiation for 8 h of the studied sample the NMR spectrum contained signals of protons of the product 14a along with signals of the starting compound 12a (Figure 1B). Complete conversion to the target compound 14a was observed after irradiation for 24 h (Figure 1C), confirming the regiospecificity of the studied reaction. It could be concluded that only the ESIPT-promoted contraction of the pyranone ring proceeds under UV irradiation of compound 12a, while the 6π-electrocyclization products were not detected in the reaction mixture. However, it should be mentioned that the resulting α-hydroxy-1,2-diketone 14a has a relatively low stability and storing of the NMR sample of compound 14a in darkness at room temperature for 5 days resulted in complete decomposition.
![[1860-5397-18-61-1]](/bjoc/content/figures/1860-5397-18-61-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: 1H NMR monitoring of the photoreaction of compound 12a under UV irradiation (365 nm) in DMSO-d6 solution.
Figure 1: 1H NMR monitoring of the photoreaction of compound 12a under UV irradiation (365 nm) in DMSO-d6 sol...
Despite the labile nature of α-hydroxy-1,2-diketone 14a we attempted to isolate this product in pure form. It should be noted that in contrast to the methylated derivative 16, pyrazole 12a has a low solubility in most organic solvents excluding acetic acid, DMSO and DMF. Initially, UV irradiation of compound 12a was carried out at a wavelength of 365 nm in a solution of DMSO in common glassware for 24 h. However, subsequent water treatment did not allow the isolation of the target product 14a. Due to the fact that aqueous medium accelerates the decomposition of target α-hydroxy-1,2-diketone 14a. Subsequently, we used DMF as a solvent for carrying out the studied photoreaction. Unexpectedly, the starting pyrazole 12a was isolated unchanged after 24 hours of UV irradiation, indicating that DMF also is not suitable for the considered photoprocess. Probably, the DMF molecule forms an intermolecular hydrogen bond with the hydroxy group of the allomaltol fragment leading to complete suppression of the ESIPT process. Finally, AcOH was used as a solvent for the photochemical synthesis of α-hydroxy-1,2-diketone 14a. The starting pyrazole 12a was irradiated for 24 h, the acetic acid was evaporated, and the residue was triturated with Et2O affording the α-hydroxy-1,2-diketone 14a in pure form in 47% yield. It should be noted that the presented conditions are optimal. A further increase in the photoreaction time to 48 h leads to a diminished yield of 14a (34%). This can be attributed to the low stability of α-hydroxy-1,2-diketone 14a, which undergoes degradation upon keeping in solution. Also, it was shown that irradiation at a wavelength of 312 nm for 24 h leads to a slight decrease in the yield of compound 14a (42%). Thus, in this communication we have described for the first time the phototransformation of the 3-hydroxypyran-4-one fragment with the isolation of the resulting substituted α-hydroxy-1,2-diketone in pure form. The structure of the obtained compound 14a was confirmed using 1H, 13C NMR spectroscopy as well as 2D NMR experiments (HSQC, HMBC, COSY). Interestingly, in the 1H NMR spectrum of the product 14a the aromatic protons of the 4-methoxyphenyl substituent are represented as four singlets due to the presence of an asymmetric center and the hindered rotation of the aryl substituent relative to the pyrazole ring. The nonequivalence of corresponding carbon atoms of the aromatic ring is also observed in the 13C NMR spectrum.
The plausible route for the formation of α-hydroxy-1,2-diketone 14a is presented in Scheme 4. Based on literature data we suppose that the photoreaction proceeds through an excited state intramolecular proton transfer (ESIPT) [34-36]. At first, compound 12a under UV irradiation undergoes rapid proton transfer from the excited state A* resulting in the formation of photoisomer B* followed by the conversion to intermediate C. Further transformation of zwitter-ion C to bicyclic oxirane D and its subsequent opening leads to the final α-hydroxy-1,2-diketone 14a.
![[1860-5397-18-61-i4]](/bjoc/content/inline/1860-5397-18-61-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Proposed mechanism for the photoreaction of compound 11a.
Scheme 4: Proposed mechanism for the photoreaction of compound 11a.
Using the process conditions described above for the preparation of α-hydroxy-1,2-diketone 14a, we attempted to apply the elaborated method to the synthesis of a number of similar compounds with various substituents. Indeed, the proposed approach allowed the synthesis of a variety of substituted α-hydroxy-1,2-diketone 14, however, according to NMR spectroscopy, the purity of the synthesized compounds 14b–l did not exceed 80%. Attempts to carry out additional purification of these products using recrystallization or chromatographic methods were unsuccessful due to the low stability of the resulting α-hydroxy-1,2-diketone 14 and their partial decomposition during purification. Thus, the purity of the obtained compounds 14 does not allow a complete characterization by various analytical methods.
Based on this fact we made attempts to convert the labile α-hydroxy-1,2-diketone 14 into stable derivatives. A convenient method for the transformation of α-hydroxy-1,2-diketone into the corresponding substituted quinoxalines by reaction with 1,2-phenylenediamine was described in the literature [22]. The application of this protocol allowed the conversion of previously obtained unstable α-hydroxy-1,2-diketone 14a–l into the corresponding quinoxalines 15a–l (Scheme 5). As it was shown earlier, acetic acid is the optimal solvent for the studied compounds and this solvent is also widely used for the condensation of α-diketones with 1,2-phenylenediamine [37-40]. We suggested that it is possible to exclude isolation of unstable α-hydroxy-1,2-diketones 14 and thereby reduce losses at this step. Indeed, UV irradiation (365 nm) of the starting 12a–l pyrazoles and subsequent thermal condensation with 1,2-phenylenediamine (17) leads to the corresponding quinoxalines 15a–l in good yields (Scheme 5).
![[1860-5397-18-61-i5]](/bjoc/content/inline/1860-5397-18-61-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Synthesis of compounds 15a–l. Reaction conditions: 1) 12a–l (0.5 mmol), AcOH (25 mL), UV irradiation, 365 nm, 24 h. 2) 17 (0.6 mmol, 0.07 g), reflux, 2 h.
Scheme 5: Synthesis of compounds 15a–l. Reaction conditions: 1) 12a–l (0.5 mmol), AcOH (25 mL), UV irradiatio...
The structure of 15a was confirmed by single-crystal X-ray diffraction (Figure 2) as a representative example. It should be noted that X-ray analysis of the final quinoxaline unambiguously proves the conversion of the allomaltol fragment into the corresponding α-hydroxy-1,2-diketone formed in situ as a result of the photoreaction.
![[1860-5397-18-61-2]](/bjoc/content/figures/1860-5397-18-61-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: The X-ray crystal structure of compound 15a.
Figure 2: The X-ray crystal structure of compound 15a.
As shown above, only the phototransformation of the allomaltol fragment occurs under UV irradiation of pyrazoles 12a–l, while the 6π-electrocyclization of the hexatriene system is not observed. Based on this fact we assumed that the structure of the pyrazole fragment should not affect the photoprocess. Therefore, isomeric pyrazoles 12m–o also can be used as starting compounds. Indeed, UV irradiation (365 nm) of these objects in acetic acid and subsequent condensation with 1,2-phenylenediamine led to quinoxalines 15m–o in good yields (Scheme 6). The structure of 15m was confirmed by single-crystal X-ray diffraction (Figure 3).
![[1860-5397-18-61-i6]](/bjoc/content/inline/1860-5397-18-61-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Synthesis of compounds 15m–o. Reaction conditions: 1) 12m–o (0.5 mmol), AcOH (25 mL), UV irradiation, 365 nm, 24 h. 2) 17 (0.6 mmol, 0.07 g), reflux, 2 h.
Scheme 6: Synthesis of compounds 15m–o. Reaction conditions: 1) 12m–o (0.5 mmol), AcOH (25 mL), UV irradiatio...
![[1860-5397-18-61-3]](/bjoc/content/figures/1860-5397-18-61-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: The X-ray crystal structure of compound 15m.
Figure 3: The X-ray crystal structure of compound 15m.
It should be mentioned that other options for trapping of photogenerated unstable α-hydroxy-1,2-diketones are also possible. For example, reduction via NaBH3CN can be used as an alternative approach [23]. Initial UV irradiation of pyrazole 12a and subsequent interaction with NaBH3CN results in the formation of the product 18 in 67% yield (Scheme 7). It should be noted that the reaction proceeds regiospecifically with the participation of one carbonyl group. Also worth mentioning is the diastereospecificity of the studied process, which leads to the formation of a trans-diastereomer (mixture of S/S- and R/R-enantiomers). The structure of vicinal diol 18 was unambiguously established by single-crystal X-ray diffraction (Figure 4).
![[1860-5397-18-61-i7]](/bjoc/content/inline/1860-5397-18-61-i7.svg?scale=2.0&max-width=1024&background=FFFFFF)
![[1860-5397-18-61-4]](/bjoc/content/figures/1860-5397-18-61-4.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: The X-ray crystal structure of compound 18.
Figure 4: The X-ray crystal structure of compound 18.
Conclusion
In summary, we have studied the photochemical behavior of terarylenes containing pyrazole and allomaltol fragments. It was shown that 6π-electrocyclization of the 1,3,5-hexatriene system does not implement for these objects compounds. At the same time UV-irradiation of the studied terarylenes regiospecifically leads to contraction of the 4-pyranone ring. As a result, labile substituted α-hydroxy-1,2-diketones can be obtained. For the first time the possibility of isolation of the final α-hydroxy-1,2-diketone in pure form was demonstrated in the case of 14a. We proposed the approach for trapping of unstable α-hydroxy-1,2-diketones in situ employing the condensation with 1,2-phenylenediamine. Based on the aforementioned protocol a method for the synthesis of a wide range of condensed quinoxalines was suggested. It was found that the photogenerated contraction of the 4-pyranone fragment does not depend on the type of pyrazole bridge. The structures of three of the synthesized products were confirmed by X-ray diffraction.
Supporting Information
| Supporting Information File 1: Experimental procedures, characterization data of all products, copies of 1H, 13C, 2D NMR, HRMS spectra of all new compounds, and X-ray crystallographic data. | ||
| Format: PDF | Size: 8.0 MB | Download |
References
-
Hoffmann, N. Chem. Rev. 2008, 108, 1052–1103. doi:10.1021/cr0680336
Return to citation in text: [1] -
Zhang, Z.; Zhou, Y.-j.; Liang, X.-W. Org. Biomol. Chem. 2020, 18, 5558–5566. doi:10.1039/d0ob01204a
Return to citation in text: [1] -
Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760
Return to citation in text: [1] -
Bach, T.; Hehn, J. P. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. doi:10.1002/anie.201002845
Return to citation in text: [1] -
Sarkar, D.; Bera, N.; Ghosh, S. Eur. J. Org. Chem. 2020, 1310–1326. doi:10.1002/ejoc.201901143
Return to citation in text: [1] -
Kaur, N. Curr. Org. Synth. 2018, 15, 298–320. doi:10.2174/1570179414666171011160355
Return to citation in text: [1] -
Di Filippo, M.; Bracken, C.; Baumann, M. Molecules 2020, 25, 356. doi:10.3390/molecules25020356
Return to citation in text: [1] -
Albini, A.; Fagnoni, M. Green Chem. 2004, 6, 1–6. doi:10.1039/b309592d
Return to citation in text: [1] -
Protti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Green Chem. 2009, 11, 239–249. doi:10.1039/b810594d
Return to citation in text: [1] -
Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d
Return to citation in text: [1] -
Matsuda, K.; Irie, M. J. Photochem. Photobiol., C 2004, 5, 169–182. doi:10.1016/j.jphotochemrev.2004.07.003
Return to citation in text: [1] -
Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Chem. Rev. 2014, 114, 12174–12277. doi:10.1021/cr500249p
Return to citation in text: [1] -
Fukaminato, T.; Ishida, S.; Métivier, R. NPG Asia Mater. 2018, 10, 859–881. doi:10.1038/s41427-018-0075-9
Return to citation in text: [1] -
Xie, N.-H.; Chen, Y.; Ye, H.; Li, C.; Zhu, M.-Q. Front. Optoelectron. 2018, 11, 317–332. doi:10.1007/s12200-018-0839-4
Return to citation in text: [1] -
Ou, P.; Huang, X. Chin. J. Org. Chem. 2020, 40, 1074–1075. doi:10.6023/cjoc202000016
Return to citation in text: [1] -
Wu, Y.; Lin, Y.-W.; He, W.-M. Chin. Chem. Lett. 2020, 31, 2999–3000. doi:10.1016/j.cclet.2020.09.005
Return to citation in text: [1] -
Kutsunugi, Y.; Kawai, S.; Nakashima, T.; Kawai, T. New J. Chem. 2009, 33, 1368–1373. doi:10.1039/b823413b
Return to citation in text: [1] -
Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074
Return to citation in text: [1] -
Taguchi, M.; Nakagawa, T.; Nakashima, T.; Kawai, T. J. Mater. Chem. 2011, 21, 17425–17432. doi:10.1039/c1jm12993g
Return to citation in text: [1] -
Dela Cruz Calupitan, J. P.; Galangau, O.; Guillermet, O.; Coratger, R.; Nakashima, T.; Rapenne, G.; Kawai, T. Eur. J. Org. Chem. 2017, 2451–2461. doi:10.1002/ejoc.201601657
Return to citation in text: [1] -
Galangau, O.; Nakashima, T.; Maurel, F.; Kawai, T. Chem. – Eur. J. 2015, 21, 8471–8482. doi:10.1002/chem.201500647
Return to citation in text: [1] -
Shiozaki, M.; Hiraoka, T. Tetrahedron Lett. 1972, 13, 4655–4658. doi:10.1016/s0040-4039(01)94390-4
Return to citation in text: [1] [2] -
Barton, D. H. R.; Hulshof, L. A. J. Chem. Soc., Perkin Trans. 1 1977, 1103–1106. doi:10.1039/p19770001103
Return to citation in text: [1] [2] -
Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Minyaev, M. E. Org. Lett. 2021, 23, 5266–5270. doi:10.1021/acs.orglett.1c01814
Return to citation in text: [1] [2] -
Milyutin, C. V.; Galimova, R. D.; Komogortsev, A. N.; Lichitskii, B. V.; Melekhina, V. G.; Migulin, V. A.; Fakhrutdinov, A. N.; Minyaev, M. E. Org. Biomol. Chem. 2021, 19, 9975–9985. doi:10.1039/d1ob01871j
Return to citation in text: [1] -
Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902
Return to citation in text: [1] [2] [3] [4] -
Melekhina, V. G.; Komogortsev, A. N.; Lichitsky, B. V.; Mityanov, V. S.; Fakhrutdinov, A. N.; Dudinov, A. A.; Migulin, V. A.; Nelyubina, Y. V.; Melnikova, E. K.; Krayushkin, M. M. Tetrahedron Lett. 2019, 60, 151080. doi:10.1016/j.tetlet.2019.151080
Return to citation in text: [1] -
Melekhina, V. G.; Mityanov, V. S.; Fakhrutdinov, A. N.; Lyssenko, K. A.; Barachevsky, V.; Valova, T.; Martynov, I.; Ayt, A.; Krayushkin, M. M. J. Photochem. Photobiol., A 2019, 369, 34–43. doi:10.1016/j.jphotochem.2018.10.017
Return to citation in text: [1] -
Melekhina, V. G.; Mityanov, V. S.; Lichitsky, B. V.; Komogortsev, A. N.; Lyssenko, K. A.; Krayushkin, M. M. Eur. J. Org. Chem. 2019, 1335–1340. doi:10.1002/ejoc.201801664
Return to citation in text: [1] -
Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G.; Komogortsev, A. N.; Fakhrutdinov, A. N.; Minyaev, M. E.; Krayushkin, M. M. Tetrahedron Lett. 2020, 61, 152469. doi:10.1016/j.tetlet.2020.152469
Return to citation in text: [1] -
Lichitskii, B. V.; Melekhina, V. G.; Komogortsev, A. N.; Milyutin, C. V.; Fakhrutdinov, A. N.; Gorbunov, Y. O.; Krayushkin, M. M. Org. Biomol. Chem. 2020, 18, 2501–2509. doi:10.1039/d0ob00149j
Return to citation in text: [1] -
Lichitsky, B. V.; Milyutin, C. V.; Melekhina, V. G.; Fakhrutdinov, A. N.; Komogortsev, A. N.; Krayushkin, M. M. Chem. Heterocycl. Compd. 2021, 57, 13–19. doi:10.1007/s10593-021-02861-2
Return to citation in text: [1] -
Komogortsev, A. N.; Melekhina, V. G.; Lichitsky, B. V.; Dudinov, A. A.; Fakhrutdinov, A. N.; Krayushkin, M. M. Russ. Chem. Bull. 2020, 69, 758–762. doi:10.1007/s11172-020-2829-0
Return to citation in text: [1] -
Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. doi:10.1039/c2cp23144a
Return to citation in text: [1] -
Joshi, H. C.; Antonov, L. Molecules 2021, 26, 1475. doi:10.3390/molecules26051475
Return to citation in text: [1] -
Wang, J.; Liu, Q.; Yang, D. Sci. Rep. 2020, 10, 5119. doi:10.1038/s41598-020-61804-7
Return to citation in text: [1] -
More, N. Y.; Jeganmohan, M. Chem. Commun. 2017, 53, 9616–9619. doi:10.1039/c7cc04829g
Return to citation in text: [1] -
Zhang, H.; Fan, J.; Wang, J.; Zhang, S.; Dou, B.; Peng, X. J. Am. Chem. Soc. 2013, 135, 11663–11669. doi:10.1021/ja4056905
Return to citation in text: [1] -
Lu, G.-p.; Cai, C. J. Heterocycl. Chem. 2014, 51, 1595–1602. doi:10.1002/jhet.1704
Return to citation in text: [1] -
Mahadik, S. S.; Garud, D. R.; Pinjari, R. V.; Kamble, R. M. J. Mol. Struct. 2022, 1248, 131541. doi:10.1016/j.molstruc.2021.131541
Return to citation in text: [1]
| 23. | Barton, D. H. R.; Hulshof, L. A. J. Chem. Soc., Perkin Trans. 1 1977, 1103–1106. doi:10.1039/p19770001103 |
| 1. | Hoffmann, N. Chem. Rev. 2008, 108, 1052–1103. doi:10.1021/cr0680336 |
| 2. | Zhang, Z.; Zhou, Y.-j.; Liang, X.-W. Org. Biomol. Chem. 2020, 18, 5558–5566. doi:10.1039/d0ob01204a |
| 3. | Kärkäs, M. D.; Porco, J. A., Jr.; Stephenson, C. R. J. Chem. Rev. 2016, 116, 9683–9747. doi:10.1021/acs.chemrev.5b00760 |
| 4. | Bach, T.; Hehn, J. P. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. doi:10.1002/anie.201002845 |
| 5. | Sarkar, D.; Bera, N.; Ghosh, S. Eur. J. Org. Chem. 2020, 1310–1326. doi:10.1002/ejoc.201901143 |
| 6. | Kaur, N. Curr. Org. Synth. 2018, 15, 298–320. doi:10.2174/1570179414666171011160355 |
| 7. | Di Filippo, M.; Bracken, C.; Baumann, M. Molecules 2020, 25, 356. doi:10.3390/molecules25020356 |
| 22. | Shiozaki, M.; Hiraoka, T. Tetrahedron Lett. 1972, 13, 4655–4658. doi:10.1016/s0040-4039(01)94390-4 |
| 22. | Shiozaki, M.; Hiraoka, T. Tetrahedron Lett. 1972, 13, 4655–4658. doi:10.1016/s0040-4039(01)94390-4 |
| 18. | Nakashima, T.; Atsumi, K.; Kawai, S.; Nakagawa, T.; Hasegawa, Y.; Kawai, T. Eur. J. Org. Chem. 2007, 3212–3218. doi:10.1002/ejoc.200700074 |
| 19. | Taguchi, M.; Nakagawa, T.; Nakashima, T.; Kawai, T. J. Mater. Chem. 2011, 21, 17425–17432. doi:10.1039/c1jm12993g |
| 20. | Dela Cruz Calupitan, J. P.; Galangau, O.; Guillermet, O.; Coratger, R.; Nakashima, T.; Rapenne, G.; Kawai, T. Eur. J. Org. Chem. 2017, 2451–2461. doi:10.1002/ejoc.201601657 |
| 21. | Galangau, O.; Nakashima, T.; Maurel, F.; Kawai, T. Chem. – Eur. J. 2015, 21, 8471–8482. doi:10.1002/chem.201500647 |
| 37. | More, N. Y.; Jeganmohan, M. Chem. Commun. 2017, 53, 9616–9619. doi:10.1039/c7cc04829g |
| 38. | Zhang, H.; Fan, J.; Wang, J.; Zhang, S.; Dou, B.; Peng, X. J. Am. Chem. Soc. 2013, 135, 11663–11669. doi:10.1021/ja4056905 |
| 39. | Lu, G.-p.; Cai, C. J. Heterocycl. Chem. 2014, 51, 1595–1602. doi:10.1002/jhet.1704 |
| 40. | Mahadik, S. S.; Garud, D. R.; Pinjari, R. V.; Kamble, R. M. J. Mol. Struct. 2022, 1248, 131541. doi:10.1016/j.molstruc.2021.131541 |
| 10. | Irie, M. Chem. Rev. 2000, 100, 1685–1716. doi:10.1021/cr980069d |
| 11. | Matsuda, K.; Irie, M. J. Photochem. Photobiol., C 2004, 5, 169–182. doi:10.1016/j.jphotochemrev.2004.07.003 |
| 12. | Irie, M.; Fukaminato, T.; Matsuda, K.; Kobatake, S. Chem. Rev. 2014, 114, 12174–12277. doi:10.1021/cr500249p |
| 13. | Fukaminato, T.; Ishida, S.; Métivier, R. NPG Asia Mater. 2018, 10, 859–881. doi:10.1038/s41427-018-0075-9 |
| 14. | Xie, N.-H.; Chen, Y.; Ye, H.; Li, C.; Zhu, M.-Q. Front. Optoelectron. 2018, 11, 317–332. doi:10.1007/s12200-018-0839-4 |
| 15. | Ou, P.; Huang, X. Chin. J. Org. Chem. 2020, 40, 1074–1075. doi:10.6023/cjoc202000016 |
| 16. | Wu, Y.; Lin, Y.-W.; He, W.-M. Chin. Chem. Lett. 2020, 31, 2999–3000. doi:10.1016/j.cclet.2020.09.005 |
| 17. | Kutsunugi, Y.; Kawai, S.; Nakashima, T.; Kawai, T. New J. Chem. 2009, 33, 1368–1373. doi:10.1039/b823413b |
| 26. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
| 8. | Albini, A.; Fagnoni, M. Green Chem. 2004, 6, 1–6. doi:10.1039/b309592d |
| 9. | Protti, S.; Dondi, D.; Fagnoni, M.; Albini, A. Green Chem. 2009, 11, 239–249. doi:10.1039/b810594d |
| 34. | Zhao, J.; Ji, S.; Chen, Y.; Guo, H.; Yang, P. Phys. Chem. Chem. Phys. 2012, 14, 8803–8817. doi:10.1039/c2cp23144a |
| 35. | Joshi, H. C.; Antonov, L. Molecules 2021, 26, 1475. doi:10.3390/molecules26051475 |
| 36. | Wang, J.; Liu, Q.; Yang, D. Sci. Rep. 2020, 10, 5119. doi:10.1038/s41598-020-61804-7 |
| 27. | Melekhina, V. G.; Komogortsev, A. N.; Lichitsky, B. V.; Mityanov, V. S.; Fakhrutdinov, A. N.; Dudinov, A. A.; Migulin, V. A.; Nelyubina, Y. V.; Melnikova, E. K.; Krayushkin, M. M. Tetrahedron Lett. 2019, 60, 151080. doi:10.1016/j.tetlet.2019.151080 |
| 28. | Melekhina, V. G.; Mityanov, V. S.; Fakhrutdinov, A. N.; Lyssenko, K. A.; Barachevsky, V.; Valova, T.; Martynov, I.; Ayt, A.; Krayushkin, M. M. J. Photochem. Photobiol., A 2019, 369, 34–43. doi:10.1016/j.jphotochem.2018.10.017 |
| 29. | Melekhina, V. G.; Mityanov, V. S.; Lichitsky, B. V.; Komogortsev, A. N.; Lyssenko, K. A.; Krayushkin, M. M. Eur. J. Org. Chem. 2019, 1335–1340. doi:10.1002/ejoc.201801664 |
| 30. | Milyutin, C. V.; Lichitsky, B. V.; Melekhina, V. G.; Komogortsev, A. N.; Fakhrutdinov, A. N.; Minyaev, M. E.; Krayushkin, M. M. Tetrahedron Lett. 2020, 61, 152469. doi:10.1016/j.tetlet.2020.152469 |
| 31. | Lichitskii, B. V.; Melekhina, V. G.; Komogortsev, A. N.; Milyutin, C. V.; Fakhrutdinov, A. N.; Gorbunov, Y. O.; Krayushkin, M. M. Org. Biomol. Chem. 2020, 18, 2501–2509. doi:10.1039/d0ob00149j |
| 32. | Lichitsky, B. V.; Milyutin, C. V.; Melekhina, V. G.; Fakhrutdinov, A. N.; Komogortsev, A. N.; Krayushkin, M. M. Chem. Heterocycl. Compd. 2021, 57, 13–19. doi:10.1007/s10593-021-02861-2 |
| 24. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Minyaev, M. E. Org. Lett. 2021, 23, 5266–5270. doi:10.1021/acs.orglett.1c01814 |
| 26. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
| 26. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
| 26. | Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Nasyrova, D. I.; Milyutin, C. V. J. Org. Chem. 2021, 86, 15345–15356. doi:10.1021/acs.joc.1c01902 |
| 24. | Milyutin, C. V.; Komogortsev, A. N.; Lichitsky, B. V.; Melekhina, V. G.; Minyaev, M. E. Org. Lett. 2021, 23, 5266–5270. doi:10.1021/acs.orglett.1c01814 |
| 25. | Milyutin, C. V.; Galimova, R. D.; Komogortsev, A. N.; Lichitskii, B. V.; Melekhina, V. G.; Migulin, V. A.; Fakhrutdinov, A. N.; Minyaev, M. E. Org. Biomol. Chem. 2021, 19, 9975–9985. doi:10.1039/d1ob01871j |
| 23. | Barton, D. H. R.; Hulshof, L. A. J. Chem. Soc., Perkin Trans. 1 1977, 1103–1106. doi:10.1039/p19770001103 |
| 33. | Komogortsev, A. N.; Melekhina, V. G.; Lichitsky, B. V.; Dudinov, A. A.; Fakhrutdinov, A. N.; Krayushkin, M. M. Russ. Chem. Bull. 2020, 69, 758–762. doi:10.1007/s11172-020-2829-0 |
© 2022 Milyutin et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.