Abstract
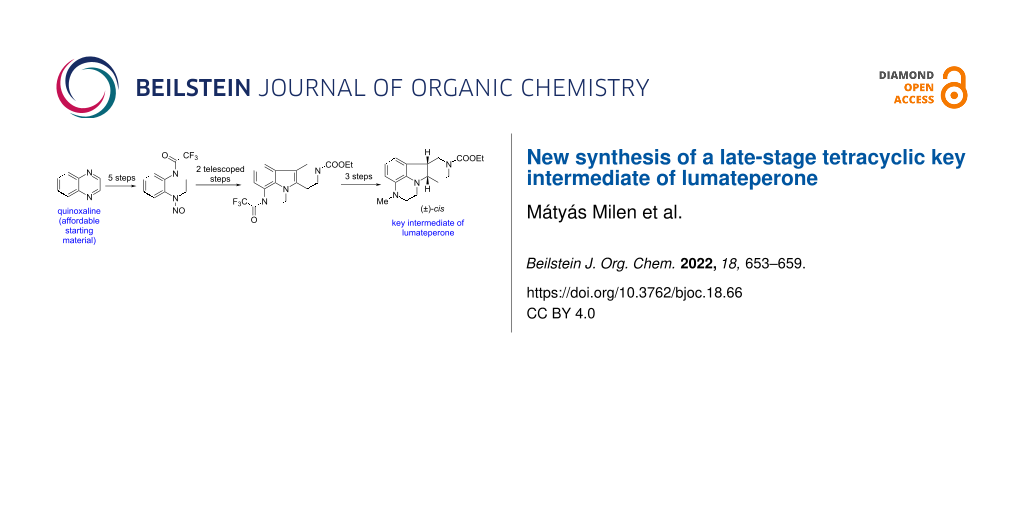
New approaches have been tested for the synthesis of lumateperone intermediates. As a result of these efforts, a novel synthesis of the late-stage tetracyclic key intermediate of lumateperone starting from the commercially available quinoxaline is described. The tetracyclic skeleton was constructed by the reaction of 1-trifluoroacetyl-4-aminoquinoxaline with ethyl 4-oxopiperidine-1-carboxylate in a Fischer indole synthesis. The inexpensive starting material, the efficient synthetic steps, and the avoidance of the borane-based reduction step provide a reasonable potential for scalability.

Graphical Abstract
Introduction
Lumateperone (1) is a recently launched drug marketed for the treatment of schizophrenia and developed for further neuropsychiatric and neurological disorders [1-6] (Figure 1).
![[1860-5397-18-66-1]](/bjoc/content/figures/1860-5397-18-66-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
The first synthesis of lumateperone (1, Caplyta®), developed by Intra-Cellular Therapies, was disclosed in the basic patent and also discussed in a later scientific publication [7,8] (Scheme 1). According to this procedure, treatment of the commercially available, but quite expensive 3,4-dihydroquinoxalin-2(1H)-one (2) with sodium nitrite gave the N-nitroso derivative 3. Reduction of the latter with zinc to the hydrazine derivative 4, followed by a Fischer indole synthesis with ethyl 4-oxopiperidine-1-carboxylate (5) provided tetracyclic compound 6. Its reduction with sodium cyanoborohydride in trifluoroacetic acid (TFA) to cis-indoline derivative (±)-7, followed by N-methylation [(±)-8] and reduction of the oxo group with borane·THF complex in THF led to compound (±)-9a. Subsequent desethoxycarbonylation afforded compound (±)-10, which was N-alkylated with 4-chloro-1-(4-fluorophenyl)butan-1-one (11) to give the racemic form of lumateperone [(±)-1]. Lumateperone (1) was finally obtained by chiral chromatography.
![[1860-5397-18-66-i1]](/bjoc/content/inline/1860-5397-18-66-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: First synthetic route leading to lumateperone (1).
Scheme 1: First synthetic route leading to lumateperone (1).
Later, alternate syntheses of lumateperone were described, also by the researchers of the originator company [8-10] (Scheme 2). The reaction of (2-bromophenyl)hydrazine (12) with 4-piperidone monohydrate hydrochloride (13) gave pyrido[4,3-b]indole congener 14, which was reduced with triethylsilane in TFA to cis-racemate 15. The key step of the syntheses is the resolution of this intermediate with (R)-mandelic acid. The enantiomerically pure (R)-mandelate salt 16 thus obtained was reacted with ethyl chloroformate to give 17. It was then transformed to tetracyclic derivative 8 in various ways (we only show here the path that seems the most advantageous). According to this, compound 17 was N-alkylated at the indoline nitrogen atom with N-methylchloroacetamide to give 18, then cyclized in one pot to derivative 8. Alternate methods for the 17→8 transformation require more reaction steps [8-10], e.g., because of the use of chloroacetamide instead of N-methylchloroacetamide, necessitating an additional N-methylation. The tetracycle 8 was finally subjected to the same reaction sequence as the corresponding racemate (see also Scheme 1), lumateperone (1) was thus prepared via intermediates 9a and 10.
![[1860-5397-18-66-i2]](/bjoc/content/inline/1860-5397-18-66-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Alternate synthesis of lumateperone.
Scheme 2: Alternate synthesis of lumateperone.
Further synthetic variants resulting in racemic precursors (±)-9a,b, which were transformed to racemic lumateperone [(±)-1] via (±)-10, were also described [11,12] (Scheme 3). The main features of these syntheses are as follows: (i) in addition to the ethoxycarbonyl group (19a), a benzyloxycarbonyl group (19b) was also used for N-protection; (ii) compounds 19a,b were N-alkylated at the indole nitrogen atom with N-methylchloroacetamide to derivatives 20a,b; (iii) the 20→21 cyclization was realized by a Xantphos/Pd-catalyzed C–N bond-forming reaction; (iv) synthetic steps of the 21a,b→(±)-9a,b transformation were swapped when starting from 21a compared to the reaction starting from 21b. In the former case, the reduction of the double bond resulting in (±)-8a was followed by the reduction of the lactam moiety, while in the case of 21b the reduction of the lactam carbonyl (21b→22b) preceded the reduction of the C–C double bond of 22b. Both the hydrolytic desethoxycarbonylation of (±)-9a as well as the removal of the benzyloxycarbonyl group of (±)-9b by catalytic hydrogenation afforded (±)-10 which was N-alkylated with 4-chloro-1-(4-fluorophenyl)butan-1-one (11) to give the racemic form of lumateperone [(±)-1]. It has to be mentioned that the resolution of none of the racemic intermediates of this synthetic route was disclosed [11,12].
![[1860-5397-18-66-i3]](/bjoc/content/inline/1860-5397-18-66-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Alternate synthetic approaches leading to racemic lumateperone ((±)-1)).
Scheme 3: Alternate synthetic approaches leading to racemic lumateperone ((±)-1)).
Results and Discussion
From the technological point of view, the obvious common disadvantage of the methods described above for the synthesis of lumateperone is the use of borane·THF complex for the reduction of the lactam carbonyl group, which may cause difficulties in industrial scale production (Schemes 1–3, step 8→9a and Scheme 3, step 21b→22b). Considering that we have developed a method and filed a patent application [13] for the resolution of compound (±)-10, a direct intermediate of lumateperone, easily available from (±)-9a, we aimed to elaborate a new, practical synthesis of the latter.
First, we envisaged a new synthetic route to the racemic key intermediate (±)-9a, significantly shorter than those described, which would have been based on the 1-methyl-4-amino-1,2,3,4-tetrahydroquinoxaline (23) intermediate (Scheme 4). We planned to convert the latter into (±)-9a via compound 22a by known methods.
![[1860-5397-18-66-i4]](/bjoc/content/inline/1860-5397-18-66-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Planned new synthesis of key intermediate (±)-9a.
Scheme 4: Planned new synthesis of key intermediate (±)-9a.
The synthesis of compound 23 was attempted as follows. N-Methylation of quinoxaline (24) with methyl p-toluenesulfonate followed by reduction of the N-methylated quaternary ammonium salt 25 with sodium borohydride gave tetrahydroquinoxaline 26. Since N-amination of the latter with hydroxylamine-O-sulfonic acid [14] had been unsuccessful, we tried to achieve our target via the N-nitroso derivative 27, which was obtained from compound 26 by a conventional procedure [7,8]. Several methods have then been tried to convert 27 to the N-amino derivative 23 (Zn dust, AcOH [8]; Zn dust, NH4Cl [15]; H2, Pd/C [16]; Na2S2O4 [17,18]; Mg, TiCl4 [19]), but to our regret, in all cases the N-nitroso group was removed and compound 26 was recovered instead of the expected product 23.
So we were forced to go on a longer new way to key intermediate (±)-9a. Quinoxaline (24), the price of which is ca. 3% of that of 3,4-dihydroquinoxalin-2(1H)-one (2), remained our starting compound [20-23] (Scheme 5). N-Benzylation of 24 was accomplished in neat conditions within 3 days of reaction time using a literature procedure [5]. Attempts in acetonitrile as solvent were unsuccessful: after 1 day at room temperature, product 28 was isolated in only 20% yield, while at reflux temperature, a decomposition has been observed. Reduction of the quaternary ammonium salt 28 with sodium borohydride gave tetrahydroquinoxaline 29. Its reaction with trifluoroacetic anhydride (TFAA) to give 30 and removal of the benzyl group by catalytic hydrogenation afforded N-trifluoroacetyl-1,2,3,4-tetrahydroquinoxaline (31). Compound 31 was then transformed into N-nitroso derivative 32 by treatment with sodium nitrite in aqueous acetic acid. The next two steps, reduction of the nitroso group to hydrazine derivative 33 and its coupling with piperidinone 5 in a Fischer indole synthesis, were telescoped to form tetracyclic product 34. The latter was reduced with sodium cyanoborohydride in acetic acid to cis-indoline derivative (±)-35. Removal of the trifluoroacetyl group to (±)-36 followed by N-methylation with formaldehyde and sodium cyanoborohydride gave target compound (±)-9a.
![[1860-5397-18-66-i5]](/bjoc/content/inline/1860-5397-18-66-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: New synthesis of key intermediate (±)-9a.
Scheme 5: New synthesis of key intermediate (±)-9a.
It has to be mentioned that the preparation of compound 31 has been recently described in a patent application [24] by a direct trifluoroacetylation of tetrahydroquinoxaline (37) with 0.92 equiv of TFAA, followed by chromatographic product separation. We carried out the same reaction (Scheme 6) and in addition to the expected compound 31 (39%), a significant amount of bis(trifluoroacetyl) derivative 38 (25%) and starting material 37 (25%) were isolated as well (see Supporting Information File 1 for details). It has become clear that the formation of substantial amounts of the diacylated product 38 could not be avoided, thus turning the simple scaled up production of compound 31 unfeasible.
![[1860-5397-18-66-i6]](/bjoc/content/inline/1860-5397-18-66-i6.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 6: Trifluoroacetylation of tetrahydroquinoxaline (37).
Scheme 6: Trifluoroacetylation of tetrahydroquinoxaline (37).
The main indicators of the synthetic approaches known from the literature and those of the new synthesis disclosed here are summarized in Table 1.
Table 1: Summary of known synthetic methods leading to tetracyclic compound 9a.
| No. of scheme | Starting material |
Target
product |
Overall yield
(%) |
No. of isolated intermediates | Advantages | Disadvantages |
| Scheme 1 |
3,4-dihydroquinoxalin-
2(1H)-one (2) |
(±)-9a | 20 | 5 | short synthesis |
expensive starting material,
BH3·THF reagent is sensitive to air and flammable, reduction in highly corrosive TFA solvent, low overall yield |
| Scheme 2 |
(2-bromophenyl)-
hydrazine (12) |
9a | 24 | 6 | synthesis of enantiopure product |
expensive starting material,
BH3·THF is sensitive to air and flammable, reduction in highly corrosive TFA solvent, low overall yield |
| Scheme 3a |
(2-bromophenyl)-
hydrazine (12) |
(±)-9a | 65/60 | 5/6 |
short synthesis,
high overall yield |
expensive starting material,
BH3·THF is sensitive to air and flammable, reduction in highly corrosive TFA solvent |
| Scheme 3b |
(2-bromophenyl)-
hydrazine (12) |
(±)-9a | 46 | 6 | ||
| Scheme 5 | quinoxaline (24) | (±)-9a | 14 | 9 |
inexpensive starting material,
avoidance of BH3·THF reagent and TFA solvent |
use of protecting groups,
higher number of steps, lower overall yield |
Conclusion
In summary, after the experimental study of various approaches, a new and efficient synthesis has been developed at laboratory scale for the synthesis of racemic key intermediate (±)-9a of the new drug lumateperone. The novel approach is based on the cheaply available starting material quinoxaline and leads to the tetracyclic compound via chemical steps characterized with good to excellent yields. The method described here does not require a reduction step carried out with borane·THF complex that is used in the syntheses described in the literature but would make it difficult to scale up the process. The significance of compound (±)-9a lies in the fact that it can be converted in a known one-step reaction to compound (±)-10. The resolution of the latter and the conversion of the suitable enantiomer to the drug lumateperone is disclosed in our patent application.
Supporting Information
| Supporting Information File 1: General information, synthetic procedures, and spectral data. | ||
| Format: PDF | Size: 3.0 MB | Download |
References
-
Syed, A. B.; Brašić, J. R. Ther. Adv. Psychopharmacol. 2021, 11, 1–14. doi:10.1177/20451253211034019
Return to citation in text: [1] -
Mei, H.; Remete, A. M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2020, 31, 2401–2413. doi:10.1016/j.cclet.2020.03.050
Return to citation in text: [1] -
Blair, H. A. Drugs 2020, 80, 417–423. doi:10.1007/s40265-020-01271-6
Return to citation in text: [1] -
Vyas, P.; Hwang, B. J.; Brašić, J. R. Expert Opin. Pharmacother. 2020, 21, 139–145. doi:10.1080/14656566.2019.1695778
Return to citation in text: [1] -
Greenwood, J.; Acharya, R. B.; Marcellus, V.; Rey, J. A. Ann. Pharmacother. 2021, 55, 98–104. doi:10.1177/1060028020936597
Return to citation in text: [1] [2] -
Snyder, G. L.; Vanover, K. E.; Davis, R. E.; Li, P.; Fienberg, A.; Mates, S. A review of the pharmacology and clinical profile of lumateperone for the treatment of schizophrenia. In A Tribute to Paul Greengard (1925–2019); Zorn, S. H., Ed.; Advances in Pharmacology, Vol. 90; Academic Press: Cambridge, MA, USA, 2021; pp 253–276. doi:10.1016/bs.apha.2020.09.001
Return to citation in text: [1] -
Robichaud, A. J.; Lee, T.; Deng, W.; Mitchell, I. S.; Yang, M. G.; Chen, W.; Chung, C. D.; Calvello, E. J. B.; Zawrotny, D. M. Preparation of substituted heterocycle fused gamma-carbolines. WO Patent WO2000077002, Dec 21, 2000.
Return to citation in text: [1] [2] -
Li, P.; Zhang, Q.; Robichaud, A. J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J. D.; Snyder, G. L.; Zhu, H.; Peng, Y.; Hendrick, J. P.; Vanover, K. E.; Davis, R. E.; Mates, S.; Wennogle, L. P. J. Med. Chem. 2014, 57, 2670–2682. doi:10.1021/jm401958n
Return to citation in text: [1] [2] [3] [4] [5] -
Tomesch, J. C.; Li, P.; Yao, W.; Zhang, Q.; Beard, J. D.; Thompson, A. S.; Cheng, H. Process for preparation of heterocycle fused gamma-carbolines. WO Patent 2008112280, Sept 18, 2008.
Return to citation in text: [1] [2] -
Li, P.; Zhang, Q. Improved methods for preparing substituted heterocycle fused gamma-carbolines. WO Patent WO2019241278, Dec 19, 2019.
Return to citation in text: [1] [2] -
Process for preparation of 1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)butan-1-one. IPCOM000257454D; Chem. Abstr. 2019, 170, 547735.
Return to citation in text: [1] [2] -
Process for preparation of 1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)butan-1-one. IPCOM000258304D; Chem. Abstr. 2019, 171, 163843.
Return to citation in text: [1] [2] -
Berecz, G.; Nyulasi, B.; Milen, M.; Simig, G.; Mravik, A.; Németh, G.; Keszthelyi, A.; Bali, B.; Volk, B.; Szlávik, L.; Varga, Z.; Ulej, D. Method for the manufacture of lumateperone and its salts. WO Patent WO2019102240, May 31, 2019.
Return to citation in text: [1] -
Wallace, R. G. Aldrichimica Acta 1980, 13, 3–11.
Return to citation in text: [1] -
Song, C.; Yang, C.; Zhang, F.; Wang, J.; Zhu, J. Org. Lett. 2016, 18, 4510–4513. doi:10.1021/acs.orglett.6b02103
Return to citation in text: [1] -
Thompson, R. D.; Beauglehole, A. R.; Wang, G. Xanthine-substituted alkynyl carbamates/reverse carbamates as A2b antagonists. U.S. Patent US20160297819, Oct 13, 2016.
Return to citation in text: [1] -
Nishida, A.; Tsuji, R.; Nakagawa, M. Heterocycles 2002, 58, 587–593. doi:10.3987/com-02-s(m)56
Return to citation in text: [1] -
Dhainant, A.; Régnier, G.; Atassi, G.; Pierré, A.; Léonce, S.; Kraus-Berthier, L.; Prost, J.-F. J. Med. Chem. 1992, 35, 2481–2496. doi:10.1021/jm00091a017
Return to citation in text: [1] -
Landriani, L.; Barlocco, D.; Pinna, G. A.; Demontis, M. P.; Miele, M.; Enrico, P.; Anania, V. Farmaco 1989, 44, 1059–1068.
Return to citation in text: [1] -
https://www.chemicalbook.com/ChemicalProductProperty_EN_CB5193734.htm. (accessed April 5, 2022).
See for 3,4-dihydro-1H-quinoxalin-2-one.
Return to citation in text: [1] -
https://www.scbt.com/p/3-4-dihydro-1h-quinoxalin-2-one-59564-59-9. (accessed April 5, 2022).
See for 3,4-dihydro-1H-quinoxalin-2-one.
Return to citation in text: [1] -
https://www.chemicalbook.com/Price/Quinoxaline.htm. (accessed April 5, 2022).
See for quinoxaline.
Return to citation in text: [1] -
https://www.sigmaaldrich.com/HU/en/product/aldrich/q1603. (accessed April 5, 2022).
See for quinoxaline.
Return to citation in text: [1] -
Murphy, B. P.; Zhang, G.; Zhao, J. Stable violet-blue to blue imidazolium compounds. WO Patent WO2018052725, March 22, 2018.
Return to citation in text: [1]
| 24. | Murphy, B. P.; Zhang, G.; Zhao, J. Stable violet-blue to blue imidazolium compounds. WO Patent WO2018052725, March 22, 2018. |
| 1. | Syed, A. B.; Brašić, J. R. Ther. Adv. Psychopharmacol. 2021, 11, 1–14. doi:10.1177/20451253211034019 |
| 2. | Mei, H.; Remete, A. M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V. A.; Han, J. Chin. Chem. Lett. 2020, 31, 2401–2413. doi:10.1016/j.cclet.2020.03.050 |
| 3. | Blair, H. A. Drugs 2020, 80, 417–423. doi:10.1007/s40265-020-01271-6 |
| 4. | Vyas, P.; Hwang, B. J.; Brašić, J. R. Expert Opin. Pharmacother. 2020, 21, 139–145. doi:10.1080/14656566.2019.1695778 |
| 5. | Greenwood, J.; Acharya, R. B.; Marcellus, V.; Rey, J. A. Ann. Pharmacother. 2021, 55, 98–104. doi:10.1177/1060028020936597 |
| 6. | Snyder, G. L.; Vanover, K. E.; Davis, R. E.; Li, P.; Fienberg, A.; Mates, S. A review of the pharmacology and clinical profile of lumateperone for the treatment of schizophrenia. In A Tribute to Paul Greengard (1925–2019); Zorn, S. H., Ed.; Advances in Pharmacology, Vol. 90; Academic Press: Cambridge, MA, USA, 2021; pp 253–276. doi:10.1016/bs.apha.2020.09.001 |
| 11. | Process for preparation of 1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)butan-1-one. IPCOM000257454D; Chem. Abstr. 2019, 170, 547735. |
| 12. | Process for preparation of 1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)butan-1-one. IPCOM000258304D; Chem. Abstr. 2019, 171, 163843. |
| 20. |
https://www.chemicalbook.com/ChemicalProductProperty_EN_CB5193734.htm. (accessed April 5, 2022).
See for 3,4-dihydro-1H-quinoxalin-2-one. |
| 21. |
https://www.scbt.com/p/3-4-dihydro-1h-quinoxalin-2-one-59564-59-9. (accessed April 5, 2022).
See for 3,4-dihydro-1H-quinoxalin-2-one. |
| 22. |
https://www.chemicalbook.com/Price/Quinoxaline.htm. (accessed April 5, 2022).
See for quinoxaline. |
| 23. |
https://www.sigmaaldrich.com/HU/en/product/aldrich/q1603. (accessed April 5, 2022).
See for quinoxaline. |
| 8. | Li, P.; Zhang, Q.; Robichaud, A. J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J. D.; Snyder, G. L.; Zhu, H.; Peng, Y.; Hendrick, J. P.; Vanover, K. E.; Davis, R. E.; Mates, S.; Wennogle, L. P. J. Med. Chem. 2014, 57, 2670–2682. doi:10.1021/jm401958n |
| 9. | Tomesch, J. C.; Li, P.; Yao, W.; Zhang, Q.; Beard, J. D.; Thompson, A. S.; Cheng, H. Process for preparation of heterocycle fused gamma-carbolines. WO Patent 2008112280, Sept 18, 2008. |
| 10. | Li, P.; Zhang, Q. Improved methods for preparing substituted heterocycle fused gamma-carbolines. WO Patent WO2019241278, Dec 19, 2019. |
| 5. | Greenwood, J.; Acharya, R. B.; Marcellus, V.; Rey, J. A. Ann. Pharmacother. 2021, 55, 98–104. doi:10.1177/1060028020936597 |
| 8. | Li, P.; Zhang, Q.; Robichaud, A. J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J. D.; Snyder, G. L.; Zhu, H.; Peng, Y.; Hendrick, J. P.; Vanover, K. E.; Davis, R. E.; Mates, S.; Wennogle, L. P. J. Med. Chem. 2014, 57, 2670–2682. doi:10.1021/jm401958n |
| 9. | Tomesch, J. C.; Li, P.; Yao, W.; Zhang, Q.; Beard, J. D.; Thompson, A. S.; Cheng, H. Process for preparation of heterocycle fused gamma-carbolines. WO Patent 2008112280, Sept 18, 2008. |
| 10. | Li, P.; Zhang, Q. Improved methods for preparing substituted heterocycle fused gamma-carbolines. WO Patent WO2019241278, Dec 19, 2019. |
| 17. | Nishida, A.; Tsuji, R.; Nakagawa, M. Heterocycles 2002, 58, 587–593. doi:10.3987/com-02-s(m)56 |
| 18. | Dhainant, A.; Régnier, G.; Atassi, G.; Pierré, A.; Léonce, S.; Kraus-Berthier, L.; Prost, J.-F. J. Med. Chem. 1992, 35, 2481–2496. doi:10.1021/jm00091a017 |
| 7. | Robichaud, A. J.; Lee, T.; Deng, W.; Mitchell, I. S.; Yang, M. G.; Chen, W.; Chung, C. D.; Calvello, E. J. B.; Zawrotny, D. M. Preparation of substituted heterocycle fused gamma-carbolines. WO Patent WO2000077002, Dec 21, 2000. |
| 8. | Li, P.; Zhang, Q.; Robichaud, A. J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J. D.; Snyder, G. L.; Zhu, H.; Peng, Y.; Hendrick, J. P.; Vanover, K. E.; Davis, R. E.; Mates, S.; Wennogle, L. P. J. Med. Chem. 2014, 57, 2670–2682. doi:10.1021/jm401958n |
| 19. | Landriani, L.; Barlocco, D.; Pinna, G. A.; Demontis, M. P.; Miele, M.; Enrico, P.; Anania, V. Farmaco 1989, 44, 1059–1068. |
| 7. | Robichaud, A. J.; Lee, T.; Deng, W.; Mitchell, I. S.; Yang, M. G.; Chen, W.; Chung, C. D.; Calvello, E. J. B.; Zawrotny, D. M. Preparation of substituted heterocycle fused gamma-carbolines. WO Patent WO2000077002, Dec 21, 2000. |
| 8. | Li, P.; Zhang, Q.; Robichaud, A. J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J. D.; Snyder, G. L.; Zhu, H.; Peng, Y.; Hendrick, J. P.; Vanover, K. E.; Davis, R. E.; Mates, S.; Wennogle, L. P. J. Med. Chem. 2014, 57, 2670–2682. doi:10.1021/jm401958n |
| 15. | Song, C.; Yang, C.; Zhang, F.; Wang, J.; Zhu, J. Org. Lett. 2016, 18, 4510–4513. doi:10.1021/acs.orglett.6b02103 |
| 16. | Thompson, R. D.; Beauglehole, A. R.; Wang, G. Xanthine-substituted alkynyl carbamates/reverse carbamates as A2b antagonists. U.S. Patent US20160297819, Oct 13, 2016. |
| 13. | Berecz, G.; Nyulasi, B.; Milen, M.; Simig, G.; Mravik, A.; Németh, G.; Keszthelyi, A.; Bali, B.; Volk, B.; Szlávik, L.; Varga, Z.; Ulej, D. Method for the manufacture of lumateperone and its salts. WO Patent WO2019102240, May 31, 2019. |
| 11. | Process for preparation of 1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)butan-1-one. IPCOM000257454D; Chem. Abstr. 2019, 170, 547735. |
| 12. | Process for preparation of 1-(4-fluorophenyl)-4-((6bR,10aS)-3-methyl-2,3,6b,9,10,10a-hexahydro-1H-pyrido[3',4':4,5]pyrrolo[1,2,3-de]quinoxalin-8(7H)-yl)butan-1-one. IPCOM000258304D; Chem. Abstr. 2019, 171, 163843. |
| 8. | Li, P.; Zhang, Q.; Robichaud, A. J.; Lee, T.; Tomesch, J.; Yao, W.; Beard, J. D.; Snyder, G. L.; Zhu, H.; Peng, Y.; Hendrick, J. P.; Vanover, K. E.; Davis, R. E.; Mates, S.; Wennogle, L. P. J. Med. Chem. 2014, 57, 2670–2682. doi:10.1021/jm401958n |
© 2022 Milen et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.