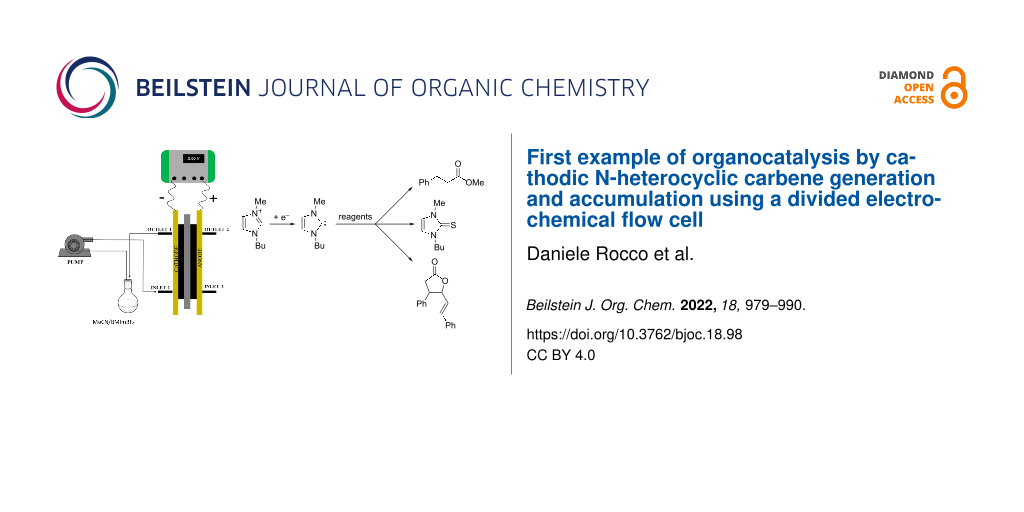
Abstract
In this paper we present the first electrochemical generation of NHC carried out in a divided flow cell. The flow cell operated in the recycle mode. The need for a divided cell derived from the anodic electroactivity of the electrogenerated carbene. In order to have NHC accumulation in the catholyte, the Nafion membrane (cell separator) was pretreated with an alkaline solution. The formation of NHC was quantified as its reaction product with elemental sulfur. The NHC was successfully used as organocatalyst in two classical umpolung reactions of cinnamaldehyde: its cyclodimerization and its oxidative esterification.

Graphical Abstract
Introduction
Ionic liquids (ILs) are well known salts, at present used in a wide variety of chemical fields. The first definition of ionic liquids was given by Paul Walden in 1914: “they are materials composed of cations and anions, that melt around 100 °C or below as an arbitrary temperature limit” [1]. ILs are salts formed by non or weakly coordinated cations and anions, usually bulky organic cations and inorganic or organic anions such as −BF4, −PF6, −N(CF3SO2)2, etc. [2,3].
The physicochemical properties of RTILs (room temperature ionic liquids) are reported in the literature and include very low vapor pressure, and thus the possibility to recycle them, good stability, low flammability, good solubility ability for many molecules, salts and gases, the possibility to use them as reagents and/or solvents, large electrochemical window, good electrical conductivity, their ionic nature sometimes increases the reactivity and the selectivity of reactions and usually the product separation is easy [4,5]. Among ILs, imidazolium derivatives are the most studied, in part due to their ease of synthesis, low cost and diverse applications from solvents and reagents in synthesis, to supporting electrolytes in electrochemistry [6].
The imidazolium cation can be modified by the presence of a base or by a single electron cathodic reduction of the C–H between nitrogen atoms of the imidazolium ring (Scheme 1), inducing the formation of a N-heterocyclic carbene (NHC) [7,8]. In recent years, NHCs have achieved great success: they have been frequently used as ligands in organometallic catalysts [9] and as versatile organocatalysts [10] in a very wide range of organic reactions such as classical benzoin condensation, transesterification, acylation, Knoevenagel reaction, Claisen condensation etc.
![[1860-5397-18-98-i1]](/bjoc/content/inline/1860-5397-18-98-i1.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 1: Electrochemical generation of NHC.
Scheme 1: Electrochemical generation of NHC.
The electrochemical generation of carbenes from ILs avoids the use of strong bases and the formation of byproducts (dimers of carbenes and nitrogen dealkylation products), where the IL acts as NHC precursor, solvent, and supporting electrolyte, needing no additional chemicals in the electrolytic cell [11]. An added attraction of this approach is that unstable NHCs are generated in situ, where they may be used as basic or nucleophilic species. Due to the difficulty isolating highly reactive NHCs, the concentration of the obtained NHC solution can be determined indirectly by addition of elemental sulfur after the electrolysis, which realizes quantitative conversion to the corresponding thione (Scheme 2) [12].
![[1860-5397-18-98-i2]](/bjoc/content/inline/1860-5397-18-98-i2.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 2: Transformation of electrochemically generated NHC into the corresponding thione by its reaction with elemental sulfur.
Scheme 2: Transformation of electrochemically generated NHC into the corresponding thione by its reaction wit...
NHCs are used as organocatalysts in many reactions of aldehydes (mainly aromatic) [13,14]. In fact, the reaction of NHCs with aldehydes can lead to the formation of the “Breslow intermediate” [15], in which the reactive character of the carbonyl carbon atom is reversed (umpolung) from electrophilic to nucleophilic (Scheme 3).
![[1860-5397-18-98-i3]](/bjoc/content/inline/1860-5397-18-98-i3.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 3: Umpolung of the aldehyde carbonyl carbon atom. Formation of the Breslow intermediate using NHCs.
Scheme 3: Umpolung of the aldehyde carbonyl carbon atom. Formation of the Breslow intermediate using NHCs.
This approach can be exploited in many organic reactions, such as: the benzoin condensation [16,17], esterification and amidation of benzaldehydes and cinnamaldehydes [18,19], synthesis of γ-butyrolactones [20], synthesis of 1,3-diketones [21], etc.
Electrosynthesis is considered as a more sustainable approach to perform chemical reactions, and an interesting alternative to conventional synthetic methods both in laboratory and industry processes. In fact, the electron may be considered to be a clean reagent, which replaces toxic chemical redox reagents and dangerous procedures [22-25]. At present, electrosynthesis in batch is more widely used and reported in literature, but some disadvantages can be encountered: the need for high concentrations of supporting electrolyte, poor performance for synthesis such as slow rates of conversion, low selectivity and reproducibility [26]. As a matter of fact, these problems can be addressed by using flow electrochemistry, usually achieving higher rates of conversion of reagents to products [27]. Moreover, electrochemical flow cells can have a very small gap between the electrodes so that lower concentrations of supporting electrolytes are needed to provide sufficient conductivity [28]. Applications of flow electrochemistry reported in the literature are mainly devoted to anodic oxidations, carried out in undivided cells, in which the counter electrode reaction at the cathode is usually H2 evolution [29]. The use of divided cells is less common in organic electrosynthesis, mainly due to complications inherent with membranes. Useful cathodic processes are less exploited in organic electrochemistry. In the context of NHC organocatalysis in flow electrochemistry, NHC instability (and anodic electroactivity) prevented its cathodic generation and subsequent use as catalyst or reagent. Instead, the NHC was generated by chemical deprotonation using a strong base (DBU) and then applied in anodic esterification [30-32], and amidation of aromatic aldehydes [33]. Flow electrochemistry was applied to oxidize the Breslow intermediate to the corresponding electrophilic acylthiazolium intermediate, which then functioned as an acyl-transfer reagent, reacting with alcohols or amines. To the best of our knowledge, only one research group reported the cathodic reduction of an imidazolium cation to NHC, in an undivided cell under flow conditions, coupled with the anodic generation of Cu(I) from a sacrificial anode to yield the corresponding N-heterocyclic carbene complex [34,35]. In this case, irreversible capture of the NHC by the metallic cation prevented NHC oxidation/degradation.
In this paper we describe the cathodic generation and accumulation of NHC in a divided flow cell and its subsequent use as organocatalyst in the self-annulation of cinnamaldehyde and in the esterification of cinnamaldehyde.
Results and Discussion
Cathodic NHC generation and accumulation using a divided flow electrochemical cell. Quantification of NHC in IL solution
The flow cell used in this work was previously described [36]. It is based on two electrode plates separated by a spacer/gasket fabricated from an PTFE sheet with its center cut away to form the electrolyte flow chamber (Figure 1). The requirement for a divided cell (a more complicated device than the undivided configuration) arises from the need to protect electrogenerated NHC from its anodic oxidation in the absence of a consumable anode.
![[1860-5397-18-98-1]](/bjoc/content/figures/1860-5397-18-98-1.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 1: Schematic representation of a plane-parallel plate flow electrochemical reactor.
Figure 1: Schematic representation of a plane-parallel plate flow electrochemical reactor.
To ensure good sealing of the electrolysis cell, the sandwich-type arrangement of cell components was compressed between two end plates using a series of bolts. This design incorporated a solution inlet and a solution outlet for the chamber to allow uniform flow over the surface of the electrodes. In the present work, where a divided cell configuration was required, a proton-permeable membrane (Nafion® 438) was inserted to separate the cathode and anode chambers, and a spacer was used on each compartment [36].
The initial goal of this work was to demonstrate the possibility to achieve NHC formation starting from 1-butyl-3-methylimidazolium tetrafluoroborate (BMImBF4), by cathodic reduction in a divided cell using flow electrochemistry technique, and to compare the results with the corresponding batch process. Once established, the flow electrochemistry NHC synthesis would be combined with applications as an organocatalyst in some organic transformations of cinnamaldehyde.
Firstly, the conditions for electrogeneration of the carbene in flow were optimized, quantifying the amount of NHC by means of its reaction with elemental sulfur. All the experiments were carried out using a 0.1 M solution of BMImBF4 in acetonitrile as catholyte in a divided cell using Nafion® 438 membrane, under N2 atmosphere, at room temperature, under galvanostatic conditions (I = 134 mA) with continuous flow rate of 36 mL/min, studying the effect of cathode material, anode solution and number of Faradays per mole of IL supplied (Table 1). At the end of the electrolysis, excess elemental sulfur was added to the catholyte and the mixture was left under ultrasound irradiation for 30 minutes. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel.
The first experiment (Table 1, entry 1) was carried out using stainless steel as cathode and a carbon-filled polyvinylidene fluoride (C/PVDF) plate as anode. BMImBF4 0.1 M in acetonitrile was the catholyte, while the anolyte was a solution of tetraethylammonium tetrafluoroborate (Et4NBF4) in acetonitrile; after only 12 minutes of electrolysis the current flow stopped. Moreover, a consumption of anode was observed (Figure 2). It is possible that, in the absence of a suitable counter electrode process, the tetrafluoroborate anion was itself oxidized [7] leading to erosion of the anode with the formation of fluorocarbons.
Table 1: Electrochemical reduction of BMImBF4,a followed by the addition of elemental sulfurb. Flow cell, in recycling mode.
![[Graphic 1]](/bjoc/content/inline/1860-5397-18-98-i6.svg?max-width=637&scale=1.0)
|
||||||
| Entry | Cathode material | Anolyte | Q (F) | Time (min) | Yield 1ac | Notes |
| 1 | SSd |
MeCN/Et4NBF4 0.05 M,
25 mL |
0.5 | 12 | <5% | anode erosion |
| 2 | SSd | MeCN–MeOH (9:1)/Et4NBF4 0.05 M, 25 mL | 1.0 | 24 | 13% | – |
| 3 | Ag | MeCN–MeOH (9:1)/Et4NBF4 0.05 M, 25 mL | 1.0 | 24 | – | – |
| 4 | Ni | MeCN–MeOH (9:1)/Et4NBF4 0.05 M, 25 mL | 1.0 | 24 | 13% | – |
| 5 | SSd | MeCN–MeOH (9.5:0.5)/Et4NBF4 0.1 M, 20 mL | 1.0 | 24 | 21% | Nafion® alkaline pretreatment |
| 6 | SSd | Dry MeCN–MeOH (9.5:0.5)/Et4NBF4 0.1 M, 20 mL | 1.0 | 24 |
32%
(1b <5%) |
Nafion® alkaline pretreatment |
aDivided cell, carbon-filled polyvinylidene fluoride (C/PVDF) anode material, Nafion® 438 membrane separator, room temperature, N2 atmosphere, galvanostatic conditions (134 mA), catholyte: BMImBF4/MeCN 0.1 M, 20 mL (2 mmol BMImBF4); flow rate: 36 mL/min. bExcess S8 (2 mmol) added at the end of the electrolysis to the catholyte. Then energy was supplied to the catholyte (ultrasound irradiation, 35 W) for 30 minutes. cIsolated yields, based on starting IL (BMImBF4). dStainless steel.
![[1860-5397-18-98-2]](/bjoc/content/figures/1860-5397-18-98-2.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 2: C/PVDF anode before (A) and after (B) the first experiment (Table 1, entry 1).
Figure 2: C/PVDF anode before (A) and after (B) the first experiment (Table 1, entry 1).
In order to avoid anode consumption/passivation, an alternative to oxidation of BF4− was required as the counter electrode reaction, and 10% of methanol was added to the anolyte solution. In this case (Table 1, entry 2) 13% of thione was obtained and the electrode did not show any signs of consumption. The same yield (13%) was obtained using nickel as cathode material (Table 1, entry 4). However, using a silver electrode the corresponding thione was not observed (Table 1, entry 3) [37]. In view of the acidic character of the perfluorosulfonic acid Nafion® membrane, and possible NHC protonation to form the imidazolium cation, the Nafion® membrane was pretreated with an alkaline solution for 24 hours. Using stainless steel as cathode and a lower amount of methanol at the anode the quantity of thione increased to 21% (Table 1, entry 5). Under the same conditions, but using dry acetonitrile, the best yield was obtained (32%, Table 1, entry 6); in this case a small amount of imidazolone 1b (<5%), probably due to the reaction with adventitious oxygen, was observed from the NMR spectrum. It should be underlined that in these electrolyzes BMImBF4 acts as both electroactive species and supporting electrolyte, and although the cathodic reduction of the imidazolium cation is a monoelectronic process, in a similar process carried out in batch the current yield usually does not exceed 50%, thus rendering necessary an excess of current [38]. The yields reported in Table 1 (entries 2 to 6) are obtained using a stoichiometric amount of electricity (1 electron per imidazolium cation). Therefore, the 32% chemical yield (with respect to starting IL) is comparable with the yield of the batch process.
Electrogenerated NHC organocatalysis. Dimerization of trans-cinnamaldehyde: synthesis of 4-phenyl-5-styryldihydrofuran-2(3H)-one
Having demonstrated that NHCs could be generated and accumulated in a continuous flow electrochemical process, the flow methodology was applied to the self-annulation of cinnamaldehyde, a classical NHC-catalyzed reaction (Scheme 4).
![[1860-5397-18-98-i4]](/bjoc/content/inline/1860-5397-18-98-i4.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 4: Electrogenerated NHC-catalyzed self-annulation of cinnamaldehyde.
Scheme 4: Electrogenerated NHC-catalyzed self-annulation of cinnamaldehyde.
All the experiments were carried out using a solution of 0.1 M BMImBF4 in acetonitrile (20 mL) as catholyte, stainless steel as cathode, C/PVDF as anode, in a divided cell, under N2 atmosphere, at room temperature, under galvanostatic conditions (I = 134 mA) with a flow rate of 36 mL/min, changing anode solution and number of Faradays per mole supplied (Table 2). Following the experimental results previously obtained, the Nafion® membrane was always pretreated with an alkaline solution. At the end of the electrolysis, 1 mmol of cinnamaldehyde was added to the catholyte and the mixture was left under stirring for two hours at room temperature. Workup and column chromatography yielded a diastereomeric mixture of γ-butyrolactones 2a and 2b.
Table 2: Electrochemical synthesis of γ-butyrolactones 2a and 2b by conjugate umpolung reaction.a
![[Graphic 2]](/bjoc/content/inline/1860-5397-18-98-i7.svg?max-width=637&scale=1.0)
|
|||||
| Entry | Anolyte | Q (F)b | Time (min) | Yieldc | |
| 2a | 2b | ||||
| 1 | MeCN–MeOH (95:5)/Et4NBF4 (0.1 M), 20 mL | 0.50 | 6 | 15% | 8% |
| 2 | MeCN–MeOH (95:5)/Et4NBF4 (0.1 M), 20 mL | 1.00 | 12 | 43% | 24% |
| 3 | MeCN–MeOH (95:5)/Et4NBF4 (0.1 M), 20 mL | 2.00 | 24 | 53% | 26% |
| 4 | MeCN–MeOH (98:2)/Et4NBF4 (0.1 M), 20 mL | 1.00 | 12 | 42% | 32% |
| 5 | MeCN–DMSO (95:5)/Et4NBF4 (0.1 M), 20 mL | 0.50 | 6 | 7% | <5% |
| 6 | MeCN–DMSO (95:5)/Et4NBF4 (0.1 M), 20 mL | 1.00 | 12 | 20% | 12% |
aDivided cell, carbon-filled polyvinylidene fluoride (C/PVDF) anode, stainless steel cathode, alkaline pretreated Nafion® 438 membrane as compartments separator, room temperature, N2 atmosphere, galvanostatic conditions (134 mA), catholyte: BMImBF4/MeCN 0.1 M, 20 mL (2 mmol BMImBF4); flow rate: 36 mL/min; 1 mmol of cinnamaldehyde added at the end of the electrolysis to the catholyte. bWith respect to the starting BMImBF4. cIsolated yields, based on starting cinnamaldehyde.
Table 2 reports the results obtained by changing the anolyte composition and the amount of applied electricity. In all cases, with regards to the trans/cis diastereomeric ratio, we observed that the cis isomer 2a was predominantly formed. The diastereoselectivity was not high; however, trans and cis γ-butyrolactones were easily separated by column chromatography. Increasing the applied charge from 0.5 F (Table 2, entry 1) to 1.0 F (Table 2, entry 2) improved the yield of both cis (2a) and trans (2b) lactones by approximately three fold. Instead, with a charge of 2.0 F the yield of 2a improved by 10%, but the yield of 2b increased only 2% (Table 2, entry 3). However, in these experiments methyl ester 3a was isolated (24% yield) as byproduct derived from methanol, which passed through the membrane, reacting with the Breslow intermediate through a redox neutral process (Scheme 5) [19].
![[1860-5397-18-98-i5]](/bjoc/content/inline/1860-5397-18-98-i5.svg?scale=2.0&max-width=1024&background=FFFFFF)
Scheme 5: Byproduct obtained from the reaction between methanol and the Breslow intermediate.
Scheme 5: Byproduct obtained from the reaction between methanol and the Breslow intermediate.
We decided to use 1 F/mol and a lower amount of methanol at the anode, in order to minimize the formation of byproduct 3a; the same amount of 2a was obtained, but the yield of the trans diastereoisomer increased from 24% to 32% (Table 2, entry 2 vs entry 4). Finally, changing methanol with DMSO in the anolyte, a lower amount of both diastereoisomers was observed (Table 2, entries 5 and 6). Thus the best yield (79%, 67:33 cis/trans ratio) was obtained using a charge of 2.0 F and a solution of MeCN–MeOH (9.5:0.5)/Et4NBF4 (0.1 M) as anolyte (Table 2, entry 3).
Electrogenerated NHC organocatalysis. Esterification of trans cinnamaldehyde
Once the possibility of obtaining the Breslow intermediate was demonstrated, another typical reaction of N-heterocyclic carbenes was tested: the oxidative esterification of cinnamaldehyde in the presence of alcohols. To carry out these experiments three different alcohols (methyl, isopropyl and benzyl alcohols) were used and the results are shown in Table 3.
Table 3: Electrochemical synthesis of esters 3a–c from cinnamaldehyde and an alcohol.a
![[Graphic 3]](/bjoc/content/inline/1860-5397-18-98-i8.svg?max-width=637&scale=1.0)
|
|||||
| Entry | ROH | Anolyte | Q (F)b | Yieldc | |
| 1 | R3 = Me | MeCN–MeOH (98:2)/Et4NBF4 0.1 M, 20 mL | 0.5 |
3a
68% |
4a
11% |
| 2 | R3 = Bn | MeCN–:BnOH (98:2)/Et4NBF4 0.1 M, 20 mL | 0.5 |
3b
73% |
4b
13% |
| 3 | R3 = iPr | MeCN–iPrOH (98:2)/Et4NBF4 0.1 M, 20 mL | 0.5 |
3c
37% |
4c
– |
aDivided cell, carbon-filled polyvinylidene fluoride (C/PVDF) anode, stainless steel cathode, alkaline pretreated Nafion® 438 membrane separator, room temperature, N2 atmosphere, galvanostatic conditions (134 mA), catholyte: BMImBF4/MeCN 0.1 M, 20 mL (2 mmol BMImBF4); flow rate: 36 mL/min; 1 mmol of cinnamaldehyde added at the end of the electrolysis to the catholyte and, after 5 minutes 2 mmol of the corresponding alcohol were added. bWith respect to the starting BMImBF4, telectrolysis = 12 min. cIsolated yields, based on starting cinnamaldehyde.
All the experiments were carried out using a solution of 0.1 M of BMImBF4 in acetonitrile (20 mL) as catholyte, stainless steel as cathode, C/PVDF as anode, in a divided cell, under N2 atmosphere, at room temperature, under galvanostatic conditions (I = 134 mA, telectrolysis = 12 min) with a flow rate of 36 mL/min, anode solution as in Table 3, and with 1.0 Faraday per mole of aldehyde. At the end of the electrolysis, 1 mmol of cinnamaldehyde was added to the catholyte and the mixture was left under stirring for five minutes and then the corresponding alcohol was added and the reaction was stirred for two hours at room temperature. Workup and column chromatography yielded esters 3a–c and unsaturated esters 4a,b as byproducts.
Good yields were obtained using benzyl and methyl alcohols (73% and 68%, respectively), while with the isopropyl alcohol the formation of the ester was only 37%, probably due to steric hindrance. In two cases (Table 3, entries 1 and 2) oxidation byproducts (esters 4a and 4b) were obtained, where the olefinic double bond is preserved. In contrast to the internal redox reactions of cinnamaldehyde giving esters 3a–c, formation of enoate byproducts 4a and 4b invoke the involvement of an external chemical oxidant species as cinnamaldehyde is added to the cathode chamber after the electrolysis has been stopped. The presence of byproducts 4a and 4b is most likely accounted for by the presence of molecular oxygen, or electrochemically generated superoxide (cathodic reduction of O2), which oxidize the Breslow intermediate [30,39]. In fact, the presence of some reactive oxygen species in the reaction environment was previously demonstrated by the formation of compound 1b (see Table 1).
IL Recycling
To investigate the possibility to recycle BMImBF4, the experiment reported in Table 3, entry 1, was replicated, using the same reagents and conditions. Exploiting the low vapor pressure of BMImBF4, (and of ILs in general), the organic solvent was removed under reduced pressure, and after the extractive workup, the recycled BMImBF4 was resubjected to cathodic reduction (thus reused in different reactions). Although this procedure led to decreased yields of esterification products 3a, 4a (10% and 12%, respectively) and diastereoisomeric 4-phenyl-5-styryldihydrofuran-2(3H)-ones 2a,b (35%, 45:55 cis/trans mixture), the potential of the recycled BMImBF4 to function as a N-heterocyclic carbene precursor was demonstrated. Interestingly, the reaction selectivity is completely lost using the recovered IL, and experiments are undergoing in order to understand the cause of this selectivity loss.
Batch vs flow electrolysis
At this point, the preliminary results from flow electrochemistry may be compared with those reported in the literature for NHC formation using batch electrolysis. The comparison between these two techniques is not straightforward. In fact, in batch electrosynthesis, the ionic liquid is normally used as solvent, but in this work the IL was present as a 0.1 M solution in MeCN.
In regard to the results obtained in the synthesis of thione 1a (the only reaction for which a reasonable comparison is possible), we compared the current efficiencies, the current being the limiting factor (Table 4). Although the current yield (49%) for the batch electrolysis in pure IL is higher than in flow, the yields for 0.1 M solutions are comparable between batch (29%) and flow (32%). In this case, the rate of production of the NHC in the electrolysis step was higher in the flow reactor (1.60 mmol/h) than in batch using pure IL (0.37 mmol/h). However, other factors should also be highlighted, including the 8× larger electrode area in the flow reactor, which enables a higher current to be passed while maintaining the same current density (15 mA/cm2).
Table 4: Comparison between electrosynthesis in batch and flow electrochemistry.
| Product | Solvent |
Batch electrochemistry yield
(current density) |
Flow electrochemistry yield
(current density) |
![[Graphic 4]](/bjoc/content/inline/1860-5397-18-98-i9.svg?max-width=637&scale=1.0)
1a |
BMImBF4 |
49%a
(15 mA/cm2) |
– |
| solvent/BMImBF4 |
29%b
(15 mA/cm2) |
32%c
(15 mA/cm2) |
|
![[Graphic 5]](/bjoc/content/inline/1860-5397-18-98-i10.svg?max-width=637&scale=1.0)
2a,b |
MeCN/BMImBF4 | – |
79%d
(15 mA/cm2) |
![[Graphic 6]](/bjoc/content/inline/1860-5397-18-98-i11.svg?max-width=637&scale=1.0)
3a |
BMImBF4 |
91%e
(20 mA/cm2) |
– |
| MeCN/BMImBF4 | – |
73%f
(15 mA/cm2) |
|
aCurrent yield. Q = 193 C. Theoretical current for a mono-electron process: 96.5 C for 1.0 mmol substrate/product. Current yield: (experimental yield/ theoretical yield) × 100. [38]. bCurrent yield (see note a). DMF as solvent. [12]. cCurrent yield (see note a). MeCN as solvent; flow rate: 36 mL/min. dChemical yield, with respect to starting cinnamaldehyde. 2.0 F/mol cinnamaldehyde; flow rate: 36 mL/min. eChemical yield, with respect to starting cinnamaldehyde. 0.7 F/mol cinnamaldehyde. 0.97 mmol/h [40]. fChemical yield, with respect to starting cinnamaldehyde. 3.65 mmol/h. 1.0 F/mol cinnamaldehyde; flow rate: 36 mL/min.
In the case of the NHC organocatalysis, we compared chemical yields of products (Table 4), although in different solvents. As far as the literature allows comparison of the two methodologies, the data in Table 4 show that, although further optimization of the flow electrochemistry conditions is required, it provides a valid alternative to batch technique. Again, productivity for the esterification is four-fold higher (3.65 mmol/h, 0.1 M solution) in flow than in batch (0.97 mmol/h, pure IL), and in flow the reaction volume could simply be increased to allow scale-up, obviously in a longer time.
Conclusion
The results reported in this work demonstrate for the first time the possibility to synthesize and accumulate N-heterocyclic carbene starting from BMImBF4 in a divided electrochemical flow cell. Although not fully optimized, the production of NHC was confirmed indirectly by isolation of its reaction product with elemental sulfur. A solution of the electrogenerated carbene was used to promote dimerization and oxidative esterification reactions of cinnamaldehyde. Under the flow conditions investigated, a higher rate of NHC production was achieved, compared to previously reported batch reactions, which is an important consideration for scale up. In the case of the flow procedure, a 0.1 M solution of the IL is employed, which may be more convenient than using pure ILs. Further investigations of electrogeneration of NHCs and applications in organic synthesis are underway.
Experimental
Materials and methods
Chemicals were purchased from Sigma-Aldrich and Alfa Aesar and used as received. All air/moisture sensitive reactions were carried out under an inert atmosphere, in oven-dried or flame-dried glassware. TLC was performed on aluminium plates precoated with silica gel 60 with an F254 indicator; visualized under UV light (254 nm) and/or by staining with potassium permanganate. Flash column chromatography was performed using high purity silica gel, pore size 60 Å, 230–400 mesh particle size, purchased from Merck. 1H NMR and 13C NMR spectra were recorded in CDCl3 (purchased from Cambridge Isotope Laboratories) at 298 K using a Bruker DPX400 (400 and 101 MHz, respectively) spectrometer. Chemical shifts are reported on the δ scale in ppm and were referenced to residual solvent (CDCl3: 7.27 ppm for 1H NMR spectra and 77.0 ppm for 13C NMR spectra). Coupling constants (J) were given in Hz and matched where possible. The following abbreviations for the multiplicity of the peaks are used: s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet).
Parallel plate divided flow cell
See Figure 3 for an expanded view of the electrochemical cell components. The details about the dimensions of each cell component are reported elsewhere [36].
![[1860-5397-18-98-3]](/bjoc/content/figures/1860-5397-18-98-3.png?scale=2.0&max-width=1024&background=FFFFFF)
Figure 3: Expanded view of the electrochemical cell components: (a) Aluminium end plates; (b) insulating PTFE foil, (c) copper plate for electrical contact; (d) carbon electrode; (e) PTFE gasket (reaction channel and sealing); (f) Nafion® membrane; (g) stainless steel electrode; (h) stainless-steel plate/electrode for electrical contact. Figure 3 was adapted from [36] with permission from The Royal Society of Chemistry. This content is not subject to CC BY 4.0.
Figure 3: Expanded view of the electrochemical cell components: (a) Aluminium end plates; (b) insulating PTFE...
General procedure for the synthesis of 1-butyl-3-methyl-1H-imidazole-2(3H)-thione
Constant current electrolyzes (I = 134 mA) were carried out using a parallel plate divided cell. Anolyte (25 mL) and catholyte (20 mL) were separated through a Nafion® 438 membrane. The anode material was carbon-filled polyvinylidene fluoride (C/PVDF) and the cathode material is described in Table 1. Electrolyzes were carried out at room temperature, under nitrogen atmosphere, using a solution of 0.1 M of BMImBF4 in acetonitrile as catholyte. Anolyte solution is given in Table 1. Electrolyte solutions were continuously passed (recycling method) through their respective compartments at a flow rate of 36 mL/min. After consumption of the requisite amount of charge (Faradays per mol of BMImBF4 reported in Table 1), the current was switched off, the flow stopped, and elemental sulfur (2.0 mmol) was added to the catholyte. The mixture was left under ultrasound irradiation for 30 minutes. The solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel (petroleum ether/ethyl acetate 7:3), affording the corresponding pure 1a as an off-white solid.
1-Butyl-3-methyl-1H-imidazole-2(3H)-thione (1a): Spectral data are consistent with those reported in the literature [38]. 1H NMR (CDCl3) δ 6.64 (s, 2H), 3.99 (t, J = 7.4 Hz, 2H), 3.57 (s, 3H), 1.79–1.64 (m, 2H), 1.39–1.28 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H) ppm; 13C NMR (CDCl3) δ 162.3, 117.5, 116.4, 47.8, 35.0, 30.9, 19.7, 13.6 ppm.
1-Butyl-3-methyl-1H-imidazol-2(3H)-one (1b): Spectral data are consistent with those reported in the literature [40]. 1H NMR (CDCl3) δ 6.19 (d, AB, Δν/J = 2.5, Δν = 7.1 Hz, J = 2.8 Hz, 1H), 6.17 (d, AB, Δν/J = 2.5, Δν = 7.1 Hz, J = 2.8 Hz, 1H), 3.59 (t, J = 7.2 Hz, 2H), 3.24 (s, 3H), 1.70–1.59 (m, 2H), 1.42–1.24 (m, 2H), 0.92 (t, J = 7.2 Hz, 3H) ppm; 13C NMR (CDCl3) δ 153.1, 111.3, 110.2, 43.5, 31.1, 30.5, 19.9, 13.6 ppm.
General procedure for dimerization of trans cinnamaldehyde
Constant current electrolyzes (I = 134 mA) were carried out using a parallel plate divided cell. Anolyte (20 mL) and catholyte (20 mL) were separated through a Nafion® 438 membrane. The anode material was carbon-filled polyvinylidene fluoride C/PVDF and stainless steel for the cathode material. Electrolyzes were carried out at room temperature, under nitrogen atmosphere, using a solution 0.1 M of BMImBF4 in acetonitrile as catholyte and for anolyte solution see Table 2, which were passed (recycling method) in their compartments with a flow rate of 36 mL/min. After the consumption of the number of Faradays per mol of BMImBF4 reported in Table 2, the current was switched off, the flow stopped and cinnamaldehyde (1.0 mmol) was added to the catholyte. The mixture was stirred for 2 h. The solvent was removed under reduced pressure and the residue was extracted with of Et2O (15 mL × 3) and then purified by column chromatography on silica gel (petroleum ether/ethyl acetate 9:1), affording the corresponding pure products 2a and 2b.
(±) cis-4-Phenyl-5-((E)-styryl)dihydrofuran-2(3H)-one (2a): Spectral data are consistent with those reported in the literature [41]. 1H NMR (CDCl3) δ 7.38–7.25 (m, 3H), 7.29–7.24 (m, 3H), 7.23–7.15 (m, 4H), 6.63 (d, J = 15.9 Hz, 1H), 5.65 (dd, J = 15.9 Hz, 6.5 Hz, 1H), 5.42–5.38 (m, 1H), 3.99–3.93 (m, 1H), 2.98 (dd, J = 17.4 Hz, 8.1 Hz, 1H), 2.91 (dd, J = 17.4 Hz, 7.4 Hz, 1H) ppm; 13C NMR (CDCl3) δ 176.3, 136.9, 135.8, 133.4, 128.9, 128.6, 128.2, 127.9, 127.8, 126.6, 123.8, 83.5, 45.6, 34.3 ppm.
(±) trans-4-Phenyl-5-((E)-styryl)dihydrofuran-2(3H)-one (2b): Spectral data are consistent with those reported in the literature [41]. 1H NMR (CDCl3) δ 7.39–7.27 (m, 10H), 6.60 (d, J = 15.9 Hz, 1H), 6.23 (dd, J = 15.9 Hz, 6.7 Hz, 1H), 5.06–5.02 (m, 1H), 3.57–3.50 (m, 1H), 3.04 (dd, J = 17.5 Hz, 8.6 Hz, 1H), 2.87 (dd, J = 17.5 Hz, 10.5 Hz, 1H) ppm; 13C NMR (CDCl3) δ 175.2, 138.0, 135.6, 133.8, 129.2, 128.7, 128.4, 127.9, 127.3, 126.8, 124.7, 86.7, 48.4, 36.7 ppm.
General procedure for the esterification of trans-cinnamaldehyde
Constant current electrolyzes (I = 134 mA) were carried out using a parallel plates divided cell. Anolyte (20 mL) and catholyte (20 mL) were separated through a Nafion® 438 membrane. The anode material was C/PVDF and stainless steel for the cathode material. Electrolyzes were carried out at room temperature, under nitrogen atmosphere, using a solution 0.1 M of BMImBF4 in acetonitrile as catholyte. Anolyte solution is given in Table 3. Electrolyte solutions were continuously passed (recycling method) through their respective compartments at a flow rate of 36 mL/min. After the consumption of 0.5 F/mol of BMImBF4 (12 min, 96 C), the current was switched off, the flow stopped and cinnamaldehyde (1.0 mmol) was added to the catholyte. The mixture was left under stirring for 5 minutes and then the corresponding alcohol (2.0 mmol) was added and the reaction was stirred for 2 hours at room temperature. The solvent was removed under reduced pressure and the residue was extracted with of Et2O (15 mL × 3) and purified by column chromatography on silica gel (petroleum ether/ethyl acetate 9.5:0.5), affording the corresponding pure esters 3a–c and 4a,b.
BMImBF4 Recycling procedure
After extraction of product 3a from the solution, the catholyte was placed under vacuum at room temperature for 30 min to remove diethyl ether residues, then it was used as catholyte for a new electrolysis (see the general procedure for the esterification of trans-cinnamaldehyde to repeat the same procedure).
Methyl 3-phenylpropanoate (3a): Spectral data are consistent with those reported in the literature [42]. 1H NMR (CDCl3) δ 7.32–7.23 (m, 2H), 7.22–7.14 (m, 3H), 3.65 (s, 3H), 2.94 (t, J = 7.9 Hz, 2H), 2.62 (dd, J = 8.4 Hz, 7.3 Hz, 2H) ppm; 13C NMR (CDCl3) δ 173.3, 140.5, 128.5, 128.5, 128.3, 126.3, 51.6, 35.7, 30.9 ppm.
Benzyl 3-phenylpropanoate (3b): Spectral data are consistent with those reported in the literature [43]. 1H NMR (CDCl3) δ 7.19–7.39 (m, 10H), 5.13 (s, 2H), 2.99 (t, J = 7.0 Hz, 2H), 2.70 (t, J = 7.0 Hz, 2H) ppm; 13C NMR (CDCl3) δ 172.7, 140.4, 136.0, 128.6, 128.5, 128.3, 128.2, 126.3, 66.3, 35.9, 31.0 ppm.
Isopropyl 3-phenylpropanoate (3c): Spectral data are consistent with those reported in the literature [44]. 1H NMR (CDCl3) δ 7.28–7.36 (m, 2H), 7.19–7.27 (m, 3H), 4.99–5.11 (m, 1H), 2.99 (t, J = 7.5 Hz, 2H), 1.24 (d, J = 6.3 Hz, 6H) ppm; 13C NMR (75 MHz, CDCl3) δ 172.5, 140.6, 128.5, 128.4, 126.2, 67.8, 36.3, 31.1, 21.8 ppm.
Methyl cinnamate (4a): Spectral data are consistent with those reported in the literature [45]. 1H NMR (CDCl3) δ 7.69 (d, J = 16.0 Hz, 1H), 7.58–7.45 (m, 2H), 7.41–7.32 (m, 3H), 6.43 (d, J = 16.0 Hz, 1H), 3.78 (s, 3H) ppm; 13C NMR (CDCl3) δ 167.4, 144.8, 134.4, 130.3, 128.9, 128.1, 117.8, 51.7 ppm.
Benzyl cinnamate (4b): Spectral data are consistent with those reported in the literature [46]. 1H NMR (CDCl3) δ 7.73 (d, J = 16.0 Hz, 1H), 7.56–7.49 (m, 2H), 7.45–7.31 (m, 8H), 6.49 (d, J = 16.0 Hz, 1H), 5.26 (s, 2H) ppm; 13C NMR (CDCl3) δ 166.8, 145.2, 136.1, 136.0, 134.3, 130.4, 128.9, 128.6, 128.29, 128.26, 128.1, 117.9, 66.4 ppm.
References
-
Walden, P. Bull. Acad. Imp. Sci. St.-Petersbourg 1914, 1800.
Return to citation in text: [1] -
Hayes, R.; Warr, G. G.; Atkin, R. Chem. Rev. 2015, 115, 6357–6426. doi:10.1021/cr500411q
Return to citation in text: [1] -
Pandolfi, F.; Bortolami, M.; Feroci, M.; Fornari, A.; Scarano, V.; Rocco, D. Materials 2022, 15, 866. doi:10.3390/ma15030866
Return to citation in text: [1] -
Wasserscheid, P.; Welton, T., Eds. Ionic liquids in synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008; Vol. 1. doi:10.1002/9783527621194
Return to citation in text: [1] -
Feroci, M.; Orsini, M.; Inesi, A. Room Temperature Ionic Liquids (RTILs) Versus Volatile Organic Compounds (VOCs) in Organic Electrosynthesis: The Requirement of a Careful Comparison. Green Solvents II; Springer: Dordrecht, Netherlands, 2012; pp 435–471. doi:10.1007/978-94-007-2891-2_16
Return to citation in text: [1] -
Green, M. D.; Long, T. E. Polym. Rev. (Philadelphia, PA, U. S.) 2009, 49, 291–314. doi:10.1080/15583720903288914
Return to citation in text: [1] -
Xiao, L.; Johnson, K. E. J. Electrochem. Soc. 2003, 150, E307. doi:10.1149/1.1568740
Return to citation in text: [1] [2] -
Feroci, M.; Chiarotto, I.; Inesi, A. Curr. Org. Chem. 2013, 17, 204–219. doi:10.2174/1385272811317030003
Return to citation in text: [1] -
Schuster, O.; Yang, L.; Raubenheimer, H. G.; Albrecht, M. Chem. Rev. 2009, 109, 3445–3478. doi:10.1021/cr8005087
Return to citation in text: [1] -
Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511–3522. doi:10.1039/c2cs15333e
Return to citation in text: [1] -
Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39–92. doi:10.1021/cr940472u
Return to citation in text: [1] -
Rocco, D.; Chiarotto, I.; D'Anna, F.; Mattiello, L.; Pandolfi, F.; Rizzo, C.; Feroci, M. ChemElectroChem 2019, 6, 4275–4283. doi:10.1002/celc.201900099
Return to citation in text: [1] [2] -
Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z
Return to citation in text: [1] -
Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380
Return to citation in text: [1] -
Breslow, R. J. Am. Chem. Soc. 1958, 80, 3719–3726. doi:10.1021/ja01547a064
Return to citation in text: [1] -
Xu, L.-W.; Gao, Y.; Yin, J.-J.; Li, L.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 5317–5320. doi:10.1016/j.tetlet.2005.06.015
Return to citation in text: [1] -
Chiarotto, I.; Feroci, M.; Orsini, M.; Feeney, M. M. M.; Inesi, A. Adv. Synth. Catal. 2010, 352, 3287–3292. doi:10.1002/adsc.201000555
Return to citation in text: [1] -
Vora, H. U.; Wheeler, P.; Rovis, T. Adv. Synth. Catal. 2012, 354, 1617–1639. doi:10.1002/adsc.201200031
Return to citation in text: [1] -
Forte, G.; Chiarotto, I.; Inesi, A.; Loreto, M. A.; Feroci, M. Adv. Synth. Catal. 2014, 356, 1773–1781. doi:10.1002/adsc.201400163
Return to citation in text: [1] [2] -
Murauski, K. J. R.; Jaworski, A. A.; Scheidt, K. A. Chem. Soc. Rev. 2018, 47, 1773–1782. doi:10.1039/c7cs00386b
Return to citation in text: [1] -
Singh, S.; Singh, P.; Rai, V. K.; Kapoor, R.; Yadav, L. D. S. Tetrahedron Lett. 2011, 52, 125–128. doi:10.1016/j.tetlet.2010.10.175
Return to citation in text: [1] -
Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. doi:10.1002/anie.201711060
Return to citation in text: [1] -
Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843
Return to citation in text: [1] -
Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d
Return to citation in text: [1] -
Pandolfi, F.; Chiarotto, I.; Rocco, D.; Feroci, M. Electrochim. Acta 2017, 254, 358–367. doi:10.1016/j.electacta.2017.09.135
Return to citation in text: [1] -
Atobe, M.; Tateno, H.; Matsumura, Y. Chem. Rev. 2018, 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353
Return to citation in text: [1] -
Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360
Return to citation in text: [1] -
Folgueiras-Amador, A. A.; Wirth, T. J. Flow Chem. 2017, 7, 94–95. doi:10.1556/1846.2017.00020
Return to citation in text: [1] -
Luis, S. V.; García-Verdugo, E. Flow Chemistry: Integrated Approaches for Practical Applications; Royal Society of Chemistry: Cambridge, UK, 2019. doi:10.1039/9781788016094
Return to citation in text: [1] -
Green, R. A.; Pletcher, D.; Leach, S. G.; Brown, R. C. D. Org. Lett. 2015, 17, 3290–3293. doi:10.1021/acs.orglett.5b01459
Return to citation in text: [1] [2] -
Finney, E. E.; Ogawa, K. A.; Boydston, A. J. J. Am. Chem. Soc. 2012, 134, 12374–12377. doi:10.1021/ja304716r
Return to citation in text: [1] -
Ogawa, K. A.; Boydston, A. J. Org. Lett. 2014, 16, 1928–1931. doi:10.1021/ol500459x
Return to citation in text: [1] -
Green, R. A.; Brown, R. C. D.; Pletcher, D.; Harji, B. Electrochem. Commun. 2016, 73, 63–66. doi:10.1016/j.elecom.2016.11.004
Return to citation in text: [1] -
Chapman, M. R.; Shafi, Y. M.; Kapur, N.; Nguyen, B. N.; Willans, C. E. Chem. Commun. 2015, 51, 1282–1284. doi:10.1039/c4cc08874c
Return to citation in text: [1] -
Schotten, C.; Bourne, R. A.; Kapur, N.; Nguyen, B. N.; Willans, C. E. Adv. Synth. Catal. 2021, 363, 3189–3200. doi:10.1002/adsc.202100264
Return to citation in text: [1] -
Folgueiras-Amador, A. A.; Teuten, A. E.; Pletcher, D.; Brown, R. C. D. React. Chem. Eng. 2020, 5, 712–718. doi:10.1039/d0re00019a
Return to citation in text: [1] [2] [3] [4] -
Gurjar, S.; Sharma, S. K.; Sharma, A.; Ratnani, S. Appl. Surf. Sci. Adv. 2021, 6, 100170. doi:10.1016/j.apsadv.2021.100170
We do not have a conclusive answer for this behavior. As the reaction of electro-generated NHC with the elemental sulfur was carried out “ex-cell” we can exclude the formation of silver sulfide. It is possible however, that either the over potential for hydrogen evolution from the imidazolium is higher at the silver cathode, or that some electrode passivation takes place on the silver electrode. While we do not have direct evidence to support the latter proposal, imidazolium species are well-known as anti-corrosion additives (usually for steel) and electrochemically generated NHCs may form a stable layer on silver.
Return to citation in text: [1] -
Feroci, M.; Orsini, M.; Inesi, A. Adv. Synth. Catal. 2009, 351, 2067–2070. doi:10.1002/adsc.200900359
Return to citation in text: [1] [2] [3] -
De Sarkar, S.; Biswas, A.; Samanta, R. C.; Studer, A. Chem. – Eur. J. 2013, 19, 4664–4678. doi:10.1002/chem.201203707
Return to citation in text: [1] -
AlNashef, I. M.; Hashim, M. A.; Mjalli, F. S.; Ali, M. Q. A.-h.; Hayyan, M. Tetrahedron Lett. 2010, 51, 1976–1978. doi:10.1016/j.tetlet.2010.02.030
Return to citation in text: [1] [2] -
Sohn, S. S.; Rosen, E. L.; Bode, J. W. J. Am. Chem. Soc. 2004, 126, 14370–14371. doi:10.1021/ja044714b
Return to citation in text: [1] [2] -
Ficker, M.; Svenningsen, S. W.; Larribeau, T.; Christensen, J. B. Tetrahedron Lett. 2018, 59, 1125–1129. doi:10.1016/j.tetlet.2018.02.026
Return to citation in text: [1] -
Feroci, M.; Chiarotto, I.; Orsini, M.; Pelagalli, R.; Inesi, A. Chem. Commun. 2012, 48, 5361–5363. doi:10.1039/c2cc30371j
Return to citation in text: [1] -
Salomé, C.; Kohn, H. Tetrahedron 2009, 65, 456–460. doi:10.1016/j.tet.2008.10.062
Return to citation in text: [1] -
Dahiya, A.; Das, B.; Sahoo, A. K.; Patel, B. K. Adv. Synth. Catal. 2022, 364, 966–973. doi:10.1002/adsc.202101431
Return to citation in text: [1] -
Sato, H.; Hosokawa, S. Synthesis 2018, 50, 1343–1349. doi:10.1055/s-0036-1589162
Return to citation in text: [1]
| 38. | Feroci, M.; Orsini, M.; Inesi, A. Adv. Synth. Catal. 2009, 351, 2067–2070. doi:10.1002/adsc.200900359 |
| 19. | Forte, G.; Chiarotto, I.; Inesi, A.; Loreto, M. A.; Feroci, M. Adv. Synth. Catal. 2014, 356, 1773–1781. doi:10.1002/adsc.201400163 |
| 30. | Green, R. A.; Pletcher, D.; Leach, S. G.; Brown, R. C. D. Org. Lett. 2015, 17, 3290–3293. doi:10.1021/acs.orglett.5b01459 |
| 39. | De Sarkar, S.; Biswas, A.; Samanta, R. C.; Studer, A. Chem. – Eur. J. 2013, 19, 4664–4678. doi:10.1002/chem.201203707 |
| 7. | Xiao, L.; Johnson, K. E. J. Electrochem. Soc. 2003, 150, E307. doi:10.1149/1.1568740 |
| 8. | Feroci, M.; Chiarotto, I.; Inesi, A. Curr. Org. Chem. 2013, 17, 204–219. doi:10.2174/1385272811317030003 |
| 21. | Singh, S.; Singh, P.; Rai, V. K.; Kapoor, R.; Yadav, L. D. S. Tetrahedron Lett. 2011, 52, 125–128. doi:10.1016/j.tetlet.2010.10.175 |
| 40. | AlNashef, I. M.; Hashim, M. A.; Mjalli, F. S.; Ali, M. Q. A.-h.; Hayyan, M. Tetrahedron Lett. 2010, 51, 1976–1978. doi:10.1016/j.tetlet.2010.02.030 |
| 6. | Green, M. D.; Long, T. E. Polym. Rev. (Philadelphia, PA, U. S.) 2009, 49, 291–314. doi:10.1080/15583720903288914 |
| 22. | Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S. R. Angew. Chem., Int. Ed. 2018, 57, 5594–5619. doi:10.1002/anie.201711060 |
| 23. | Yoshida, J.-i.; Kataoka, K.; Horcajada, R.; Nagaki, A. Chem. Rev. 2008, 108, 2265–2299. doi:10.1021/cr0680843 |
| 24. | Frontana-Uribe, B. A.; Little, R. D.; Ibanez, J. G.; Palma, A.; Vasquez-Medrano, R. Green Chem. 2010, 12, 2099–2119. doi:10.1039/c0gc00382d |
| 25. | Pandolfi, F.; Chiarotto, I.; Rocco, D.; Feroci, M. Electrochim. Acta 2017, 254, 358–367. doi:10.1016/j.electacta.2017.09.135 |
| 41. | Sohn, S. S.; Rosen, E. L.; Bode, J. W. J. Am. Chem. Soc. 2004, 126, 14370–14371. doi:10.1021/ja044714b |
| 4. | Wasserscheid, P.; Welton, T., Eds. Ionic liquids in synthesis, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2008; Vol. 1. doi:10.1002/9783527621194 |
| 5. | Feroci, M.; Orsini, M.; Inesi, A. Room Temperature Ionic Liquids (RTILs) Versus Volatile Organic Compounds (VOCs) in Organic Electrosynthesis: The Requirement of a Careful Comparison. Green Solvents II; Springer: Dordrecht, Netherlands, 2012; pp 435–471. doi:10.1007/978-94-007-2891-2_16 |
| 18. | Vora, H. U.; Wheeler, P.; Rovis, T. Adv. Synth. Catal. 2012, 354, 1617–1639. doi:10.1002/adsc.201200031 |
| 19. | Forte, G.; Chiarotto, I.; Inesi, A.; Loreto, M. A.; Feroci, M. Adv. Synth. Catal. 2014, 356, 1773–1781. doi:10.1002/adsc.201400163 |
| 36. | Folgueiras-Amador, A. A.; Teuten, A. E.; Pletcher, D.; Brown, R. C. D. React. Chem. Eng. 2020, 5, 712–718. doi:10.1039/d0re00019a |
| 2. | Hayes, R.; Warr, G. G.; Atkin, R. Chem. Rev. 2015, 115, 6357–6426. doi:10.1021/cr500411q |
| 3. | Pandolfi, F.; Bortolami, M.; Feroci, M.; Fornari, A.; Scarano, V.; Rocco, D. Materials 2022, 15, 866. doi:10.3390/ma15030866 |
| 20. | Murauski, K. J. R.; Jaworski, A. A.; Scheidt, K. A. Chem. Soc. Rev. 2018, 47, 1773–1782. doi:10.1039/c7cs00386b |
| 38. | Feroci, M.; Orsini, M.; Inesi, A. Adv. Synth. Catal. 2009, 351, 2067–2070. doi:10.1002/adsc.200900359 |
| 12. | Rocco, D.; Chiarotto, I.; D'Anna, F.; Mattiello, L.; Pandolfi, F.; Rizzo, C.; Feroci, M. ChemElectroChem 2019, 6, 4275–4283. doi:10.1002/celc.201900099 |
| 40. | AlNashef, I. M.; Hashim, M. A.; Mjalli, F. S.; Ali, M. Q. A.-h.; Hayyan, M. Tetrahedron Lett. 2010, 51, 1976–1978. doi:10.1016/j.tetlet.2010.02.030 |
| 11. | Bourissou, D.; Guerret, O.; Gabbaï, F. P.; Bertrand, G. Chem. Rev. 2000, 100, 39–92. doi:10.1021/cr940472u |
| 16. | Xu, L.-W.; Gao, Y.; Yin, J.-J.; Li, L.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 5317–5320. doi:10.1016/j.tetlet.2005.06.015 |
| 17. | Chiarotto, I.; Feroci, M.; Orsini, M.; Feeney, M. M. M.; Inesi, A. Adv. Synth. Catal. 2010, 352, 3287–3292. doi:10.1002/adsc.201000555 |
| 36. | Folgueiras-Amador, A. A.; Teuten, A. E.; Pletcher, D.; Brown, R. C. D. React. Chem. Eng. 2020, 5, 712–718. doi:10.1039/d0re00019a |
| 10. | Bugaut, X.; Glorius, F. Chem. Soc. Rev. 2012, 41, 3511–3522. doi:10.1039/c2cs15333e |
| 38. | Feroci, M.; Orsini, M.; Inesi, A. Adv. Synth. Catal. 2009, 351, 2067–2070. doi:10.1002/adsc.200900359 |
| 9. | Schuster, O.; Yang, L.; Raubenheimer, H. G.; Albrecht, M. Chem. Rev. 2009, 109, 3445–3478. doi:10.1021/cr8005087 |
| 13. | Enders, D.; Niemeier, O.; Henseler, A. Chem. Rev. 2007, 107, 5606–5655. doi:10.1021/cr068372z |
| 14. | Marion, N.; Díez-González, S.; Nolan, S. P. Angew. Chem., Int. Ed. 2007, 46, 2988–3000. doi:10.1002/anie.200603380 |
| 12. | Rocco, D.; Chiarotto, I.; D'Anna, F.; Mattiello, L.; Pandolfi, F.; Rizzo, C.; Feroci, M. ChemElectroChem 2019, 6, 4275–4283. doi:10.1002/celc.201900099 |
| 28. | Folgueiras-Amador, A. A.; Wirth, T. J. Flow Chem. 2017, 7, 94–95. doi:10.1556/1846.2017.00020 |
| 26. | Atobe, M.; Tateno, H.; Matsumura, Y. Chem. Rev. 2018, 118, 4541–4572. doi:10.1021/acs.chemrev.7b00353 |
| 41. | Sohn, S. S.; Rosen, E. L.; Bode, J. W. J. Am. Chem. Soc. 2004, 126, 14370–14371. doi:10.1021/ja044714b |
| 27. | Pletcher, D.; Green, R. A.; Brown, R. C. D. Chem. Rev. 2018, 118, 4573–4591. doi:10.1021/acs.chemrev.7b00360 |
| 42. | Ficker, M.; Svenningsen, S. W.; Larribeau, T.; Christensen, J. B. Tetrahedron Lett. 2018, 59, 1125–1129. doi:10.1016/j.tetlet.2018.02.026 |
| 43. | Feroci, M.; Chiarotto, I.; Orsini, M.; Pelagalli, R.; Inesi, A. Chem. Commun. 2012, 48, 5361–5363. doi:10.1039/c2cc30371j |
| 7. | Xiao, L.; Johnson, K. E. J. Electrochem. Soc. 2003, 150, E307. doi:10.1149/1.1568740 |
| 37. |
Gurjar, S.; Sharma, S. K.; Sharma, A.; Ratnani, S. Appl. Surf. Sci. Adv. 2021, 6, 100170. doi:10.1016/j.apsadv.2021.100170
We do not have a conclusive answer for this behavior. As the reaction of electro-generated NHC with the elemental sulfur was carried out “ex-cell” we can exclude the formation of silver sulfide. It is possible however, that either the over potential for hydrogen evolution from the imidazolium is higher at the silver cathode, or that some electrode passivation takes place on the silver electrode. While we do not have direct evidence to support the latter proposal, imidazolium species are well-known as anti-corrosion additives (usually for steel) and electrochemically generated NHCs may form a stable layer on silver. |
| 36. | Folgueiras-Amador, A. A.; Teuten, A. E.; Pletcher, D.; Brown, R. C. D. React. Chem. Eng. 2020, 5, 712–718. doi:10.1039/d0re00019a |
| 36. | Folgueiras-Amador, A. A.; Teuten, A. E.; Pletcher, D.; Brown, R. C. D. React. Chem. Eng. 2020, 5, 712–718. doi:10.1039/d0re00019a |
| 33. | Green, R. A.; Brown, R. C. D.; Pletcher, D.; Harji, B. Electrochem. Commun. 2016, 73, 63–66. doi:10.1016/j.elecom.2016.11.004 |
| 46. | Sato, H.; Hosokawa, S. Synthesis 2018, 50, 1343–1349. doi:10.1055/s-0036-1589162 |
| 34. | Chapman, M. R.; Shafi, Y. M.; Kapur, N.; Nguyen, B. N.; Willans, C. E. Chem. Commun. 2015, 51, 1282–1284. doi:10.1039/c4cc08874c |
| 35. | Schotten, C.; Bourne, R. A.; Kapur, N.; Nguyen, B. N.; Willans, C. E. Adv. Synth. Catal. 2021, 363, 3189–3200. doi:10.1002/adsc.202100264 |
| 29. | Luis, S. V.; García-Verdugo, E. Flow Chemistry: Integrated Approaches for Practical Applications; Royal Society of Chemistry: Cambridge, UK, 2019. doi:10.1039/9781788016094 |
| 44. | Salomé, C.; Kohn, H. Tetrahedron 2009, 65, 456–460. doi:10.1016/j.tet.2008.10.062 |
| 30. | Green, R. A.; Pletcher, D.; Leach, S. G.; Brown, R. C. D. Org. Lett. 2015, 17, 3290–3293. doi:10.1021/acs.orglett.5b01459 |
| 31. | Finney, E. E.; Ogawa, K. A.; Boydston, A. J. J. Am. Chem. Soc. 2012, 134, 12374–12377. doi:10.1021/ja304716r |
| 32. | Ogawa, K. A.; Boydston, A. J. Org. Lett. 2014, 16, 1928–1931. doi:10.1021/ol500459x |
| 45. | Dahiya, A.; Das, B.; Sahoo, A. K.; Patel, B. K. Adv. Synth. Catal. 2022, 364, 966–973. doi:10.1002/adsc.202101431 |
© 2022 Rocco et al.; licensee Beilstein-Institut.
This is an open access article licensed under the terms of the Beilstein-Institut Open Access License Agreement (https://www.beilstein-journals.org/bjoc/terms), which is identical to the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0). The reuse of material under this license requires that the author(s), source and license are credited. Third-party material in this article could be subject to other licenses (typically indicated in the credit line), and in this case, users are required to obtain permission from the license holder to reuse the material.