Abstract
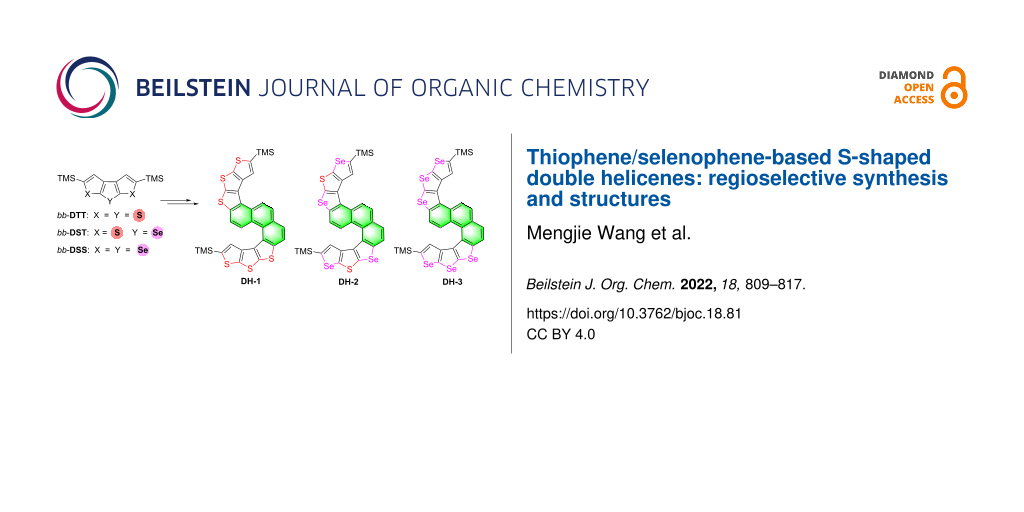
2,5-Di(trimethylsilanyl)dithieno[2,3-b:3′,2′-d]thiophene ((TMS)2-bb-DTT), 2,5-di(trimethylsilanyl)diseleno[2,3-b:3′,2′-d]thiophene ((TMS)2-bb-DST), and 2,5-di(trimethylsilanyl)diseleno[2,3-b:3′,2′-d] selenophene ((TMS)2-bb-DSS) were used as starting materials to synthesize three S-shaped double helicenes (i.e., DH-1, DH-2, and DH-3) through monobromination, formylation, the Wittig reaction, and double oxidative photocyclization. The photocyclization was a highly regioselective process. The molecular structures of DH-1 and DH-2 were confirmed by X-ray single-crystal analysis. Multiple intermolecular interactions, such as C–S, C–Se, S–S, S–Se, and Se–Se, were observed in the crystal packing structures of these compounds. Spectroscopic results and our previous work showed that the combination of molecular structure change and heteroatom replacement from S to Se could precisely modulate molecular energy levels.

Graphical Abstract
Introduction
Given their esthetically pleasing helical structures, inherent helical chirality, and extended π-conjugation, helicenes have attracted extensive research attention. Helicenes are generally divided into carbohelicenes and heterohelicenes. The rapid development of carbohelicenes has led to the synthesis of double, triple, quadruple, quintuple, and sextuple molecules, the chiro-optical properties of these molecules, such as their circular dichroism (CD) and circularly polarized luminescence (CPL), have also been widely studied [1-8]. The development of thiahelicene, a class of typical heterohelicenes, has led to the preparation of symmetric thiophene-based [5]-, [7]-, [9]-, and [11]helicenes [9-16], unsymmetric thiophene-based [7]helicenes [17], and thiophene-based double helicenes with spiro-silicon atoms [18], “saddle” formed 8π annulene [12], and twisted naphthalene as central spacers [19].
As its close analogue, selenophene has properties very similar to those of thiophene. Fused aromatic compounds containing selenophene units show favorable optical and electrochemical properties and improved charge transport characteristics in the solid state mainly because such fused aromatic compounds often undergo increased Se–Se interactions, which confer ordering at the molecular scale and, thus, lead to well-aligned solid-state packing and excellent charge-transport properties [20,21].
However, as an important type of heteroacenes, fused selenophenes have rarely been reported in the literature because their synthesis is extremely challenging. The first seven-ring-fused heteroacene containing selenophene was synthesized through the intramolecular triple cyclization of bis(o-haloaryl)diacetylene by Yamaguchi in 2005 [22]. Using a similar method, Takimiya reported the synthesis of six-ring-fused and four-ring-fused heteroacenes containing selenopheno[3,2-b]selenophene in 2007 and 2009, respectively [23,24]. Five years later, Cheng reported the synthesis of two types of five-ring-fused isomers of diselenopheno[2,3-b:7,6-b′]fluorene and diselenopheno[3,2-b:6,7-b′]fluorene through the cyclization of terminal acetylene as well as six types of biselenophene-based fused tricyclic derivatives [25,26]. In 2017, we reported the first member of diselenoselenophenes (DSSs), 2,5-di(trimethylsilanyl)diseleno[2,3-b:3′,2′-d]selenophene ((TMS)2-bb-DSS), from which the TMS group could easily be removed by trifluoroacetic acid and replaced by bromine [27]. Another isomer of DSS, diseleno[3,2-b:2′,3′-d]selenophene (tt-DSS) has been successively synthesized from selenophene [28,29].
Among the limited fused aromatic compounds containing selenophene currently available, helicenes have received relatively little attention [27]. In our previous work, bull horn-shaped selenophene-based heteroacenes (1, Figure 1) and selenophene-based [7]helicene (2, Figure 1) were synthesized from the (TMS)2-substituted selenophene triacenes, 2,5-di(trimethylsilanyl)diseleno[3,2-b:2′,3′-d]selenophene ((TMS)2-tt-DSS) and (TMS)2-bb-DSS, respectively. In this work, the novel S-shaped double helicene DH-3 (Figure 1), which is based on selenophene units, was constructed as a new member of the selenohelicene family (Figure 1).
![[1860-5397-18-81-1]](/bjoc/content/figures/1860-5397-18-81-1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Molecular structures of bull horn-shaped heteroacene 1, selenophene-based [7]helicene 2 and novel compound DH-3.
Figure 1: Molecular structures of bull horn-shaped heteroacene 1, selenophene-based [7]helicene 2 and novel c...
In this synthetic study, regioselective double oxidative photocyclization was observed during the construction of three S-shaped double helicenes DH-1–3 based on thiophene/selenophene. From DH-1 to DH-3, sulfur atoms in the molecular framework were gradually replaced by selenium atoms. The crystal structures of DH-1 and DH-2 and spectroscopic features of DH-1–3 were then studied. Finally, the reaction sites of oxidative photocyclization, energy levels, and the electron cloud distribution of the highest occupied molecular orbitals (HOMOs) and the lowest unoccupied molecular orbitals (LUMOs) are predicted.
Results and Discussion
Synthesis of 5a–c and DH-1–3
The synthetic route of 1,3-bis(2-(5-(trimethylsilyl)dithieno-[2,3-b:3',2'-d]thiophen-2-yl)vinyl)benzene (5a), 1,3-bis(2-(5-(trimethylsilyl)diselenopheno[2,3-b:3',2'-d]thiophen-2-yl)vinyl)benzene (5b), 1,3-bis(2-(5-(trimethylsilyl)diselenopheno[2,3-b:3',2'-d]selenophen-2-yl)vinyl)benzene (5c), and S-shaped double helicenes DH-1–3 is shown in Scheme 1. The double oxidative photocyclization of 5a–c is the key step in the synthesis of DH-1–3 because oxidative photocyclization induces double radicals on a double bond, which led to the C=C bond rotation along the resulting single C(radical)–C(radical) bond and randomly directed annelated products [30]. Moreover, compounds 5a–c bear two C=C bonds, which may lead to more complex photocyclization products.
![[1860-5397-18-81-i1]](/bjoc/content/inline/1860-5397-18-81-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Synthetic route to S-shaped double helicenes DH-1–3.
Scheme 1: Synthetic route to S-shaped double helicenes DH-1–3.
5-(Trimethylsilyl)dithieno[2,3-b:3′,2′-d]thiophene-2-carbaldehyde (4a) [19], 5-(trimethylsilyl)diseleno[2,3-b:3′,2′-d]thiophene-2-carbaldehyde (4b) [27], and 5-(trimethylsilyl)diseleno[2,3-b:3′,2′-d]selenophene-2-carbaldehyde (4c) [27] were prepared via monobromination and formylation reactions with (TMS)2-bb-DTT, (TMS)2-bb-DST, and (TMS)2-bb-DSS as starting materials according to the literature. In (TMS)2-bb-DTT, (TMS)2-bb-DST, and (TMS)2-bb-DSS, the notation bb means that the positions of the heteroatoms of the outer two heterocycles are on the same side as those of the middle heterocycle. Taking dithienothiophene as example, according to the positions of sulfur atoms, there are six isomers of dithienothiophene, in which the sulfur atoms on the same side is defined as bb by us. The double Wittig reaction of 4a and 4b with 1,3-phenyldimethyltriphenylphosphonium bromide afforded compounds 5a and 5b with yields of 46% and 64%, respectively. After the double Wittig reaction of 4c we obtained a mixture of cis and trans isomers of 5c with the total yield of 50%. According to the results of 1H NMR the ratio of cis and trans isomers was approximately 1:0.25 (Scheme 1).
Compounds 5 had five isomers and three reaction sites (2, 4, and 6-positions in the benzene moiety) during oxidative photocyclization. Irradiation of 5a–c resulted in oxidative photocyclization products with two types of configurations wherein two benzene rings were closed in the same and opposite direction, such as DH-1–3 and 6 (Figure 2). However, after the double oxidative photocyclizations of 5a–c in the presence of iodine and propylene oxide in dry toluene through irradiation by a 450 W Hg medium-pressure lamp for 1.5 h, only one type of ring-closing product with two benzene rings formed in the same direction, i.e., S-shaped double helicenes DH-1–3 were obtained in yields of 62%, 30%, and 53%, respectively (Figure 2).
![[1860-5397-18-81-2]](/bjoc/content/figures/1860-5397-18-81-2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: Five kinds of isomer structures of 5 and two kinds of possible oxidative photocyclization product structures of 6 and DH-1–3.
Figure 2: Five kinds of isomer structures of 5 and two kinds of possible oxidative photocyclization product s...
The double oxidative photocyclization reaction sites of 5 were predicted by the orbital-weighted Fukui function in Multiwfn using Gaussian 09 [31,32] at the B3LYP/6-31G** [33] level of theory to verify the reaction-site selectivity of oxidative photocyclization further. Results showed that the conformations of 5a are varied, but the orbital-weighted Fukui function is not connected to conformation. In the orbital-weighted Fukui function, the larger the isosurface distribution, the higher the activity of reactions. Thus the 4 and 6-positions of benzene are the most likely sites for reaction (see Supporting Information File 1, Figure S21). However, after the formation of the naphthalene ring, the α-position is the most likely site for reaction (Supporting Information File 1, Figure S22). Therefore, after the double oxidative photocyclization of compound 5a, product DH-1 is mainly obtained. The predicted result of the double oxidative photocyclizations of 5a is consistent with the experimental result, that is, an S-shaped double helicene can be selectively obtained through the double oxidative photocyclization of compound 5a.
Crystallographic analyses of DH-1 and DH-2
The molecular structures of DH-1 and DH-2 were confirmed by single-crystal analysis (Figure 3). Both DH-1 and DH-2 belong to the triclinic space group P-1. After double oxidative photocyclizations of 5a and 5b, DH-1 and DH-2 are compressed into S-shaped double helical structures (Figure 3A and B), which consist of one [5]helicene and one [6]helicene. The two helicenes have the same configuration and bend toward the same direction on the same side of the shared naphthalene ring (Figure 3C and D). Both products DH-1 or DH-2 feature a pair of enantiomers MM and PP in their unit cell (see Supporting Information File 2, Figures S2 and S6). The crystal parameters of DH-1 and DH-2 are shown in Table 1. The replacement of sulfur with selenium in DH-1 and DH-2 leads to turn angles in-plane and helix climbs of [5]helicene and [6]helicene of DH-1 and DH-2 significantly change (Table 1, Figure 3).
![[1860-5397-18-81-3]](/bjoc/content/figures/1860-5397-18-81-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Molecular structures and side view for DH-1 and DH-2. A and B are molecular structures for DH-1 and DH-2. C and D are side views for DH-1 and DH-2. Carbon, sulfur, selenium, and silicon atoms are depicted with thermal ellipsoids set at the 30% probability level and all hydrogen atoms are omitted for clarity.
Figure 3: Molecular structures and side view for DH-1 and DH-2. A and B are molecular structures for DH-1 and ...
Table 1: Crystal parameters of helicenes DH-1 and DH-2.
| Compound |
Dihedral angle
(o)a |
Torsion angle
(o)b |
Turn angle in-plane
(o)c |
Helix climbs
(Å)d |
|
| DH-1 | [5]helicene | 25.00 | 38.64 | 231.1 | 0.932 |
| [6]helicene | 50.61 | 64.38 | 273.6 | 2.424 | |
| DH-2 | [5]helicene | 25.01 | 37.27 | 230.9 | 0.879 |
| [6]helicene | 48.16 | 64.12 | 279.3 | 2.462 | |
aDihedral angle between two terminal rings of helicene. bThe sum of the three ([5]helicene) or four ([6]helicene) distortion angles. cThe angle of turns in-plane. dInternal helix climb height.
Each of the two DH-1 molecules (blue and red molecules in Figure S3 of Supporting Information File 2) in the unit cell interacts with six adjacent molecules. For example, in Figure S3 (Supporting Information File 2), the blue molecule interacts with six adjacent molecules through multiple interactions, such as, S···S (3.44 Å), S···H (2.94 Å, 2.91 Å, 2.97 Å), H···H (2.31 Å), C···C (3.31 Å), S···C (3.45 Å), and C···H (2.90 Å). Each of the two DH-2 molecules (blue and red molecules, Supporting Information File 2, Figure S7) in the unit cell interact with eight adjacent molecules through multiple interactions, such as, C···C (3.40 Å), Se···H (3.09 Å), H···H (2.26 Å), C···H (2.90 Å), Se···Se (3.62 Å), and Se···S (3.58 Å). However, in contrast to DH-1, DH-2 exhibits multiple S···H (2.88 Å), S···C (3.45 Å), and Se···H (2.94 Å) interactions between two molecules in the unit cell. These multiple interactions confer DH-1 and DH-2 with a regular arrangement featuring multi channels (Figures S4 and S8 in Supporting Information File 2), which is a suitable characteristic for helicene compounds used as supramolecular self-assembly units [34-41].
Spectroscopic features of DH-1–3
The UV–vis absorption spectra of DH-1–3 in dichloromethane are shown in Figure 4. In general, the UV–vis absorption spectra of DH-1–3 are generally similar in shape and exhibit three major absorption bands within 230–280 nm (band-I), 280–330 nm (band-II), and 304–414 nm (band-III) (Figure 4). Progressive red-shifts in the absorption spectra of band-I, band-II, and band-III occur with increasing number of selenium atoms. In band-I, compounds DH-1–3 show a maximum absorption peak at 232, 240, and 242 nm, respectively. In band-II and band-III, helical distortion and possible conjugation through heteroatoms (e.g., sulfur and selenium atoms) in DH-1–3 may increase π-electron delocalization, leading to a red-shifted broad absorption. The maximum absorption peaks of DH-1–3 appear at 268, 275, and 279 nm, respectively, in band-II and at 323, 331, and 336 nm, respectively, in band-III. Thus, the optical band gaps estimated from the absorption edges gradually decrease from DH-1 to DH-2 to DH-3, and are equal to 3.08, 3.01, and 2.98 eV, respectively. This change trend is consistent with the calculated results, which are 3.97, 3.83, and 3.81 eV for DH-1, DH-2, and DH-3, respectively (see Table S2 in Supporting Information File 1). However, the optical band gaps of 1, 2, and DH-3 obviously differ because of changes in their molecular configuration and equal to 2.86, 3.15, and 3.81 eV, respectively [27,28]. As the number of selenium atoms increases from DH-1 to DH-3, the fluorescence intensity (Figure S19 in Supporting Information File 1) and the fluorescence quantum yield (ΦF, Figure S20, Table S1 in Supporting Information File 1) of the molecules also decrease (Figure 4).
![[1860-5397-18-81-4]](/bjoc/content/figures/1860-5397-18-81-4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Figure 4: UV–vis absorption spectra of DH-1–3 in CH2Cl2 ([c] = 1 × 10−5 M).
Figure 4: UV–vis absorption spectra of DH-1–3 in CH2Cl2 ([c] = 1 × 10−5 M).
Conclusion
In summary, the key step of regioselective double oxidative photocyclization was successfully employed in the preparation of three S-shaped double helicenes, namely, DH-1, DH-2 and DH-3 with (TMS)2-bb-DTT, (TMS)2-bb-DST, and (TMS)2-bb-DSS as starting materials. The synthetic method described in this research not only provides a method for the synthesis of S- shaped double helicenes but also enriches the family of selenophene helicenes. Multiple intermolecular interactions and regular arrangement in the crystal packing structures of DH-1 and DH-2 indicate that these compounds may be used as supramolecular self-assembly units. Changes in molecular structure may substantially modulate the optical band gap of heteroacenes, and the replacement of heteroatoms from S to Se could fine-tune their optical band gap [18,27,28]. Thus, the combination of molecular structure modification and atom replacement could be a viable strategy, for the precise modulation molecular energy levels and yield molecules with strong application potential in organic functional materials, such as OFETs, and CPLs, among others.
Experimental
General procedures and materials
Tetrahydrofuran (THF) for use on vacuum line was freshly distilled from sodium/benzophenone prior to use. n-BuLi (hexane) were obtained from Energy Chemical; prior to use, its concentration was determined by titration with N-pivaloyl-o-toluidine [42]. Column chromatography was carried out on silica gel (300–400 mesh). Analytical thin-layer chromatography was performed on glass plates of silica gel GF-254 with detection by UV. Standard techniques for synthesis under inert atmosphere and Schlenk glassware equipped with an 8 mm PTFE vacuum stopcock, were employed. All starting materials and reagents were commercially available.
1H NMR and 13C{1H} NMR spectra were recorded on 300 or 400 MHz NMR instruments using CDCl3 as solvent. The chemical shift references were as follows: (1H) CDCl3, 7.26 ppm (CHCl3); (13C{1H}) CDCl3, 77.00 ppm (CDCl3). IR spectra were obtained using an FTIR instrument. MS analysis was carried out on mass spectrometers equipped with EI (70 eV). HRMS analysis was carried out on a mass spectrometer equipped with DART-FT-ICR and MALDI-TOF-CHCA. Melting-point determination was taken on a Melt-Temp apparatus and mp are uncorrected. The X-ray crystallographic analyses were performed using crystals of compounds DH-1 and DH-2 with sizes of 0.14 × 0.12 × 0.08, 0.21 × 0.17 × 0.12 mm3, respectively. The intensity data were collected with the ω scan mode (296 K) on a diffractometer with a CCD detector using Cu Kα radiation (λ = 1.54184 Å). The data were corrected for Lorentz and polarization effects, and absorption corrections were performed using the SADABS program [43]. The crystal structures were solved using the SHELXTL program and refined using full-matrix least-squares [44]. Further details are in the deposited CIF files. Slow evaporation of solutions of DH-1 and DH-2 in CHCl3/CH3OH 5:1 (v/v) were employed for growing single crystals. The fluorescence quantum yields (ΦF) of DH-1–3 are characterized in dichloromethane with quinine sulfate in 0.1 N H2SO4 as the control.
Synthesis of 5a–c and DH-1–3
Synthesis of 1,3-bis(2-(5-(trimethylsilyl)dithieno[2,3-b:3',2'-d]thiophen-2-yl)vinyl)benzene (5a)
n-BuLi (0.06 mL, 2.50 M in hexane, 0.168 mmol, 2.5 equiv) was added dropwise to 1,3-bis[methyl(bromotriphenylphosphonium)] (53.3 mg, 0.067 mmol) in THF (40 mL) at −78 °C. After 2 h at −78 °C, a solution of 4a (41.0 mg, 0.138 mmol, 2.05 equiv) in THF (5 mL) was added at −78 °C, the mixture kept for 1 h, and then the reaction mixture was warmed up slowly to ambient temperature overnight. The reaction mixture was quenched with CH3OH and extracted with CH2Cl2 (3 × 10 mL). The organic layer was washed with saturated NaCl (20 mL) and water (2 × 20 mL), and then dried over MgSO4. The residue was purified by column chromatography (eluent: hexane/CH2Cl2 3:1 (v/v) and recrystallized from CHCl3/CH3OH to yield 5a (20.9 mg, 46%) as a yellow solid; mp > 300 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 7.58 (s, 1H), 7.45 (s, 2H), 7.41–7.36 (m, 3H), 7.34 (s, 2H), 7.31 (d, J = 16.0 Hz, 2H), 6.93 (d, J = 16.0 Hz, 2H), 0.39 (s, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ (ppm) 145.3, 144.7, 143.7, 140.7, 138.4, 137.8, 137.2, 129.1, 127.8, 125.5, 124.9, 124.4, 122.8, 118.0, −0.1; IR (KBr): 3018, 2958, 2849, 1631, 1408, 1360, 945, 837 cm−1; EIMS (70 eV) m/z: [M]+ 662.18 (40); HRMS-MALDI (m/z): [M]+ calcd for C32H30S6Si2, 662.0216; found, 662.0205.
1,3-Bis(2-(5-(trimethylsilyl)diselenopheno[2,3-b:3',2'-d]thiophen-2-yl)vinyl)benzene (5b) was synthesized according to the procedure described for compound 5a.
5b: yellow solid in yield of 64% (46.2 mg); mp > 300 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 7.73 (s, 2H), 7.55 (d, J = 12.9 Hz, 3H), 7.38–7.32 (m, 5 H), 6.79 (d, J = 15.6 Hz, 2H), 0.38 (s, 18H); 13C{1H} NMR (100 MHz, CDCl3) δ (ppm) 151.6, 149.7, 144.5, 142.6, 141.1, 137.01, 136.98, 129.2, 128.6, 127.5, 125.6, 124.9, 124.4, 121.0, 0.2; IR (KBr): 3011, 2949, 2891, 1627, 1427, 1367, 920, 831 cm−1; DARTMS (m/z): [M + H]+ 854.8 (75); HRMS-DART (m/z): [M + H]+ calcd for C32H31S2Se4Si2, 854.8071; found, 854.8067.
1,3-Bis(2-(5-(trimethylsilyl)diselenopheno[2,3-b:3',2'-d]selenophen-2-yl)vinyl)benzene (5c) was synthesized according to the procedure described for compound 5a.
Mixture of cis and trans isomers 5c: yellow solid in yield of 50% (24.4 mg); mp > 300 °C; 1H NMR (400 MHz, CDCl3) δ (ppm): 7.76 (s, trans-), 7.73 (s, cis- and trans-), 7.72 (s, cis- and trans-), 7.57 (s, cis- and trans-), 7.56 (s, trans-), 7.49–7.33 (m, trans-, cis- and trans-), 7.30 (d, J = 16.0 Hz, cis- and trans-), 6.90 (d, J = 12.0 Hz, cis- and trans-), 6.79 (d, J = 16.0 Hz, trans-), 6.78 (d, J = 16.0 Hz, trans- and trans-), 6.65 (d, J = 12.0 Hz, cis- and trans-), 0.38 (s, trans-), 0.36 (s, cis- and trans-), 0.35 (s, cis- and trans-), the ratio of integral areas of the peaks at 7.76, 7.73, and 7.72 ppm is 1.0:0.25; 13C{1H} NMR (125 MHz, CDCl3) δ (ppm): 152.78, 152.70, 152.62, 151.28, 151.23, 147.31, 146.49, 146.34, 143.49, 143.47, 141.84, 140.87, 140.81, 140.72, 139.02, 137.66, 137.50, 137.43, 135.39, 135.36, 129.74, 129.30, 129.28, 129.23, 129.05, 128.83, 128.42, 128.35, 127.50, 126.97, 126.28, 125.68, 125.20, 125.10, 125.04, 124.85, 124.63, 122.61, 122.51, 0.43, 0.41; IR (KBr): 3010, 2951, 2889, 1624, 1413, 1369, 908, 833 cm−1; MALDIMS (m/z): [M]+ 949.8; HRMS-DART-FT (m/z): [M]+ calcd for C32H30Se6Si2, 949.6894; found, 949.6887.
Synthesis of DH-1
To a solution of 5a (9.6 mg, 0.014 mmol) in dry toluene (6 mL), iodine (7.3 mg, 0.028 mmol, 2.0 equiv) and excess propylene oxide were added. The reaction solution was irradiated with a 450 W unfiltered Hg medium-pressure lamp for 1.5 h. The reaction was quenched with saturated Na2SO3 solution (5 mL). The reaction mixture was extracted with CH2Cl2 (3 × 5 mL), washed with H2O (3 × 10 mL), and then dried over MgSO4. After removing the solvent in vacuum, the crude product was purified by PTLC with petrol ether (60–90 °C) and hexane/CH2Cl2 5:1 (v/v) as developer to yield DH-1 (5.9 mg, 62%) as a light-yellow solid; mp > 300 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 9.09 (d, J = 8.8 Hz, 1H), 8.51 (d, J = 8.8 Hz, 1H), 8.22 (s, 1H), 8.11 (d, J = 9.2 Hz, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 8.8 Hz, 1H), 6.62 (s, 1H), 0.48 (s, 9H), 0.14 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ (ppm) 144.6, 144.1, 143.19, 143.18, 142.1, 141.7, 141.6, 141.4, 141.3, 141.1, 135.0, 133.4, 130.6, 129.8, 129.0, 128.5, 127.53, 127.46, 127.3, 127.0, 126.7, 126.4, 124.3, 123.7, 121.5, 119. 7, 0.0, −0.4; IR (KBr): 3045, 2952, 2860, 1645, 1487, 1410, 972, 837 cm−1; EIMS (70 eV) m/z: [M]+ 657.87 (30); HRMS-MALDI (m/z): [M]+ calcd for C32H26S6Si2 657.9897; found, 657.9892.
DH-2 was synthesized according to the procedure described for compound DH-1.
DH-2: light-yellow solid in yield of 30% (15.8 mg); mp > 300 °C; 1H NMR (400 MHz, CDCl3) δ (ppm) 8.92 (d, J = 8.8 Hz, 1H), 8.54 (s, 1H), 8.32 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H), 6.66 (s, 1H), 0.47 (s, 9H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ (ppm) 149.9, 146.9, 145.7, 145.1, 144.8, 144.3, 142.7, 142.6, 139.8, 139.4, 139.2, 137.5, 132.8, 130.8, 130.6, 130.2, 129.59, 129.58, 128.4, 128.1, 126.7, 125.6, 124.5, 124.3, 124.2, 122.2, 0.3, −0.2; IR (KBr): 3060, 2949, 2896, 1632, 1489, 1402, 933, 829 cm−1; DARTMS (m/z): [M + H]+ 850.8 (45); HRMS-DART (m/z): [M + H]+ calcd for C32H27S2Si2Se4, 850.7753; found, 850.7743.
DH-3 was synthesized according to the procedure described for DH-1.
DH-3: light yellow solid in yield of 53% (19.5 mg); mp 265.1–266.9 °C; 1H NMR (300 MHz, CDCl3) δ (ppm) 8.85 (d, J = 8.7 Hz, 1H), 8.56 (s, 1H), 8.22 (d, J = 8.7 Hz, 1H), 8.06 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 9.0 Hz, 1H), 7.81 (d, J = 8.7 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H), 6.68 (s, 1H), 0.45 (s, 9H), −0.11 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ (ppm) 151.0, 148.0, 147.4, 147.0, 145.9, 143.9, 142.6, 141.1, 140.9, 139.34, 139.26, 138.9, 134.1, 132.3, 131.7, 130.7, 130.2, 129.3, 128.8, 128.0, 126.7, 125.0, 124.9, 124.1, 123.9, 121.9, 0.4, −0.3; IR (KBr): 3047, 2947, 2895, 1633, 1504, 1409, 925, 831 cm−1; DARTMS (m/z): [M]+ 945.8; HRMS-DART-FT (m/z): [M]+ calcd for C32H26Se6Si2, 945.6564; found, 945.6564.
Supporting Information
| Supporting Information File 1: Spectral and computational data. | ||
| Format: PDF | Size: 2.0 MB | Download |
| Supporting Information File 2: Supporting crystallographic information for compounds DH-1 and DH-2. | ||
| Format: PDF | Size: 885.1 KB | Download |
| Supporting Information File 3: Title CIF file for compound DH-1. | ||
| Format: CIF | Size: 738.3 KB | Download |
| Supporting Information File 4: Title CIF file for compound DH-2. | ||
| Format: CIF | Size: 738.6 KB | Download |
References
-
Mori, T. Chem. Rev. 2021, 121, 2373–2412. doi:10.1021/acs.chemrev.0c01017
Return to citation in text: [1] -
Dhbaibi, K.; Favereau, L.; Crassous, J. Chem. Rev. 2019, 119, 8846–8953. doi:10.1021/acs.chemrev.9b00033
Return to citation in text: [1] -
Rickhaus, M.; Mayor, M.; Juríček, M. Chem. Soc. Rev. 2016, 45, 1542–1556. doi:10.1039/c5cs00620a
Return to citation in text: [1] -
Hoffmann, N. J. Photochem. Photobiol., C 2014, 19, 1–19. doi:10.1016/j.jphotochemrev.2013.11.001
Return to citation in text: [1] -
Gingras, M. Chem. Soc. Rev. 2013, 42, 968–1006. doi:10.1039/c2cs35154d
Return to citation in text: [1] -
Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r
Return to citation in text: [1] -
Urbano, A. Angew. Chem., Int. Ed. 2003, 42, 3986–3989. doi:10.1002/anie.200301667
Return to citation in text: [1] -
Katz, T. J. Angew. Chem., Int. Ed. 2000, 39, 1921–1923. doi:10.1002/1521-3773(20000602)39:11<1921::aid-anie1921>3.0.co;2-f
Return to citation in text: [1] -
Miyasaka, M.; Rajca, A. Synlett 2004, 177–181. doi:10.1055/s-2003-43335
Return to citation in text: [1] -
Zhang, C.; Ma, Z.; Wang, G.; Li, C.; Wang, H. Org. Chem. Front. 2020, 7, 3926–3934. doi:10.1039/d0qo00957a
Return to citation in text: [1] -
Rajca, A.; Wang, H.; Pink, M.; Rajca, S. Angew. Chem., Int. Ed. 2000, 39, 4481–4483. doi:10.1002/1521-3773(20001215)39:24<4481::aid-anie4481>3.0.co;2-g
Return to citation in text: [1] -
Li, C.; Shi, J.; Xu, L.; Wang, Y.; Cheng, Y.; Wang, H. J. Org. Chem. 2009, 74, 408–411. doi:10.1021/jo802080g
Return to citation in text: [1] [2] -
Liu, X.; Sun, H.; Xu, W.; Wan, S.; Shi, J.; Li, C.; Wang, H. Org. Chem. Front. 2018, 5, 1257–1261. doi:10.1039/c7qo01049d
Return to citation in text: [1] -
Rajca, A.; Miyasaka, M.; Pink, M.; Wang, H.; Rajca, S. J. Am. Chem. Soc. 2004, 126, 15211–15222. doi:10.1021/ja0462530
Return to citation in text: [1] -
Miyasaka, M.; Pink, M.; Olankitwanit, A.; Rajca, S.; Rajca, A. Org. Lett. 2012, 14, 3076–3079. doi:10.1021/ol3011437
Return to citation in text: [1] -
Miyasaka, M.; Rajca, A.; Pink, M.; Rajca, S. J. Am. Chem. Soc. 2005, 127, 13806–13807. doi:10.1021/ja055414c
Return to citation in text: [1] -
Wang, J.; Wang, G.; Li, C.; Dong, Y.; Ma, Z.; Wang, H. J. Org. Chem. 2021, 86, 4413–4422. doi:10.1021/acs.joc.0c02629
Return to citation in text: [1] -
Li, C.; Wu, L.; Xu, W.; Song, J.; Shi, J.; Yu, P.; Kan, Y.; Wang, H. J. Org. Chem. 2015, 80, 11156–11161. doi:10.1021/acs.joc.5b01910
Return to citation in text: [1] [2] -
Wang, Z.; Shi, J.; Wang, J.; Li, C.; Tian, X.; Cheng, Y.; Wang, H. Org. Lett. 2010, 12, 456–459. doi:10.1021/ol902613g
Return to citation in text: [1] [2] -
Hollinger, J.; Gao, D.; Seferos, D. S. Isr. J. Chem. 2014, 54, 440–453. doi:10.1002/ijch.201400011
Return to citation in text: [1] -
Patra, A.; Bendikov, M. J. Mater. Chem. 2010, 20, 422–433. doi:10.1039/b908983g
Return to citation in text: [1] -
Okamoto, T.; Kudoh, K.; Wakamiya, A.; Yamaguchi, S. Org. Lett. 2005, 7, 5301–5304. doi:10.1021/ol0523650
Return to citation in text: [1] -
Yamamoto, T.; Takimiya, K. J. Am. Chem. Soc. 2007, 129, 2224–2225. doi:10.1021/ja068429z
Return to citation in text: [1] -
Izawa, T.; Miyazaki, E.; Takimiya, K. Chem. Mater. 2009, 21, 903–912. doi:10.1021/cm8030126
Return to citation in text: [1] -
Pao, Y.-C.; Chen, Y.-L.; Chen, Y.-T.; Cheng, S.-W.; Lai, Y.-Y.; Huang, W.-C.; Cheng, Y.-J. Org. Lett. 2014, 16, 5724–5727. doi:10.1021/ol502793e
Return to citation in text: [1] -
Lee, C.-H.; Lai, Y.-Y.; Cheng, S.-W.; Cheng, Y.-J. Org. Lett. 2014, 16, 936–939. doi:10.1021/ol4036787
Return to citation in text: [1] -
Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362
Return to citation in text: [1] [2] [3] [4] [5] [6] -
Xu, W.; Wang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2018, 83, 12154–12163. doi:10.1021/acs.joc.8b02107
Return to citation in text: [1] [2] [3] -
Schwartz, P.-O.; Förtsch, S.; Vogt, A.; Mena-Osteritz, E.; Bäuerle, P. Beilstein J. Org. Chem. 2019, 15, 1379–1393. doi:10.3762/bjoc.15.138
Return to citation in text: [1] -
Liu, X.; Yu, P.; Xu, L.; Yang, J.; Shi, J.; Wang, Z.; Cheng, Y.; Wang, H. J. Org. Chem. 2013, 78, 6316–6321. doi:10.1021/jo400691s
Return to citation in text: [1] -
Pino-Rios, R.; Yañez, O.; Inostroza, D.; Ruiz, L.; Cardenas, C.; Fuentealba, P.; Tiznado, W. J. Comput. Chem. 2017, 38, 481–488. doi:10.1002/jcc.24699
Return to citation in text: [1] -
Fu, R.; Lu, T.; Chen, F.-W. Acta Phys.-Chim. Sin. 2014, 30, 628–639. doi:10.3866/pku.whxb201401211
Return to citation in text: [1] -
Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, 2013.
Return to citation in text: [1] -
Ernst, K.-H.; Böhringer, M.; McFadden, C. F.; Hug, P.; Müller, U.; Ellerbeck, U. Nanotechnology 1999, 10, 355–361. doi:10.1088/0957-4484/10/3/320
Return to citation in text: [1] -
Taniguchi, M.; Nakagawa, H.; Yamagishi, A.; Yamada, K. Surf. Sci. 2000, 454-456, 1005–1009. doi:10.1016/s0039-6028(00)00174-6
Return to citation in text: [1] -
Taniguchi, M.; Nakagawa, H.; Yamagishi, A.; Yamada, K. J. Mol. Catal. A: Chem. 2003, 199, 65–71. doi:10.1016/s1381-1169(03)00019-0
Return to citation in text: [1] -
Fasel, R.; Parschau, M.; Ernst, K.-H. Angew. Chem., Int. Ed. 2003, 42, 5178–5181. doi:10.1002/anie.200352232
Return to citation in text: [1] -
Fasel, R.; Parschau, M.; Ernst, K.-H. Nature 2006, 439, 449–452. doi:10.1038/nature04419
Return to citation in text: [1] -
Lovinger, A. J.; Nuckolls, C.; Katz, T. J. J. Am. Chem. Soc. 1998, 120, 264–268. doi:10.1021/ja973366t
Return to citation in text: [1] -
Li, P.; Lai, Y.; Wang, Y.; Qian, Y.; Duan, W.; Li, C.; Wang, Z.; Fang, Q.; Wang, H.; Tu, B.; Geng, Y.; Zeng, Q. Sci. China: Chem. 2018, 61, 844–849. doi:10.1007/s11426-017-9233-2
Return to citation in text: [1] -
Zhang, G.; Tan, J.; Zhou, L.; Liu, C.; Liu, J.; Zou, Y.; Narita, A.; Hu, Y. Org. Lett. 2021, 23, 6183–6188. doi:10.1021/acs.orglett.1c00678
Return to citation in text: [1] -
Pieken, W. A.; Kozarich, J. W. J. Org. Chem. 1989, 54, 510–512. doi:10.1021/jo00263a053
Return to citation in text: [1] -
SADABS; University of Göttingen: Göttingen, Germany, 1996.
Return to citation in text: [1] -
SHELXTL, Version 5.1; Bruker Analytical Xray Systems, Inc.: Madison, WI, 1997.
Return to citation in text: [1]
| 1. | Mori, T. Chem. Rev. 2021, 121, 2373–2412. doi:10.1021/acs.chemrev.0c01017 |
| 2. | Dhbaibi, K.; Favereau, L.; Crassous, J. Chem. Rev. 2019, 119, 8846–8953. doi:10.1021/acs.chemrev.9b00033 |
| 3. | Rickhaus, M.; Mayor, M.; Juríček, M. Chem. Soc. Rev. 2016, 45, 1542–1556. doi:10.1039/c5cs00620a |
| 4. | Hoffmann, N. J. Photochem. Photobiol., C 2014, 19, 1–19. doi:10.1016/j.jphotochemrev.2013.11.001 |
| 5. | Gingras, M. Chem. Soc. Rev. 2013, 42, 968–1006. doi:10.1039/c2cs35154d |
| 6. | Shen, Y.; Chen, C.-F. Chem. Rev. 2012, 112, 1463–1535. doi:10.1021/cr200087r |
| 7. | Urbano, A. Angew. Chem., Int. Ed. 2003, 42, 3986–3989. doi:10.1002/anie.200301667 |
| 8. | Katz, T. J. Angew. Chem., Int. Ed. 2000, 39, 1921–1923. doi:10.1002/1521-3773(20000602)39:11<1921::aid-anie1921>3.0.co;2-f |
| 12. | Li, C.; Shi, J.; Xu, L.; Wang, Y.; Cheng, Y.; Wang, H. J. Org. Chem. 2009, 74, 408–411. doi:10.1021/jo802080g |
| 19. | Wang, Z.; Shi, J.; Wang, J.; Li, C.; Tian, X.; Cheng, Y.; Wang, H. Org. Lett. 2010, 12, 456–459. doi:10.1021/ol902613g |
| 18. | Li, C.; Wu, L.; Xu, W.; Song, J.; Shi, J.; Yu, P.; Kan, Y.; Wang, H. J. Org. Chem. 2015, 80, 11156–11161. doi:10.1021/acs.joc.5b01910 |
| 27. | Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362 |
| 17. | Wang, J.; Wang, G.; Li, C.; Dong, Y.; Ma, Z.; Wang, H. J. Org. Chem. 2021, 86, 4413–4422. doi:10.1021/acs.joc.0c02629 |
| 27. | Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362 |
| 9. | Miyasaka, M.; Rajca, A. Synlett 2004, 177–181. doi:10.1055/s-2003-43335 |
| 10. | Zhang, C.; Ma, Z.; Wang, G.; Li, C.; Wang, H. Org. Chem. Front. 2020, 7, 3926–3934. doi:10.1039/d0qo00957a |
| 11. | Rajca, A.; Wang, H.; Pink, M.; Rajca, S. Angew. Chem., Int. Ed. 2000, 39, 4481–4483. doi:10.1002/1521-3773(20001215)39:24<4481::aid-anie4481>3.0.co;2-g |
| 12. | Li, C.; Shi, J.; Xu, L.; Wang, Y.; Cheng, Y.; Wang, H. J. Org. Chem. 2009, 74, 408–411. doi:10.1021/jo802080g |
| 13. | Liu, X.; Sun, H.; Xu, W.; Wan, S.; Shi, J.; Li, C.; Wang, H. Org. Chem. Front. 2018, 5, 1257–1261. doi:10.1039/c7qo01049d |
| 14. | Rajca, A.; Miyasaka, M.; Pink, M.; Wang, H.; Rajca, S. J. Am. Chem. Soc. 2004, 126, 15211–15222. doi:10.1021/ja0462530 |
| 15. | Miyasaka, M.; Pink, M.; Olankitwanit, A.; Rajca, S.; Rajca, A. Org. Lett. 2012, 14, 3076–3079. doi:10.1021/ol3011437 |
| 16. | Miyasaka, M.; Rajca, A.; Pink, M.; Rajca, S. J. Am. Chem. Soc. 2005, 127, 13806–13807. doi:10.1021/ja055414c |
| 30. | Liu, X.; Yu, P.; Xu, L.; Yang, J.; Shi, J.; Wang, Z.; Cheng, Y.; Wang, H. J. Org. Chem. 2013, 78, 6316–6321. doi:10.1021/jo400691s |
| 23. | Yamamoto, T.; Takimiya, K. J. Am. Chem. Soc. 2007, 129, 2224–2225. doi:10.1021/ja068429z |
| 24. | Izawa, T.; Miyazaki, E.; Takimiya, K. Chem. Mater. 2009, 21, 903–912. doi:10.1021/cm8030126 |
| 27. | Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362 |
| 22. | Okamoto, T.; Kudoh, K.; Wakamiya, A.; Yamaguchi, S. Org. Lett. 2005, 7, 5301–5304. doi:10.1021/ol0523650 |
| 28. | Xu, W.; Wang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2018, 83, 12154–12163. doi:10.1021/acs.joc.8b02107 |
| 29. | Schwartz, P.-O.; Förtsch, S.; Vogt, A.; Mena-Osteritz, E.; Bäuerle, P. Beilstein J. Org. Chem. 2019, 15, 1379–1393. doi:10.3762/bjoc.15.138 |
| 20. | Hollinger, J.; Gao, D.; Seferos, D. S. Isr. J. Chem. 2014, 54, 440–453. doi:10.1002/ijch.201400011 |
| 21. | Patra, A.; Bendikov, M. J. Mater. Chem. 2010, 20, 422–433. doi:10.1039/b908983g |
| 19. | Wang, Z.; Shi, J.; Wang, J.; Li, C.; Tian, X.; Cheng, Y.; Wang, H. Org. Lett. 2010, 12, 456–459. doi:10.1021/ol902613g |
| 25. | Pao, Y.-C.; Chen, Y.-L.; Chen, Y.-T.; Cheng, S.-W.; Lai, Y.-Y.; Huang, W.-C.; Cheng, Y.-J. Org. Lett. 2014, 16, 5724–5727. doi:10.1021/ol502793e |
| 26. | Lee, C.-H.; Lai, Y.-Y.; Cheng, S.-W.; Cheng, Y.-J. Org. Lett. 2014, 16, 936–939. doi:10.1021/ol4036787 |
| 27. | Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362 |
| 31. | Pino-Rios, R.; Yañez, O.; Inostroza, D.; Ruiz, L.; Cardenas, C.; Fuentealba, P.; Tiznado, W. J. Comput. Chem. 2017, 38, 481–488. doi:10.1002/jcc.24699 |
| 32. | Fu, R.; Lu, T.; Chen, F.-W. Acta Phys.-Chim. Sin. 2014, 30, 628–639. doi:10.3866/pku.whxb201401211 |
| 44. | SHELXTL, Version 5.1; Bruker Analytical Xray Systems, Inc.: Madison, WI, 1997. |
| 18. | Li, C.; Wu, L.; Xu, W.; Song, J.; Shi, J.; Yu, P.; Kan, Y.; Wang, H. J. Org. Chem. 2015, 80, 11156–11161. doi:10.1021/acs.joc.5b01910 |
| 27. | Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362 |
| 28. | Xu, W.; Wang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2018, 83, 12154–12163. doi:10.1021/acs.joc.8b02107 |
| 42. | Pieken, W. A.; Kozarich, J. W. J. Org. Chem. 1989, 54, 510–512. doi:10.1021/jo00263a053 |
| 34. | Ernst, K.-H.; Böhringer, M.; McFadden, C. F.; Hug, P.; Müller, U.; Ellerbeck, U. Nanotechnology 1999, 10, 355–361. doi:10.1088/0957-4484/10/3/320 |
| 35. | Taniguchi, M.; Nakagawa, H.; Yamagishi, A.; Yamada, K. Surf. Sci. 2000, 454-456, 1005–1009. doi:10.1016/s0039-6028(00)00174-6 |
| 36. | Taniguchi, M.; Nakagawa, H.; Yamagishi, A.; Yamada, K. J. Mol. Catal. A: Chem. 2003, 199, 65–71. doi:10.1016/s1381-1169(03)00019-0 |
| 37. | Fasel, R.; Parschau, M.; Ernst, K.-H. Angew. Chem., Int. Ed. 2003, 42, 5178–5181. doi:10.1002/anie.200352232 |
| 38. | Fasel, R.; Parschau, M.; Ernst, K.-H. Nature 2006, 439, 449–452. doi:10.1038/nature04419 |
| 39. | Lovinger, A. J.; Nuckolls, C.; Katz, T. J. J. Am. Chem. Soc. 1998, 120, 264–268. doi:10.1021/ja973366t |
| 40. | Li, P.; Lai, Y.; Wang, Y.; Qian, Y.; Duan, W.; Li, C.; Wang, Z.; Fang, Q.; Wang, H.; Tu, B.; Geng, Y.; Zeng, Q. Sci. China: Chem. 2018, 61, 844–849. doi:10.1007/s11426-017-9233-2 |
| 41. | Zhang, G.; Tan, J.; Zhou, L.; Liu, C.; Liu, J.; Zou, Y.; Narita, A.; Hu, Y. Org. Lett. 2021, 23, 6183–6188. doi:10.1021/acs.orglett.1c00678 |
| 27. | Xu, W.; Wu, L.; Fang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2017, 82, 11192–11197. doi:10.1021/acs.joc.7b01362 |
| 28. | Xu, W.; Wang, M.; Ma, Z.; Shan, Z.; Li, C.; Wang, H. J. Org. Chem. 2018, 83, 12154–12163. doi:10.1021/acs.joc.8b02107 |
© 2022 Wang et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.